")
Back to Journals » Medical Devices: Evidence and Research » Volume 8
Post-market clinical research conducted by medical device manufacturers: a cross-sectional survey
Authors Ross JS , Blount K, Ritchie J, Hodshon B, Krumholz H
Received 16 February 2015
Accepted for publication 30 April 2015
Published 27 May 2015 Volume 2015:8 Pages 241—249
DOI https://doi.org/10.2147/MDER.S82964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Joseph S Ross, Katrina L Blount, Jessica D Ritchie, Beth Hodshon, Harlan M Krumholz
Center for Outcomes Research and Evaluation, Yale-New Haven Hospital, New Haven, CT, USA
Background: In the US, once a medical device is made available for use, several requirements have been established by the US Food and Drug Administration (FDA) to ensure ongoing post-market surveillance of device safety and effectiveness. Our objective was to determine how commonly medical device manufacturers initiate post-market clinical studies or augment FDA post-market surveillance requirements for higher-risk devices that are most often approved via the FDA's pre-market approval (PMA) pathway.
Methods and results: We conducted a cross-sectional survey of 47 manufacturers with operations in California, Minnesota, and Massachusetts who market devices approved via the PMA pathway. Among 22 respondents (response rate =47%), nearly all self-reported conducting post-market clinical research studies, commonly between 1 and 5; only 1 respondent reported never conducting post-market clinical research studies. While manufacturers most often engaged in these studies to satisfy FDA requirements, other reasons were reported, including performance monitoring and surveillance and market acceptance initiatives. Risks of conducting and not conducting post-market clinical research studies were described through open-ended response to questions.
Conclusion: Medical device manufacturers commonly initiate post-market clinical studies at the request of the FDA. Clinical data from these studies should be integrated into national post-market surveillance initiatives.
Keywords: FDA, PMA pathway, post-market surveillance
Introduction
In the US, once a medical device is made available on the market for use, several requirements and regulations have been established by the US Food and Drug Administration (FDA) to ensure ongoing post-market surveillance of device safety and effectiveness.1 These include mandatory reporting of certain device-related adverse events, such as deaths and serious injuries, and product problems, such as device malfunctions. In addition, post-market surveillance studies can be required under section 522 of the Federal Food, Drug and Cosmetic Act for higher-risk devices. Lastly, post-approval studies, including clinical trials and product registries, are often required at the time of approval for medical device applications evaluated through the pre-market approval (PMA), humanitarian device exemption, or product development protocol pathways to help assure and better understand safety and effectiveness of the approved device.
Despite these rules and regulations to facilitate post-market surveillance, there are ongoing questions about whether the current system is optimally structured to ascertain sufficient information for post-market performance evaluation of medical devices.2 The FDA is actively engaged in an effort to strengthen the national post-market surveillance system, with the goal of developing a life-cycle approach to product evaluation that integrates pre-market and post-market evaluations and makes better use of existing sources of device information, including administrative claims and electronic medical record data, while simultaneously establishing registries for select products.3,4 However, unknown is whether and how commonly medical device manufacturers engage in post-market performance evaluation beyond FDA requirements, or whether these evaluations augment FDA requirements by being broader in scope, and whether this information can be better integrated into the national post-market surveillance system.
There are a number of reasons why medical device manufacturers might initiate post-market studies or augment FDA requirements, including internal performance monitoring, design and product maintenance, as well as to evaluate expanded indications for use. This information will be of particular interest for higher-risk medical devices, which are most often approved via the FDA’s PMA pathway, as these products require more intensive post-market safety surveillance. Accordingly, we conducted a cross-sectional survey of medical device manufacturers with operations in California, Minnesota, and Massachusetts, three states in which medical device manufacturers are known to cluster, to better understand post-market clinical research for their products approved via the FDA’s PMA pathway and inform ongoing FDA initiatives, as well as the public’s understanding of medical device post-market surveillance.
