")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 11
Post-marketing surveillance study of the safety and efficacy of nalfurafine hydrochloride (Remitch® capsules 2.5 μg) in 3,762 hemodialysis patients with intractable pruritus
Authors Kozono H, Yoshitani H, Nakano R
Received 7 July 2017
Accepted for publication 17 October 2017
Published 15 January 2018 Volume 2018:11 Pages 9—24
DOI https://doi.org/10.2147/IJNRD.S145720
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Hideki Kozono,* Hiroshi Yoshitani,* Ryoko Nakano*
Pharmaceutical and Medical Device Vigilance Department, Toray Industries, Inc., Tokyo, Japan
*The authors contributed equally to this work
Background: Intractable pruritus in hemodialysis patients can significantly decrease their quality of life and is also associated with poor vital prognosis. Although combined multiple causes of intractable pruritus in these patients have been identified, no existing treatments are proven to be sufficiently effective. We conducted a post-marketing surveillance to follow-up and assess the safety and efficacy of nalfurafine, a selective κ-opioid receptor agonist, for the treatment of intractable pruritus in patients undergoing hemodialysis.
Patients and methods: Hemodialysis patients with intractable pruritus from institutions in Japan who received oral nalfurafine hydrochloride between January 2010 and December 2013 were enrolled in the surveillance. Surveillance was completed in July 2015. Safety data during 1 year after nalfurafine treatment onset, and efficacy data of nalfurafine evaluating the first 12-week treatment period and the following period until 1 year after the initial dose of nalfurafine (using global assessment of the itch improvement by the physician, Visual Analog Scale, and the Shiratori’s severity scores) were collected and analyzed.
Results: In total, 3,762 patients were analyzed for safety. Adverse drug reactions were experienced by 402/3,762 (10.69%) patients. The most frequent adverse drug reactions were insomnia (127/3,762 [3.38%] patients), constipation (34 [0.90%]), somnolence (32 [0.85%]), dizziness (23 [0.61%]), nausea (13 [0.35%]), and malaise (9 [0.24%]). No patients developed dependence on nalfurafine. Nalfurafine was effective in 82.50% (2,880/3,491) of patients during the first 12 weeks and in 84.95% (2,167/2,551) on treatment during the subsequent period until 1 year after nalfurafine treatment initiation. Statistically significant decreases were reported in the Visual Analog Scale and the Shiratori’s severity scores (p<0.001).
Conclusion: Oral nalfurafine hydrochloride (from 2.5 μg/day to a maximum of 5.0 μg/day) continues to be safe and effective for the treatment of intractable pruritus in hemodialysis patients in real-world clinical settings.
Keywords: post-marketing surveillance, safety, efficacy, nalfurafine hydrochloride, pruritus, hemodialysis
Background
As of December 2015, 324,986 patients in Japan were reportedly undergoing chronic hemodialysis.1 The number of hemodialysis patients is steadily increasing, partly due to the aging population and the growing number of patients with diseases such as diabetic nephropathy.2 Dialysis technology and treatment for complications have progressed in recent years, but the complications that decrease the quality of life of patients are still present and remain an urgent unmet clinical need.
Moderate to extreme pruritus is experienced by 42% of patients undergoing hemodialysis.3 As well as significantly impacting patients’ quality of life, intractable pruritus can also result in poor prognosis.3–6 None of the current treatment options for intractable pruritus in patients with hemodialysis, which include oral antihistamines and antiallergic agents, moisturizing agents or steroids, specifically selected dialysis membranes, type B ultraviolet light (ultraviolet B) therapy, or performing hemodiafiltration, are sufficiently effective.6–9 The same is also the case for treatments that are available internationally, such as gabapentin, pregabalin, mast cell stabilizers, cholestyramine, naltrexone, or thalidomide.10–13
Several complex factors contribute to the pathogenesis of intractable pruritus, including extended nerve fibers in the epidermis, chemical mediators, and endogenous opioids.14–19 Nalfurafine hydrochloride (Remitch capsules 2.5 μg; Toray Industries, Inc., Tokyo, Japan) is a selective κ-receptor agonist that was developed in 1992. In nonclinical studies, nalfurafine suppressed scratching behavior,20,21 showed no drug dependence (unlike morphine, which agonizes the μ-receptor),21–24 and, unlike the existing κ-receptor agonists, caused no aversion.25 As such, it was considered to be a promising agent for the treatment of intractable pruritus in hemodialysis patients. Clinical trials were, therefore, conducted with nalfurafine with the aim of developing a novel anti-itch agent.
The efficacy of nalfurafine for this condition was first demonstrated in the confirmatory trial, a 14-day, multicenter, double-blind, placebo-controlled trial, in which 337 Japanese hemodialysis patients with intractable pruritus resistant to existing treatment were randomly treated with either nalfurafine (2.5 or 5.0 μg) or placebo per day.26 Nalfurafine was also demonstrated to be clinically effective in an open-label trial when administered at 5.0 μg/day for 1 year to 211 hemodialysis patients with intractable pruritus resistant to existing treatment.27 The latter long-term study found no psychological or physical dependence on nalfurafine.
Based on these results, nalfurafine was approved in Japan in 2009 for the treatment of pruritus in hemodialysis patients (for use only when sufficient efficacy is not obtained with existing therapies or treatments) and subsequently approved in Korea in 2013. A limitation of the above clinical trials was the relatively similar background of the registered patient populations. As such, we decided to conduct a post-marketing study (PMS) whose objectives were to reexamine the risks identified in the preceding trials, find hidden serious risks and missing key information, and enhance the adequate use of the agent while minimizing its risks.
Patients and methods
This surveillance was conducted in compliance with the Good Post-marketing Study Practice, or the Standard for Conducting Post-marketing Surveillance and Trials of Drugs, which is an ordinance enacted under the Japanese Pharmaceutical Affairs Law. This surveillance did not need informed consent from patients because it is not required under Japanese regulations.
Patients
Hemodialysis patients with intractable pruritus identified as receiving oral nalfurafine at any period between January 2010 and December 2013 were enrolled in the surveillance. Patients were administered nalfurafine after itch treatment with their existing therapy (ie, itch treatment approved in Japan, such as antihistamine/antiallergic agent, moisturizing agent, topical antihistamine, topical steroid, and ultraviolet B light therapy) was thought to be insufficiently effective by the physician. Surveillance was completed in July 2015. We planned to study 3,000 hemodialysis patients with intractable pruritus. The sample size was determined to enable detection of significance at the 95% CI of at least one case of a 0.1% frequency adverse drug reaction (ADR).
Design
Patients were registered at an independent patient registration center in this prospective surveillance with a 1-year observation period. Survey items included baseline patient characteristics, nalfurafine dosage regimen, hemodialysis plans, previous and concomitant treatment for pruritus, concomitant treatment for diseases other than pruritus, improvement in itch severity, laboratory test results (serum prolactin and thyroid hormones [thyroid-stimulating hormone {TSH}, free triiodothyronine, and free thyroxine] among others), dependence, and adverse events. Adverse events whose causation by nalfurafine cannot be denied were defined as ADRs.
As nonclinical studies and clinical trials had previously noted, nalfurafine induced variation of prolactin and TSH; therefore, measurements of these hormones were included in the laboratory test of this surveillance for further examination.
Also, in the preceding clinical trials, insomnia and somnolence were found in high frequency and hypomania was experienced as a psychiatric disorder; therefore, we further studied them in this surveillance.
It should be noted that the coadministration of nalfurafine with hypnotic, antianxiety, antidepressant, antipsychotic, or antiepilepsy drugs should be conducted using caution as it may enhance ADRs that affect the central nervous system. Frequencies of psychiatric and nervous system disorders were studied in this regard among the patients who received concomitant treatment using these drugs (excluding treatment drugs against adverse events).
Assessment of itch improvement
Improvement in itch severity was measured by three criteria: global assessment of the itch improvement by the physician’s diagnosis and observations from three phases (improved, stable, or aggravated), the Visual Analog Scale (VAS), and the Shiratori’s severity score.