Methods
Study design and sample
We conducted a cross-sectional survey from November 2013 to January 2014 of medical device manufacturers with at least one marketed medical device that had been approved via the FDA’s PMA pathway. To assemble a list of potential respondents (Figure 1), we contacted four state-based trade organizations that represent medical device manufacturers and advocate on their behalf: California Healthcare Institute (also known as CHI),5 based in California; Indiana Medical Device Manufacturers Council (also known as IMDMC),6 based in Indiana; LifeScience Alley (also known as LSA),7 based in Minnesota; and the Massachusetts Medical Device Industry Council (MassMEDIC),8 based in Massachusetts. Trade organizations in these four states were selected based on conversations with stakeholders, including manufacturers, the FDA, and the national trade organizations AdvaMed, the Advanced Medical Technology Association, and the Medical Device Manufacturers Association (also known as MDMA), who suggested that the preponderance of manufacturers with products approved via the FDA’s PMA pathway were based in these four states. CHI, LSA, and MassMEDIC each agreed to partner in the survey, whereas IMDMC declined participation.
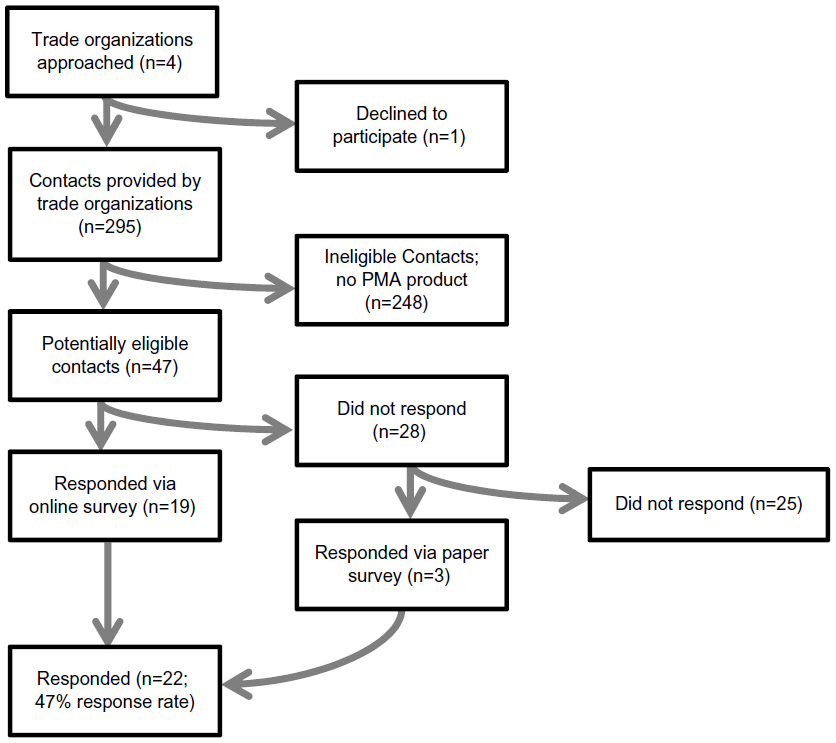 | Figure 1 Flowchart of survey respondent identification and participation. |
Each partnering trade organization provided a list of medical device manufacturers with operations in the state, including contact information. To identify manufacturers with at least one marketed medical device that had been approved via the FDA’s PMA pathway, we cross-referenced the state organization lists of manufacturers against an FDA-maintained list of manufacturers with medical devices approved via the PMA pathway.9 We limited state lists to a single contact for each manufacturer, preferentially selecting contacts whose job title suggested involvement in clinical research (such as Chief Scientific Officer). However, when manufacturers were listed on multiple state lists, one contact within each state was included to ensure representation of multiple manufacturer divisions with independent operations in each state.
For each potential survey respondent, we abstracted information from public data sources, such as manufacturers’ websites, in order to compare survey respondents and non-respondents. This information included general company characteristics, including ownership and clinical therapeutic area, and worldwide revenue in the preceding year.