The analysis of the global assessment of the itch improvement included only those patients whose status was considered to have “improved”, “stable”, or “aggravated”.
The VAS is an established technique used to assess a patient’s level of itch.28 The left end of a 100 mm horizontal line is labeled “no itch”, and the right end is labeled “itch as bad as it could be”. The patient marks on the line the point that most represents their itching sensation. Only those patients who had recorded their VAS values after breakfast and after evening meal, both prior to and after nalfurafine administration, were analyzed. Of the two values available, the larger VAS value was used in the analysis.
In the Shiratori’s severity score, which provided the basis for Kawashima’s classification scheme, the intensities of daytime and nighttime itching were reported using a 5-level scale.29,30 The benchmark descriptors for each level were as follows: 0 (ie, no symptoms) “daytime: no or almost no itch, nighttime: no or almost no itch”; 1 (ie, very slight) “daytime: occasional restless sensations that do not necessarily induce scratching behavior, nighttime: very little itch at bedtime that does not induce conscious scratching behavior and never disturbs sound sleep”; 2 (ie, slight) “daytime: itch that can be subdued by occasional reaching out and light scratching, nighttime: occasional itch that can be subdued by scratching and does not wake the patient”; 3 (ie, moderate) “daytime: significant scratching even in public or irritating itch causes ceaseless scratching, nighttime: the itch disrupts sleep, and although the patient can go back into sleep after scratching, the patient keeps scratching unconsciously”; and 4 (ie, fierce) “daytime: unbearable itch causes scratching and scratching only aggravates the itch sensation and interferes with work or study, nighttime: almost deprived of sleep, the patient keeps scratching ceaselessly, which only aggravates the itch sensation”. The analysis of the Shiratori’s severity score included only those patients who recorded daytime and nighttime scores both prior to and after nalfurafine administration; the larger value of the two scores was used for the analysis.
The assessments were made according to the following schedule: the global assessment of the itch improvement was checked at 12 weeks (or upon treatment interruption, if applicable) and at 1 year (or upon treatment interruption, if applicable) after the initial dose of nalfurafine. The VAS value and the Shiratori’s severity score were checked within 30 days before nalfurafine initiation. After the initial dose of nalfurafine, the VAS values and Shiratori’s severity scores were recorded at 12 weeks (or upon treatment interruption, if applicable) and at 1 year (or upon treatment interruption, if applicable).
Assessment of dependence
Dependence was evaluated with the Questionnaire of Drug Dependence comprising psychological dependence-related questions, physical dependence-related questions, and tolerance related questions for the periods up to 12 weeks and between 13 weeks and 1 year.27,31 For each period, dependence during nalfurafine treatment was measured by 10 questions. Additionally, in case treatment was interrupted during each observation period, dependence during the 4 weeks after the end of treatment was assessed by six questions. Each patient was assigned one of four options (“remarkable”, “moderate”, “slight”, or “none”). If a patient’s symptoms were described as either “remarkable” or “moderate”, the reasons expressed by the patient and the findings of the physician were also recorded for further consideration.
Statistical analysis
The global assessment of the itch improvement was calculated as the point estimate of the improvement rate. The incidences of ADRs were counted in accordance with the MedDRA/J (version 18.1) preferred terms. Fundamental statistics were calculated from the VAS values, Shiratori’s severity scores, and the levels of serum prolactin and thyroid hormones observed in each patient. They were examined by the one-sample t-test for the difference between the pre- and posttreatment values, with two-sided p-values <0.05 considered as statistically significant. SAS version 9.1.3 software (SAS Institute Inc., Cary, NC, USA) was used for the statistical analyses.
Ethics approval and consent to participate
In accordance with the Japanese Ministry of Health, Labor, and Welfare, this surveillance was conducted in compliance with the Good Post-marketing Study Practice and the Standard for Conducting Post-marketing Surveillance and Trials of Drugs, which is an ordinance enacted under Article 14, Section 4, Clause 4 and Article 14, Section 6, Clause 4 of the Japanese Law (ie, Pharmaceuticals and Medical Devices Law) for Ensuring the Quality, Efficacy, and Safety of Drugs and Medical Devices. Separate ethics approval for this surveillance and informed consent to participate in the surveillance were not required under Japanese law. It should also be noted that all original data have been completely anonymized such that the privacy of patients or facilities involved was ensured.