In coordination with CHI, LSA, and MassMEDIC, all potential survey respondents were sent an initial e-mail in November 2013 to describe the purpose of the study, request their participation, and provide a link to the survey, with four follow-up requests sent by e-mail in November and December 2013. Non-respondents were contacted by telephone to solicit their participation up to twice per week, but no more than once per day, until one contact was made. In January 2014, non-respondents for whom telephone contact had not been made were mailed a paper version of the survey, which was followed by one last contact attempt via telephone a week later.
Invitations to participate did not reference a specific hypothesis of the study, but stated that manufacturer participation would further the understanding of post-market clinical research activities and would be published, as well as shared with the FDA and all respondents. All internet-based responses were collected using a Web-based survey platform (Qualtrics Labs, Provo, UT, USA). Approval from the Yale University School of Medicine Human Research Protection Program was obtained prior to study conduct and consent was considered to be implied when participants completed the online survey.
Survey instrument development
The design of our 59-item survey instrument was informed by previously published surveys,10,11 a review of the literature on post-market medical device clinical research, and discussion with multiple experts and stakeholders, including representatives from industry (Medtronic, Inc., Minneapolis, MN, USA and Johnson & Johnson, New Brunswick, NJ, USA), representatives from trade organizations that advocate on behalf of industry (LSA, AdvaMed, and MDMA), representatives from the FDA, and academic investigators. Experts recommended survey topics that they considered to be compelling for the field of post-market clinical research and device surveillance. The survey was pre-tested with three representatives from cardiovascular medical device companies and modified iteratively to improve clarity, face validity, and content validity. Adaptive questioning was used to decrease response burden. Items were presented in multiple response, Likert scale, and open-ended formats. The complete instrument will be made available on request from the authors.
Survey domains
Post-market clinical research programs
We used Likert-type questions to assess the regularity with which manufacturers conduct post-market clinical research studies for devices approved via the FDA’s PMA pathway as well as multiple response questions to ascertain manufacturers’ primary and secondary reasons for engaging in research. For manufacturers who do not conduct post-market clinical research studies, we used multiple response questions to ascertain manufacturers’ primary and secondary reasons for not engaging in the research. We used yes/no questions to characterize the type of clinical research studies conducted by manufacturers in the post-market period, including clinical registries initiated at the time of, or after, market approval, randomized clinical trials with either active or placebo comparators, and single-arm clinical studies. Multiple response questions were used to ascertain the types of clinical efficacy and safety endpoints on which these studies are focused and the manufacturers’ means of disseminating study results.
Management of safety concerns identified through post-market clinical research
We used yes/no questions to determine whether manufacturers had identified a new safety concern through post-market clinical research activities in the past 3 years and multiple response questions to determine what confirmatory, communication, or other actions were subsequently taken.
Risks of doing (or not doing) post-market clinical research
Among manufacturers who do and do not conduct post-market clinical research studies, we used open-ended questions to explore what risks, if any, manufacturers perceive that they face by either engaging or not engaging in post-market clinical research studies for devices approved via the FDA’s PMA pathway.
Manufacturer characteristics
Respondents were asked to characterize the manufacturer they represented using multiple choice and multiple response questions, including the number of devices manufactured that were approved via the FDA’s PMA pathway, the broad clinical areas in which these devices are used, the geographic markets in which these devices have been approved for use, and the size and sales of the manufacturer.
Statistical analysis
To compare characteristics of survey respondents and non-respondents, we used chi-square or Fisher’s exact tests for all categorical variables. Next, we conducted descriptive analyses, calculating the proportion of manufacturers who do and do not conduct post-market clinical research studies, as well as the characteristics of these efforts. Because there were large proportions of missing responses among survey questions, ranging from 5% (1 of 22 respondents not answering a given question) to 36% (8 of 22 respondents), all proportions were estimated using the complete denominator of respondents and we reported missing responses for each question. Finally, exploratory analyses were conducted to examine differences between small and large manufacturers, stratified by worldwide number of employees. Survey data were analyzed using JMP Version 10.0 (SAS Institute Inc., Cary, NC, USA).