Results
Baseline patient characteristics
A total of 3,866 patients were enrolled at 1,023 institutions; 3,762 patients were analyzed for safety of nalfurafine (Figure 1). Excluding 2 patients who had not undergone hemodialysis at the time, a total of 3,760 patients were analyzed for efficacy. Tables 1 and 2 show the major demographic characteristics and the treatment profile of the safety analysis set, respectively. In this surveillance, compared to the proportion of all hemodialysis patients in Japan,1 the ratio of males was slightly higher at 71.00% (2,671/3,762 patients). More males were registered than females, but almost no bias in sex or age was noted among the registered patients. In this surveillance, on the basis of the mean daily dose, 87.37% (3,287/3,762 patients) were administered nalfurafine at 2.5 μg, 11.59% (436/3,762 patients) received over 2.5 μg but <5.0 μg, and 0.74% (28/3,762 patients) received 5.0 μg (Table 2). While most of the registered patients kept taking the regular mean daily dose of 2.5 μg, at least 10% of patients received the elevated dose of 5.0 μg.
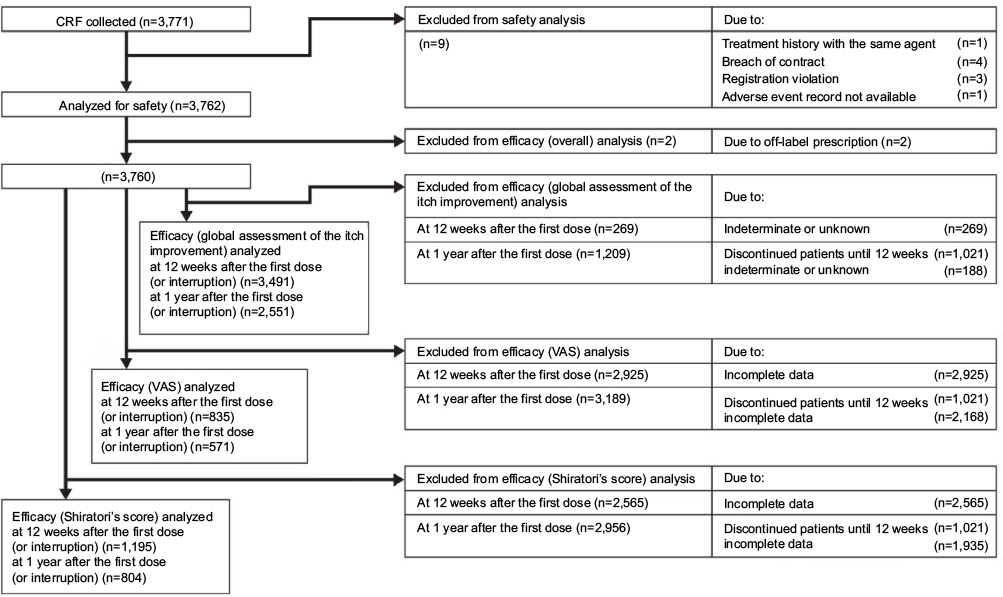 | Figure 1 Participant flow diagram. Abbreviations: CRF, case report form; VAS, Visual Analog Scale. |
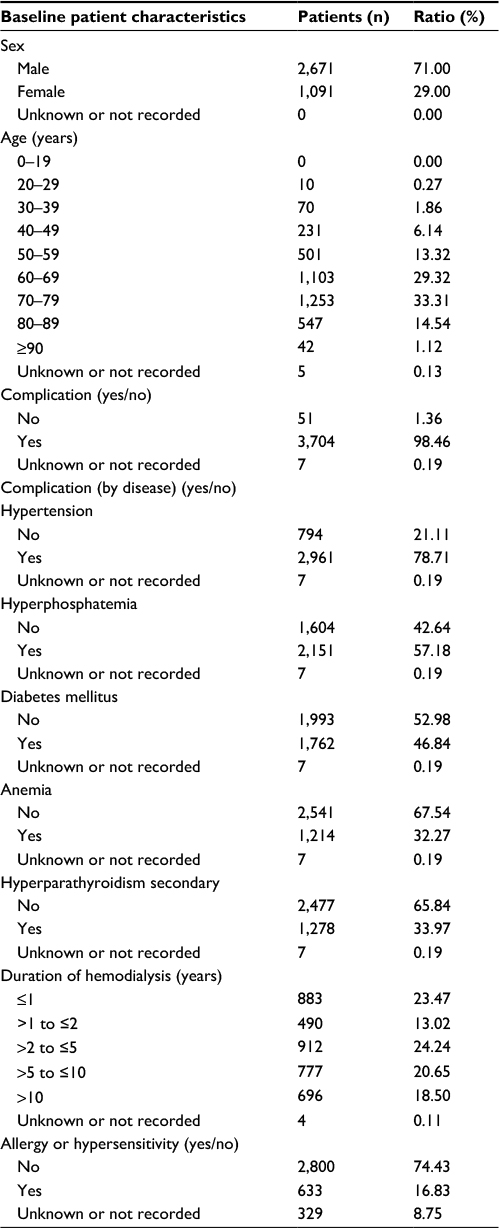 | Table 1 Baseline demography and disease characteristics |
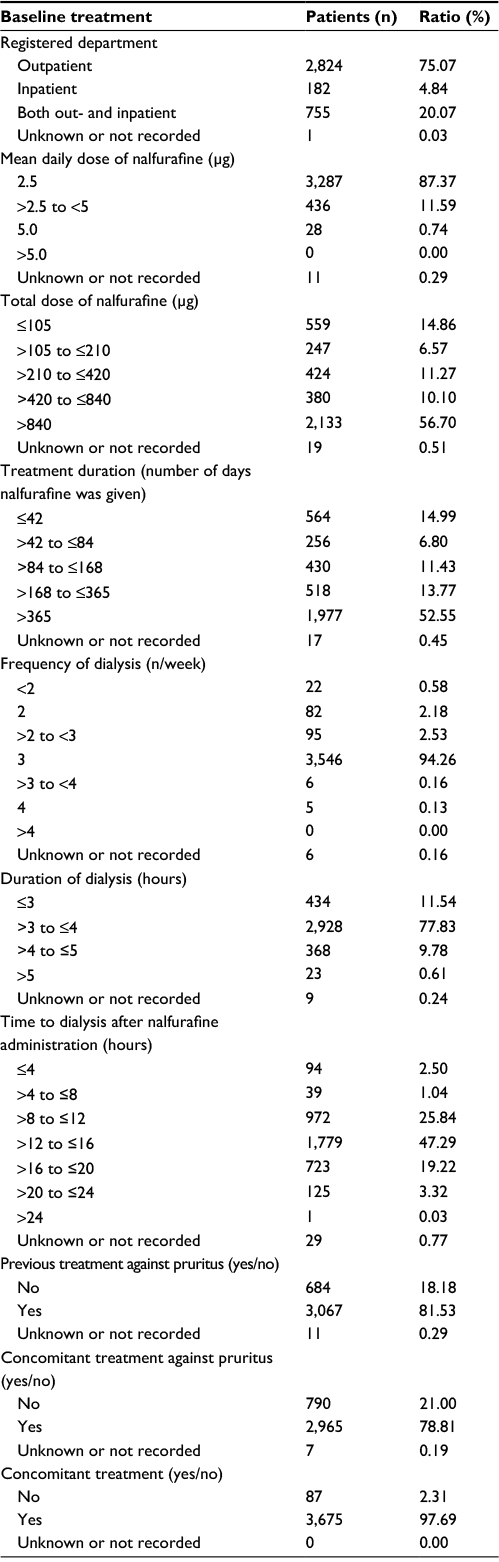 | Table 2 Summary of the treatment |
Safety
Frequency of ADRs
The frequency of all ADRs is shown in Tables 3–6. Among the 3,762 patients analyzed for safety, 402 (10.69%) patients experienced 598 incidents of ADRs. The ADRs most commonly experienced were: insomnia, constipation, somnolence, dizziness, nausea, and malaise (Tables 3–6). The ADR found among the nine patients excluded from the safety analysis was one case of pruritus. Two or more incidents of serious ADRs included cerebral infarction in six patients (0.16%), hallucinations, Alzheimer’s type dementia, and abnormal hepatic function in three patients each (0.08%), and pneumonia, gastric cancer, anemia, hyperkalemia, insomnia, dizziness, acute myocardial infarction, myocardial infarction, peripheral arterial occlusive disease, pneumonia aspiration, pulmonary edema, ileus paralytic, death, and sudden death in two patients each (0.05%).
 | Table 3 List of ADRs for infections and infestations, neoplasms benign, malignant, and unspecified (including cysts and polyps), blood and lymphatic system disorders, endocrine disorders, metabolism and nutrition disorders. Notes: ADRs are coded with the preferred terms of MedDRA/J (version 18.1). The figure beside each category of disease shows the number (%) of patients with the type of ADR, while the number of each ADR shows the number of incidents (in parentheses, the number of incidents over the total number of patients researched). Abbreviation: ADRs, adverse drug reactions. |
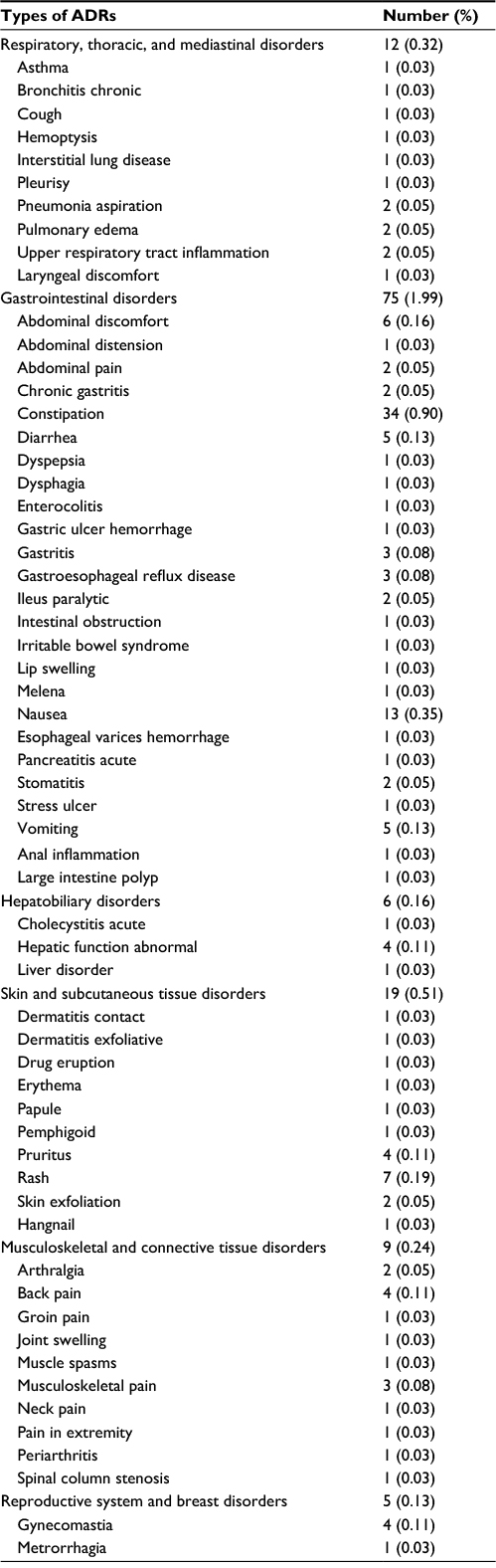 | Table 5 List of ADRs for respiratory, thoracic, and mediastinal disorders, gastrointestinal disorders, hepatobiliary disorders, skin and subcutaneous tissue disorders, musculoskeletal and connective tissue disorders, reproductive system and breast disorders. Notes: ADRs are coded with the preferred terms of MedDRA/J (version 18.1). The figure beside each category of disease shows the number (%) of patients with the type of ADR, while the number of each ADR shows the number of incidents (in parentheses, the number of incidents over the total number of patients researched). Abbreviation: ADRs, adverse drug reactions. |
 | Table 6 List of ADRs for general disorders and administration site conditions, investigations, injury, poisoning, and procedural complications. Notes: ADRs are coded with the preferred terms of MedDRA/J (version 18.1). The figure beside each category of disease shows the number (%) of patients with the type of ADR, while the number of each ADR shows the number of incidents (in parentheses, the number of incidents over the total number of patients researched). Abbreviation: ADRs, adverse drug reactions. |
Frequency of sleep disorder and other psychiatric disorders
The sleep disorders experienced included insomnia (127 [3.38%]), somnolence (32 [0.85%]), and one case (0.03%) each of initial insomnia and middle insomnia. The identified psychiatric disorders included 127 (3.38%) cases of insomnia, 7 (0.19%) cases of hallucination, 5 (0.13%) each of delirium and restlessness, and 4 (0.11%) each of depression and irritability (Table 4).
 | Table 4 List of ADRs for psychiatric disorders, nervous system disorders, eye disorders, ear and labyrinth disorders, cardiac disorders, vascular disorders. Notes: ADRs are coded with the preferred terms of MedDRA/J (version 18.1). The figure beside each category of disease shows the number (%) of patients with the type of ADR, while the number of each ADR shows the number of incidents (in parentheses, the number of incidents over the total number of patients researched). Abbreviation: ADRs, adverse drug reactions. |
Coadministration of hypnotic, antianxiety, antidepressant, antipsychotic, or antiepilepsy drugs
The number of ADRs was counted for each of the three categories of concomitant drugs (Table 7). Antidepressants are included in the residual category of “other antipsychotic drugs”. There was no major difference among the categories for the most frequent insomnia.
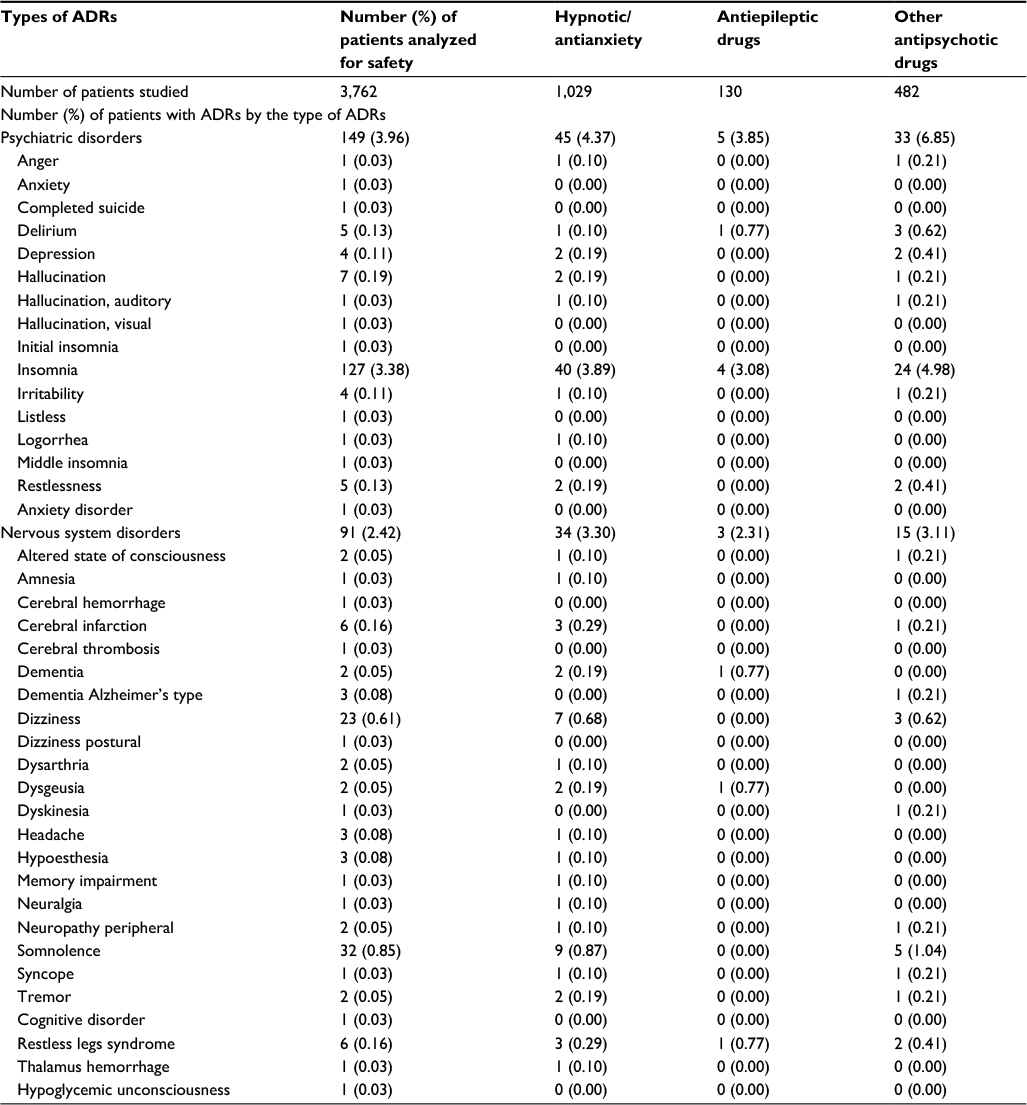 | Table 7 Safety of combination therapies with hypnotic, antianxiety, antidepressant, antipsychotic, or antiepilepsy drugs Notes: ADRs are coded with the preferred terms of MedDRA/J (version 18.1). The figure beside each category of disease shows the number (%) of patients with the type of ADR, while the number of each ADR shows the number of incidents (in parentheses, the number of incidents over the total number of patients researched). Abbreviation: ADRs, adverse drug reactions. |
Changes in serum prolactin and thyroid hormones
Measurements were obtained before and after the onset of nalfurafine treatment. A statistically significant decrease of serum TSH was observed, with the pretreatment level being 2.39±2.37 to 2.00±1.90 μIU/mL after the initiation of nalfurafine treatment (p=0.035; Table 8). Although two adverse events in two patients of lower TSH levels were observed, causal relationships between the adverse events and nalfurafine were rejected.