Results
Survey participation requests were sent to 47 eligible respondents representing medical device manufacturers with operations in the states of California, Minnesota, and Massachusetts, 22 of whom completed the survey either online (n=19) or on paper (n=3), yielding a response rate of 47%. Survey respondents did not differ from non-respondents with respect to trade organization representation, company ownership, worldwide gross revenue from medical sales, or clinical therapeutic area of medical devices approved via the FDA’s PMA pathway (P-values >0.10; Table 1).
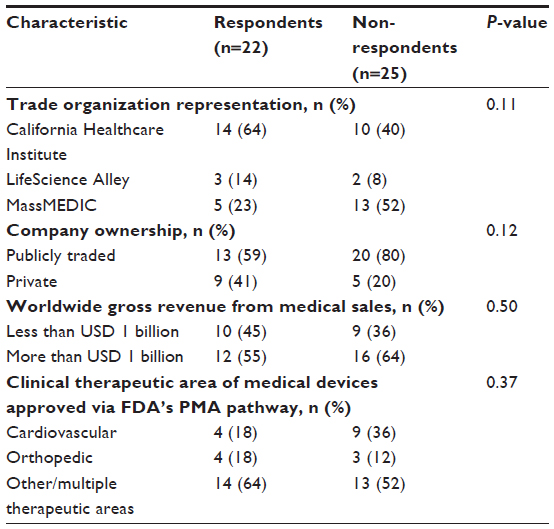 | Table 1 Characteristics of survey respondents and non-respondents |
Respondents represented a variety of medical device manufacturers (Table 2), including those manufacturing cardiovascular devices, orthopedic devices, anesthesiology devices, as well as manufacturing devices that had been approved for use in the US, Europe, and Japan. The majority of respondents represented manufacturers with more than 1,000 employees and with annual gross revenue in excess of $1 million.
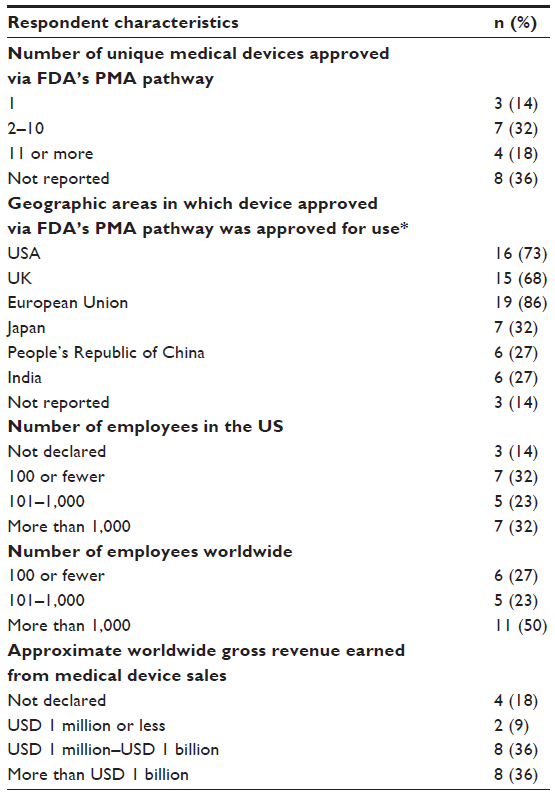 | Table 2 Self-reported characteristics of respondent manufacturers |
Post-market clinical research programs
Nearly all respondents self-reported conducting post-market clinical research studies after their medical device(s) had been approved via the FDA’s PMA pathway, 59% (13 of 22) “always” or “regularly” and 27% (6 of 22) “sometimes” or “rarely”; only 5% (1 of 22) reported never conducting post-market clinical research studies (Table 3). Among those conducting these studies, 79% (15 of 19) self-reported conducting only between 1 and 5 studies during the first 3 years the device is on the market. Exploratory analyses stratified by worldwide number of employees suggest similar self-reported rates and numbers of post-market clinical research studies conducted after approval among small and large manufacturers.