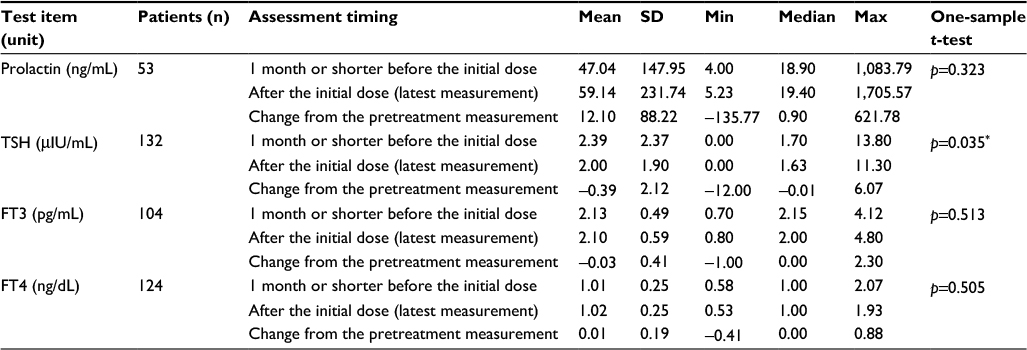 | Table 8 Assessment of serum prolactin and thyroid hormones before and after the treatment onset Notes: The difference between the measurements at 1 month or shorter before the initial dose and the latest measurement after the initial dose. *p<0.05; significance was tested by one-sample t-test. Abbreviations: FT3, free triiodothyronine; FT4, free thyroxine; max, maximum; min, minimum; TSH, thyroid-stimulating hormone. |
Dependence
We collected 3,265 “on-treatment” responses and 530 “off-treatment” responses on evaluating the first 12 weeks, and 2,324 “on-treatment” responses and 257 “off-treatment” responses on evaluating the period from 13 weeks to 1 year. None of the nine patients excluded from the safety analysis were assigned as “remarkable” or “moderate” to any of the questions. “Remarkable” or “moderate” evaluations were most often obtained for three “on-treatment” questions (ie, “Do you want to continue taking this drug?”; “Do you think this drug became less effective?”; “Do you want to take this drug in a larger dosis?”), as shown in Tables 9 and 10. Dependence was further examined by analyzing the patients’ own explanations and the physicians’ findings that accompanied “remarkable” or “moderate” responses, in cases where such concurring feedback was frequent for particular question items. First, of the patients in the category of “remarkable” or “moderate” for the psychological dependence-related question “Do you want to continue taking this drug?”, as many as 434 in the first 12 weeks and 252 from 13 weeks to 1 year mentioned reasons such as “withdrawal from the agent may aggravate pruritus”, simply anticipating symptom relief associated with use of the agent. Likewise, none of the remainder who were all rated “moderate” for the same question item had any reasons or expert observations that suggested dependence. All patients answering “remarkable” or “moderate” for the psychological dependence-related question “Do you want to take this drug in a larger dosis?” expressed hope for greater efficacy. None of the “remarkable” or “moderate” responses to any other question items were accompanied by reasons or expert findings that suggested psychological dependence. Then, very few patients were assigned as being “remarkable” or “moderate” for the physical dependence-related questions. Finally, while some of the patients who responded “remarkable” or “moderate” to the tolerance-related question “Do you think this drug became less effective?” (in the first 12 weeks: 40 patients; from 13 weeks to 1 year: 26 patients) were evaluated to be showing “weaker efficacy”, no reasons or physician findings suggesting the dependence were given.
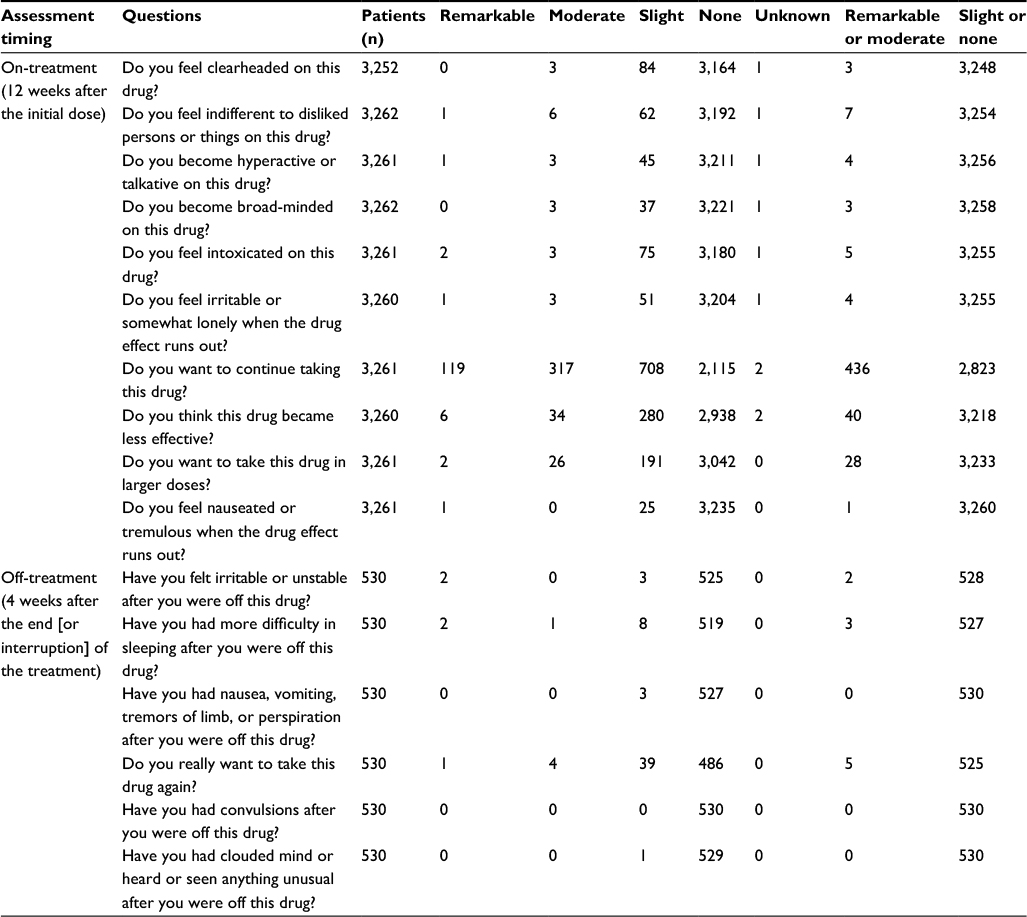 | Table 9 Summary of results of dependence (psychological dependence, physical dependence, tolerance) assessment (up to 12 weeks) |
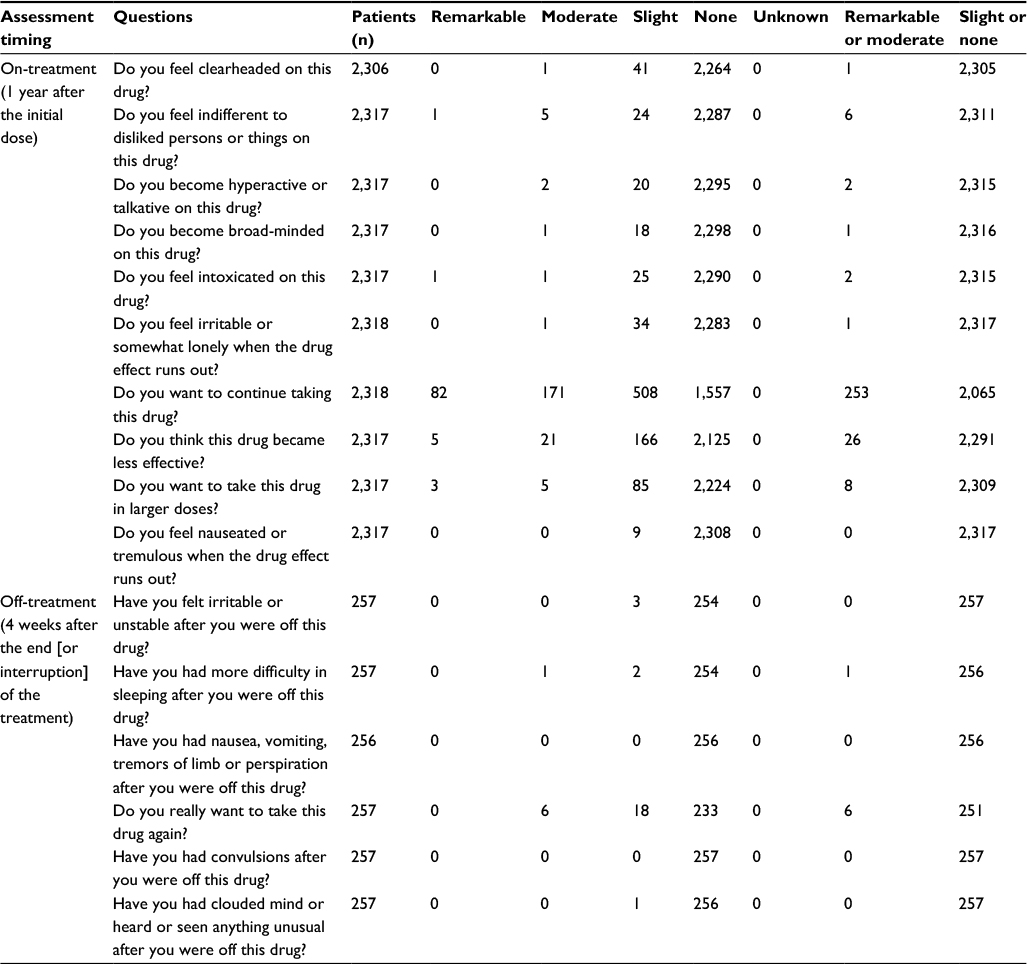 | Table 10 Summary of results of dependence (psychological dependence, physical dependence, tolerance) assessment (13 weeks–1 year) |
Efficacy
Global assessment of the itch improvement
At 12 weeks, 3,491 patients were analyzed, excluding 269 whose data were indeterminate or unknown. At 1 year, 2,551 patients were analyzed, while 1,021 who discontinued until 12 weeks and 188 whose data were indeterminate or unknown were excluded from the analysis (Figure 1). Nalfurafine was determined to be effective (ie, as evidenced by improvement in itch severity) in 82.50% (2,880/3,491 patients) of patients at 12 weeks and 84.95% (2,167/2,551 patients) of patients at 1 year.