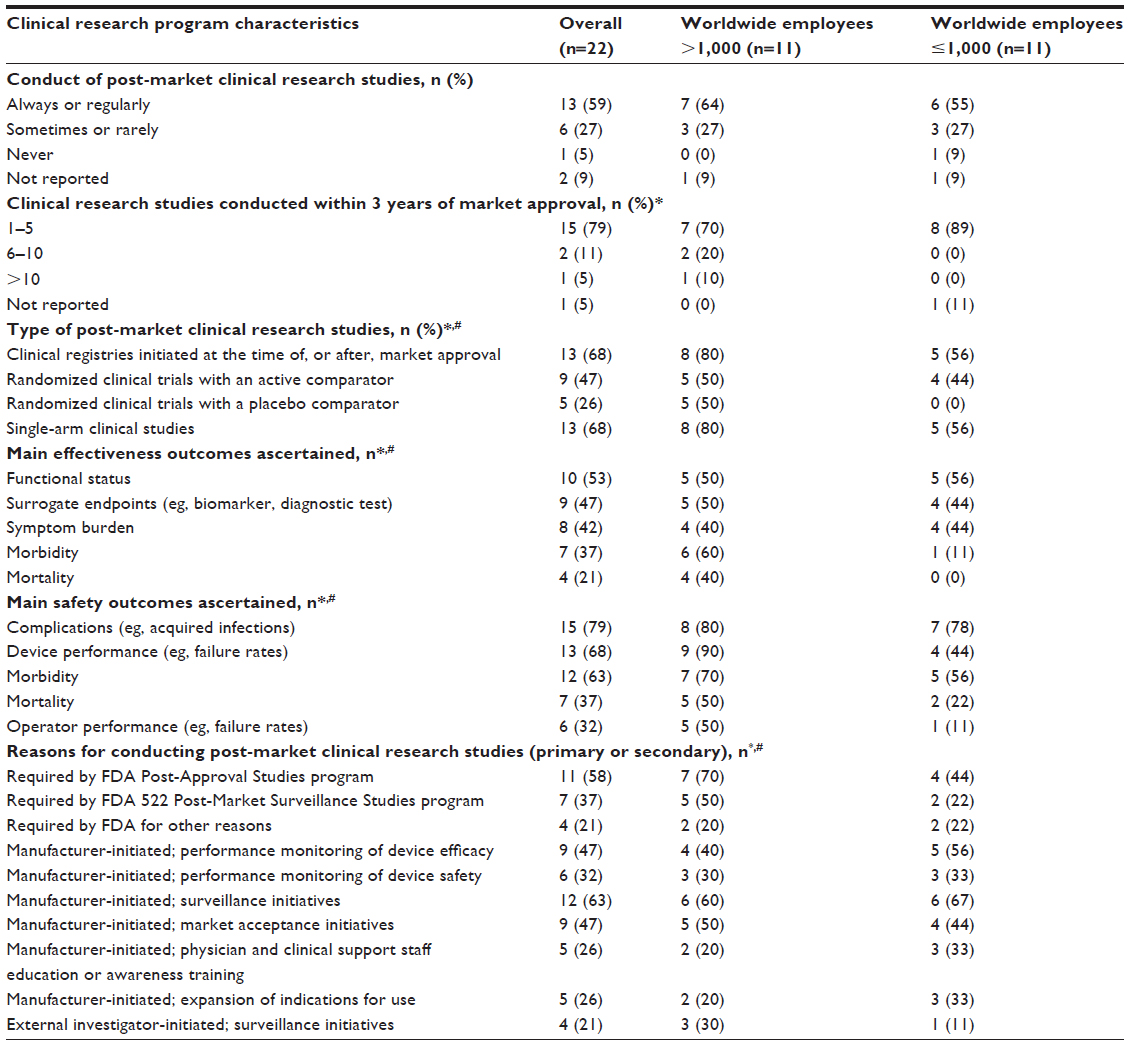 | Table 3 Characteristics of manufacturers’ post-market clinical research programs for medical devices approved via FDA’s premarket approval pathway |
Manufacturers reported engaging in a wide variety of post-market clinical research studies, most commonly clinical registries initiated at the time of, or after, market approval (13 of 19; 68%) and single-arm clinical studies (13 of 19; 68%) (Table 3). Post-market clinical registries were most commonly described as device specific (n=12), voluntary (n=10), and involving recruitment from 10 or more centers (n=12) that were most frequently located in the US (n=12) and Europe (n=10), but were most commonly conducted without partner organizations (n=7), such as professional societies. Similarly, single-arm clinical studies were most commonly described as involving recruitment from 10 or more centers (n=9) that were most frequently located in the US (n=11) and Europe (n=8). The most common effectiveness outcomes ascertained in these studies were functional status (n=10) and surrogate endpoints, such as biomarkers or diagnostic tests (n=9), whereas the most common safety outcomes ascertained were complications (n=15) and device performance, such as failure rates (n=13). Exploratory analyses stratified by worldwide number of employees suggest similar study designs and outcomes ascertained through post-market clinical research studies conducted after approval among small and large manufacturers.