Visual Analog Scale
At 12 weeks, 835 patients were analyzed, excluding 2,925 who had incomplete data. At 1 year, 571 patients were analyzed, while 1,021 who discontinued until 12 weeks and 2,168 who had incomplete data were excluded from the analysis (Figure 1). The VAS value decreased significantly from the pretreatment level of 75.8±19.2 to 38.6±26.5 mm at 12 weeks (p<0.001). Furthermore, the 34.3±26.0 mm VAS value at 1 year was also lower than the baseline level of 76.6±19.1 mm; the decrease was statistically significant (p<0.001; Figure 2).
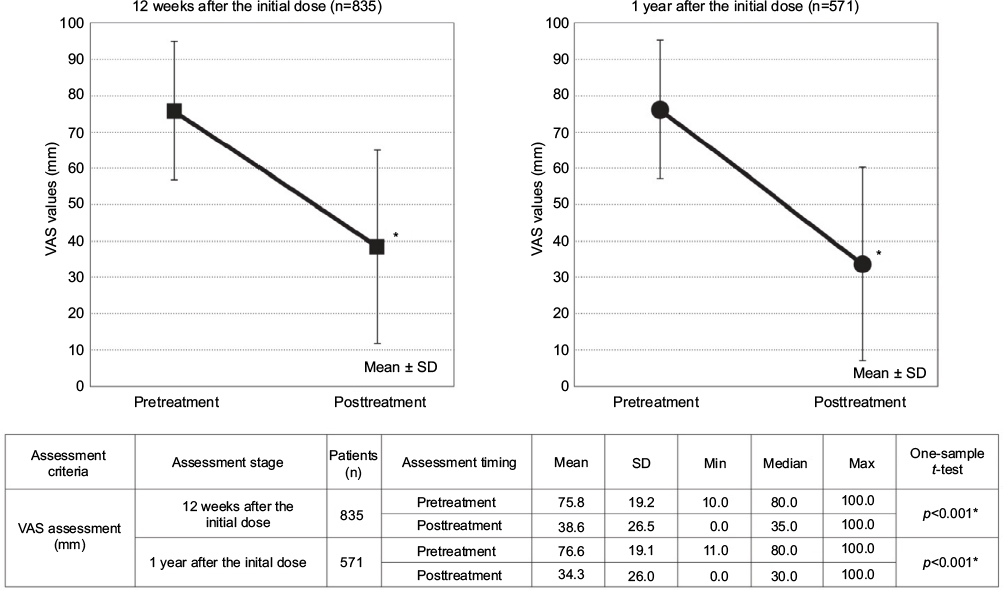 | Figure 2 VAS values (mm) scores before and after the treatment onset. Note: *p<0.001; significance was tested by one-sample t-test of the posttreatment figures against the pretreatment figures. Abbreviations: max, maximum; min, minimum; VAS, Visual Analog Scale. |
Shiratori’s severity score
At 12 weeks, 1,195 patients among the efficacy analysis population were analyzed, excluding 2,565 who had incomplete data. At 1 year, 804 patients among the efficacy analysis population were analyzed, excluding 1,021 who discontinued until 12 weeks and 1,935 who had incomplete data (Figure 1). The decrease in the Shiratori’s severity score from the pretreatment level of 3.3±0.6 to 1.8±1.0 at 12 weeks was statistically significant (p<0.001). The score of 1.6±1.0 at 1 year was also significantly lower than the pretreatment value of 3.3±0.6 (p<0.001; Figure 3).
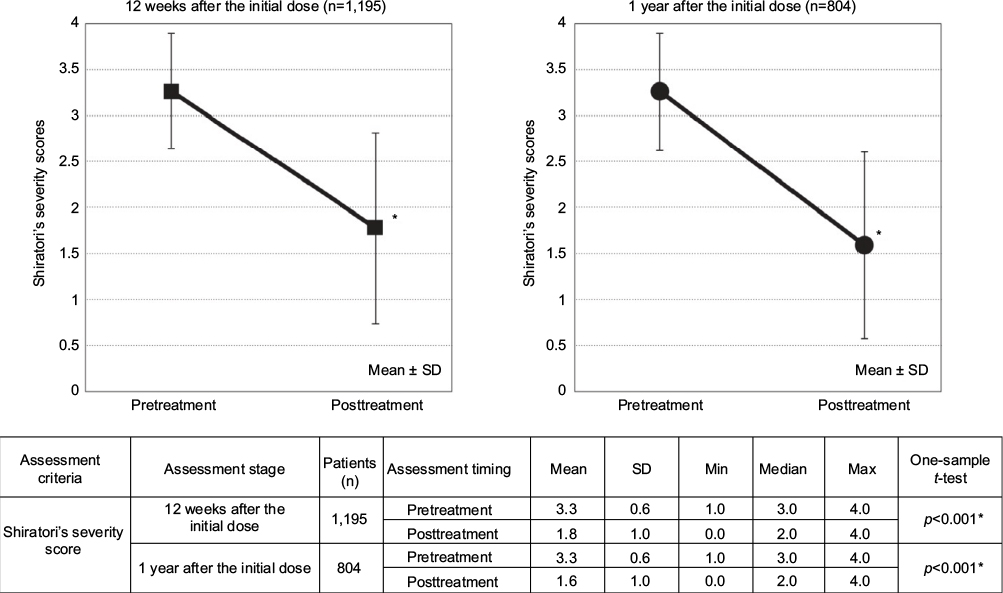 | Figure 3 Shiratori’s severity scores before and after the treatment onset. Note: *p<0.001; significance was tested by one-sample t-test of the posttreatment figures against the pretreatment figures. Abbreviations: max, maximum; min, minimum. |
Discussion
This present PMS analyzed the safety and efficacy of nalfurafine in hemodialysis patients with intractable pruritus. As some items included in this surveillance are not required prerequisites for regulatory PMS, data could not be obtained unless the specific items were routinely measured by physicians at a participating institution. Consequently, a considerable number of cases with incomplete data had to be excluded from the efficacy analysis, which included strictly those patients whose evaluation was available twice daily, both before and after administration of nalfurafine. It may be argued that such exclusion has resulted in a biased body of patients. However, it is noteworthy that the number of patients was sufficiently large to conduct the safety analysis, which is an important consideration given that the primary purposes of this surveillance were to reevaluate known risks in actual clinical settings and detect any new or hidden safety risks.