Among manufacturers conducting any post-market clinical research studies, the primary reason most commonly cited was FDA Post-Approval Studies program requirements (5 of 19; 26%) (Table 3). However, when asked to report additional reasons for conducting these studies, commonly cited reasons included FDA requirements, such as the Post-Approval Studies (n=11) and 522 Post-Market Surveillance Studies (n=7) programs, surveillance initiatives (n=12), manufacturer-initiated programs for performance monitoring of device efficacy (n=9), and market acceptance initiatives (n=9), as well as external investigator-initiated surveillance initiatives (n=4). Exploratory analyses stratified by worldwide number of employees suggest similar reasons for the conducts of post-market clinical research studies after approval among small and large manufacturers.
Management of safety concerns identified through post-market clinical research
Among manufacturers conducting any post-market clinical research studies, 11% (2 of 19) identified new safety concerns associated with their device through their research program. In both cases, the manufacturers analyzed data from company customer complaint reporting programs to confirm the finding; other confirmatory steps included analyzing data from the FDA’s adverse event reporting program (eg, Manufacturer and User Facility Device Experience) and designing and conducting a new clinical research study. In addition, in both cases where new safety concerns were identified, the safety concern was discussed directly with physicians through company representatives and by updating company outreach materials, including those intended for patient and physician education and for field support.
Risks of doing (or not doing) post-market clinical research
In open-response questions, manufacturers conducting any post-market clinical research studies reported that the risks of doing post-market research included a perception among physicians or the general public that the medical device was still experimental. Manufacturers also commented that post-market research raised the risks of both liability and criticisms from competitors. In contrast, respondents reported that the risks of not conducting post-market research included missing low event safety issues, not understanding long-term effects of the device, and losing the support of key opinion leaders, customers, and regulators. One company wrote that they viewed not monitoring product performance as an “ethical failure”.
The manufacturer who reported never conducting post-market clinical research studies indicated that the risks of doing post-market research included finding smaller treatment effect sizes than the approval trial as well as opportunity costs, as they choose to invest their research dollars on the development of next generation products rather than post-market surveillance of approved products. In contrast, the manufacturer reported that the risks of not conducting post-market research included not being able to address scientific questions that arise from clinicians and not generating data that are important to payers.
Discussion
In our survey of medical device manufacturers who have had products approved through the FDA’s PMA pathway and have operations in the states of California, Minnesota, and Massachusetts, nearly all reported conducting additional clinical research studies after FDA approval, when the products were already available for use. In addition, while manufacturers most often engaged in these research studies in order to satisfy the requirements of the FDA’s Post-Approval Studies and 522 Post-Market Surveillance Studies programs, there were clear advantages for manufacturers, as these studies provided performance monitoring of device efficacy, surveillance initiatives, and market acceptance initiatives. Given the frequency with which respondent manufacturers initiate these studies, their clinical data should be integrated into national post-market surveillance initiatives.