The recommended dose of nalfurafine for adults is 2.5 μg once daily, administered orally after an evening meal or before bedtime. The dose can be increased in accordance with the symptoms; the maximum dose is 5.0 μg once daily. The frequency of ADRs in the elevation-to-5.0 μg group was 9.68% (42/434 patients); the frequency was 10.92% (359/3,287 patients) in the 2.5 μg group. Global assessment of the itch improvement showed improvement at 1 year in 86.08% (186/2,162) and 78.77% (282/358) of patients among the regular 2.5 μg dose and the elevation-to-5.0 μg group patients, respectively. Increasing the dose from 2.5 to 5.0 μg was because of more severe pruritus and/or due to insufficient efficacy. Despite supposedly more severe baseline disease of the elevation-to-5.0 μg group, symptom improvement was evident in nearly 80% of patients with similar rate of ADRs. Dose elevation occurred more frequently during the period between 2 and 4 weeks after nalfurafine treatment onset than any other period. Some institutions are reported to switch to 5.0 μg nalfurafine if 2.5 μg dose does not prove to be sufficiently effective after a 2–4-week observation period.32 Therefore, it seems a reasonable strategy to schedule an assessment after a set period of time since initiation of treatment with nalfurafine 2.5 μg, so that the dose may be elevated to 5.0 μg if required.
Those patients who have a shorter history of hemodialysis are anticipated to be in a less stable clinical condition. In this surveillance, 23.47% (883/3,762) of patients had a history of 1 year or shorter. ADRs were experienced by 9.17% (81/883) of these “novice” patients; an improved level of itch by global assessment at 1 year was achieved by 87.09% (553/635) of this population. However, no difference in these safety and efficacy levels was noted among the patients of hemodialysis induction of <1 year and the patients of hemodialysis induction of 1 year or more. Therefore, nalfurafine is demonstrated to be safe and effective in early-stage hemodialysis patients.
The ADR incidence of 10.69% (402/3,762 patients) in this surveillance was lower than the incidences of 25.0% (28/112 patients, 2.5 μg arm), 35.1% (40/114 patients, 5 μg arm) in the placebo-controlled, double-blind study, or 48.8% (103/211patients, 5 μg arm) in the long-term study.26,27 The type of most frequently experienced ADRs was the same as previous studies, and the incidence of these ADRs was essentially no different from those noted in previous studies. We also investigated the onset timing of the three most frequently experienced ADRs (ie, insomnia, constipation, and somnolence). Insomnia was experienced within 3 days by 26.77% (34/127 patients) and within 2 weeks by 55.90% (71/127 patients). Constipation began within 3 days in 20.58% (7/34 patients) and within 2 weeks in 44.12% (15/34 patients). The onset of somnolence was noted within 3 days in 21.88% (7/32 patients) and within 2 weeks in 56.25% (18/32 patients). It, therefore, seems that the common ADRs of nalfurafine may set in at a relatively early stage of treatment.
When we examined the frequency of sleep disorders and psychiatric disorders among the ADRs in comparison with the frequency of such ADRs in preceding clinical trials for hemodialysis patients, no significant difference or noteworthy issues were found. The only ADR with a ≥3% frequency was insomnia (3.38%). Its frequency was lower than that of 7.1% (8/112 patients, 2.5 μg arm), 14.0% (16/114 patients, 5 μg arm) in the placebo-controlled, double-blind study, or 19.4% (41/211 patients, 5 μg arm) in the long-term study.26,27 This surveillance found no previously unknown hidden risks related to sleep disorders or psychiatric disorders.
The frequencies of psychiatric disorders and nervous system disorders associated with nalfurafine coadministration with central nervous system agent were both lower than the values found in preceding domestic clinical trials for hemodialysis patients, namely, 16.42% (100/609, at approval) for psychiatric disorders and 6.73% (41/609, at approval) for nervous system disorders. A relatively higher frequency of psychiatric disorders was found in patients who used “other antipsychotic drugs”, but the number of patients who experienced each specific type of ADRs, excluding insomnia, was not more than three. Insomnia was the most common ADR associated with coadministration (4.98% [24/482] of patients). However, the frequency was lower than that observed in the preceding domestic clinical trials for hemodialysis patients at 15.27% (93/609, at approval). This surveillance found no previously unknown latent risks related to psychiatric disorders or nervous system disorders.
Preceding clinical trials had noted one patient who experienced temporary gynecomastia. This surveillance also found four patients with gynecomastia, urging sufficient attention to increased levels of serum prolactin. We noticed a few types of ADRs that could possibly manifest a lower TSH level. Three incidents in two patients of Graves’ disease, hyperthyroidism, and thyrotoxic crisis (one each) were reported, but they could not be included in the analysis comparing the pre- and posttreatment levels because their serum TSH levels had not been recorded. There was also one such adverse event whose association with nalfurafine administration had been rejected; one incident of goiter was reported in a patient, but the patient was excluded from the analysis because serum TSH level of the patient was not available. Therefore, while there appears to be no previously unknown or hidden risks in relation to serum prolactin, the investigation of nalfurafine’s potential effect on thyroid hormones can be considered to be insufficient. The effect of nalfurafine on the endocrine system had been confirmed in previous nonclinical and clinical trials. The agent appears to induce endocrinal changes by actions common to those of opioids, which are known to act on the central nervous system and may trigger endocrinal disorders.32 Therefore, tests as appropriate are recommended during administration of nalfurafine.
In this Questionnaire of Drug Dependence, none of the patients’ own explanations or physicians’ observations suggested possibilities of dependence (psychological dependence, physical dependence, or tolerance). Although this surveillance noted no previously unknown risks of developing dependence on the agent, long-term administration of the agent seems to increase the risk of a patient developing dependence. It is, therefore, important that we continue to collect clinical data related to dependence issues.
Efficacy was shown by the significantly decreased VAS values at both 12 weeks and 1 year. Since this surveillance lacks a placebo arm, comparison with previous clinical trials cannot be made. Efficacy assessment in the placebo-controlled, double-blind study was conducted by checking the VAS values before and 14 days after the initial dose of nalfurafine.26 The changes in VAS were 15.31 mm (n=111; 95% CI, 11.53–19.09), 24.45 mm (n=112; 95% CI, 20.68–28.21), and 23.44 mm (n=114; 95% CI, 19.78–27.11) in the placebo arm, nalfurafine 2.5 μg arm, and 5 μg arm, respectively. The long-term study found a VAS change of 43.88 mm (n=145; 95% CI, 39.60–48.16) between pretreatment and 52 weeks after nalfurafine treatment onset.27 The change of VAS in this surveillance is equivalent to those of above trials. Moreover, a similar tendency was found in the Shiratori’s severity score. The results of the global assessment of the itch improvement showed improvement both at 12 weeks and 1 year. In contrast to the VAS and the Shiratori’s severity score that reflected the patient’s subjective self-assessment, the global assessment of the itch improvement was determined by physicians evaluating the degree of patients’ pruritus. Interestingly, this surveillance demonstrated the itch-reducing efficacy of nalfurafine from the perspective of both patients and experts.
Limitations
This surveillance is a prospective observation based on predetermined survey items. It lacks a control arm. As such, interpretation of the survey results has certain limitations inherent in this standard approach. The limitation of this surveillance is that the number of the patients between safety analysis and efficacy analysis was very different. In particular, the number of patients whose prolactin was measured was very small. Furthermore, efficacy was not examined for each dose at 12 weeks and 1 year after treatment.
Conclusion
This surveillance found sufficient safety and efficacy of nalfurafine as an indication for improvement of intractable pruritus in hemodialysis patients. Regarding the safety profile of this agent, no previously unknown hidden risks were identified. In May 2015, nalfurafine was approved in Japan for the additional indication for improvement of pruritus in chronic liver disease patients (use only when sufficient efficacy is not obtained with existing therapies or treatments). Application for another indication, that is, improvement of pruritus in peritoneal dialysis patients, has been filed, and the approval process is ongoing. In order to promote the proper and appropriate use of nalfurafine, we will continue to accumulate further safety and efficacy data, including the incidents of ADRs.
Acknowledgments
We would like to thank the physicians, nurses, and staff of the participating institutions for their cooperation and assistance in this post-marketing surveillance. This post-marketing surveillance was funded by Toray Industries, Inc.
The surveillance was sponsored by Toray Industries, Inc., and Torii Pharmaceutical Co., Ltd. The statistical analyses were jointly planned by Toray Industries, Inc., and EPS Corporation, and performed by the EPS Corporation. Medcore Associates, Inc., provided editorial support. Toray Industries, Inc., covered all financial costs for the aforementioned services.
Author contributions
Hideki Kozono contributed to the design of this surveillance, conducted the surveillance, planned the statistical analyses, and prepared a draft of the present manuscript. Hiroshi Yoshitani conducted the surveillance and planned the statistical analyses. Ryoko Nakano contributed to designing this surveillance and conducting the surveillance. All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Hideki Kozono, Hiroshi Yoshitani, and Ryoko Nakano are employees of Toray Industries, Inc., and report no other conflicts of interest in this work.
References
An overview of regular dialysis treatment in Japan as of Dec. 31, 2015. Japanese Society for Dialysis Therapy (in Japanese). Available from: http://docs.jsdt.or.jp/overview/. Accessed August 1, 2017. Japanese. | ||
Masakane I, Nakai S, Ogata S, et al. An overview of regular dialysis treatment in Japan (as of December 31, 2013). Ther Apher Dial. 2015;19(6):540–574. | ||
Pisoni RL, Wikström B, Elder SJ, et al. Pruritus in hemodialysis patients: international results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Nephrol Dial Transpant. 2006;21(12):3495–3505. | ||
Yamada S, Sakurai H, Kasuga H, Kawahara H. Investigation of the status of uremic pruritus in hemodialysis patients and the efficacy of nalfurafine hydrochloride – questionnaire administered to 1936 patients from 17 clinics in Tokai area of Japan. J Jpn Soc Dial Ther. 2012;45:1133–1140. Japanese. | ||
Omori K, Aoike I, Aoyagi H, et al. Risk factors for uremic pruritus in long-term hemodialysis patients. J Jpn Soc Dial Ther. 2001;34:1469–1477. Japanese. | ||
Narita I, Aichi B, Omori K, et al. Etiology and prognostic significance of severe uremic pruritus in chronic hemodialysis patients. Kidney Int. 2006;69(9):1626–1632. | ||
Danno K. A perfect guide of itch therapy indispensable to dialysis institutions. Kinpodo. 2008:1–45. Japanese. | ||
Krajnik M, Zylicz Z. Understanding pruritus in systemic disease. J Pain Symptom Manage. 2001;21(2):151–168. | ||
Szepietowski JC, Schwartz RA. Uremic pruritus. Int J Dermatol. 1998;37(4):247–253. | ||
Simonsen E, Komenda P, Lerner B, et al. Treatment of uremic pruritus: a systematic review. Am J Kidney Dis. 2017;6386(17):30781–30783. | ||
Combs SA, Teixeira JP, Germain MJ. Pruritus in kidney disease. Semin Nephrol. 2015;35(4):383–391. | ||
Mettang T, Kremer AE. Uremic pruritus. Kidney Int. 2015; 87(4):685–691. | ||
Manenti L, Tansinda P, Vaglio A. Uraemic pruritus: clinical characteristics, pathophysiology and treatment. Drugs. 2009;69(3):251–263. | ||
Kumagai H, Saruta T, Matsukawa S, Utsumi J. Prospects for a novel κ-opioid receptor agonist, TRK-820, in uremic pruritus. In Yosipovitch G, Greaves MW, Fleischer JA, Mc-Glone F, editors. Itch: Basic Mechanisms and Therapy. New York, NY: Marcel Dekker; 2004:279–286. | ||
Taneda K, Tominaga M, Negi O, et al. Evaluation of epidermal nerve density and opioid receptor levels in psoriatic itch. Br J Dermatol. 2011;165(2):277–284. | ||
Thornton JR, Losowsky MS. Plasma beta endorphin in cirrhosis and renal failure. Gut. 1991;32(3):306–308. | ||
Kumagai H, Hayashi M, Saruta T. Contribution of endogenous opioid to itch in chronic hemodialysis patients. Rinsho Yakuri/Japanese J Clin Pharmacology and Therapeutics. 2000; doi:10.3999/jscpt.31.457. Japanese. | ||
Peer G, Kivity S, Agami O, et al. Randomized crossover trial of naltrexone in uraemic pruritus. Lancet. 1996;348(9041):1552–1554. | ||
Pauli-Magnus C, Mikus G, Alscher DM, et al. Naltrexone does not relieve uremic pruritus: results of a randomized, double-blind, placebo-controlled crossover study. J Am Soc Nephrol. 2000;11(3):514–519. | ||
Togashi Y, Umeuchi H, Okano K, et al. Antipruritic activity of the kappa-opioid receptor agonist, TRK-820. Eur J Pharmacol. 2002;435(2–3):259–264. | ||
Umeuchi H, Togashi Y, Honda T, et al. Involvement of central mu-opioid system in the scratching behavior in mice, and the suppression of it by the activation of kappa-opioid system. Eur J Pharmacol. 2003;477(1):29–35. | ||
Nakao K, Hirakata M, Miyamoto Y, Kainoh M, Wakasa Y, Yanagita T. Nalfurafine hydrochloride, a selective κ opioid receptor agonist, has no reinforcing effect on intravenous self-administration in rhesus monkeys. J Pharmacol Sci. 2016;130(1):8–14. | ||
Kawai K, Hayakawa J, Miyamoto T, et al. Design, synthesis, and structure-activity relationship of novel opioid kappa-agonists. Bioorg Med Chem. 2008;16(20):9188–9201. | ||
Nagase H, Hayakawa J, Kawamura K, et al. Discovery of a structurally novel opioid kappa-agonist derived from 4,5-epoxymorphinan. Chem Pharm Bull (Tokyo). 1998;46(2):366–369. | ||
Tsuji M, Takeda H, Matsumiya T, Nagase H, Narita M, Suzuki T. The novel kappa-opioid receptor agonist TRK-820 suppresses the rewarding and locomotor-enhancing effects of morphine in mice. Life Sci. 2001;68(15):1717–1725. | ||
Kumagai H, Ebata T, Takamori K, Muramatsu T, Nakamoto H, Suzuki H. Effect of a novel kappa-receptor agonist, nalfurafine hydrochloride, on severe itch in 337 haemodialysis patients: a Phase III, randomized, double-blind, placebo-controlled study. Nephrol Dial Transplant. 2010;25(4):1251–1257. | ||
Kumagai H, Ebata T, Takamori K, et al. Efficacy and safety of a novel κ-agonist for managing intractable pruritus in dialysis patients. Am J Nephrol. 2012;36(2):175–183. | ||
Wahlgren CF, Ekblom A, Hägermark O. Some aspects of the experimental induction and measurement of itch. Acta Derm Venereol. 1989;69(3):185–189. | ||
Kawashima M, Tango T, Noguchi T, Inagi M, Nakagawa H, Harada S. Addition of fexofenadine to a topical corticosteroid reduces the pruritus associated with atopic dermatitis in a 1-week randomized, multicentre, double-blind, placebo-controlled, parallel-group study. Br J Dermatol. 2003;148(6):1212–1221. | ||
Kawashima M, Harada S, Tango T. Evaluation of itch based on a new rating scale using patient diary. Jpn J Clin Dermatol. 2002;56:692–697. Japanese. | ||
Kurihara M, Jimbo M, Hirose T, et al. Double-blind comparison of clinical effects of ID-540, (fludiazepam) diazepam and placebo on psychoneurotic patients and a tentative draft of dependency questionnaire. Rinsho Hyoka (Clinical Evaluation). 1977;5:341–368. apanese. | ||
Takahashi N, Yoshizawa T, Kumagai J, et al. Response of patients with hemodialysis-associated pruritus to new treatment algorithm with nalfurafine hydrochloride: a retrospective survey-based study. Renal Replacement Therapy. 2016;2:27. | ||
Grossman A. Brain opiates and neuroendocrine function. Clin Endocrinol Metab. 1983;12(3):725–746. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.