To our knowledge, no prior studies have examined whether and how commonly medical device manufacturers engage in post-market performance evaluation beyond FDA requirements. However, our study has important limitations that should be discussed before interpreting our results. Our survey was limited to medical device manufacturers with operations in the states of California, Minnesota, and Massachusetts. As the FDA does not maintain a comprehensive public listing of medical device manufacturers who are currently marketing products approved through the PMA pathway, there was not a feasible method to survey this information at the national level. We targeted these three states, along with Indiana, because the preponderance and diversity of manufacturers with products approved via the FDA’s PMA pathway are based there, ensuring the generalizability of our research. In addition, our study was limited because only half of potential respondents identified by the trade organizations with which we partnered completed our questionnaire. Because non-respondents may be less likely to perform post-market studies, influencing their decision to participate in our survey, our results may estimate higher rates of post-market clinical research than exist in practice across the industry. Moreover, our sample sizes were small and our study was likely underpowered to detect differences across medical device manufacturer respondent characteristics. Finally, despite pre-testing the survey with three representatives from cardiovascular medical device companies and iteratively modifying the text to improve clarity, face validity, and content validity, there were several missing responses among survey questions. We would only be able to speculate as to why respondents did not answer specific survey questions, as no concerns with question clarity or content sensitivity were raised.
There are other important potential explanations for our low response rate, including concerns that any stated non-compliance with FDA regulatory requirements would lead to citation or fines, manufacturers’ concerns with publicly reporting their post-market clinical research strategies, which could place the company at a competitive disadvantage, or simply that contacted respondents were unaware of the scope of their company’s post-market clinical research activities, and decided not to complete the survey. These potential explanations should be taken into consideration when interpreting our main finding that nearly all manufacturers, including large, publicly traded companies with ample resources and smaller, private companies with fewer resources, self-reported conducting additional clinical research studies after FDA approval. The only manufacturer who reported not conducting post-market clinical research was a smaller company that explained that it invests its research dollars on the development of next generation products rather than post-market surveillance of already approved products. Moreover, among those manufacturers engaging in post-market clinical research of their medical devices, most commonly these studies were single-arm trials and clinical registries, not large-scale randomized trials, and nearly half were focused on surrogate endpoints. The limitations of pre-market clinical testing of medical devices have been well described,12–16 particularly for higher-risk devices that are expected to require more intensive safety evaluation. Further work needs to be done to characterize the strength of these clinical studies to ensure that they are sufficiently robust to be used for post-market safety surveillance.
Despite potential benefits for manufacturers engaging in post-market clinical research, these studies appear to be largely driven by FDA requirements. In part this may be a consequence of perceived risk, as several respondents explained that post-market clinical research studies potentially exposed their companies to criticism by clinicians, the public, and competitors. Since the FDA is actively re-envisioning its national post-market surveillance system, with the goal of developing a life-cycle approach to product evaluation that integrates pre-market and post-market evaluations,3,4 the potential implications for manufacturers’ post-market clinical research activities, and incentives to engage in this work, should be considered. Currently, the FDA is proposing to make better use of existing sources of device information, including administrative claims and electronic medical record data, while simultaneously establishing registries for select products.3,4 The role of post-market research studies, particularly clinical trials to further enhance our understanding of medical device efficacy and safety, has not been articulated. As current FDA surveillance efforts are largely based on voluntary complaint rates that are known to be under-reported,17 there is a clear need for a broader system of evaluation, including both existing data sources and robust clinical trials to address specific efficacy and safety objectives.
Conclusion
Nearly all medical device manufacturer survey respondents who have had products approved through the FDA’s PMA pathway and have operations in the states of California, Minnesota, and Massachusetts self-reported conducting post-market clinical research studies, after FDA approval. Despite limitations in their design, the frequency with which these studies are being conducted suggests there is clear potential to integrate these initiatives with current FDA efforts to strengthen the national post-market surveillance system. A broad and robust system of post-market medical device evaluation, including both existing data sources and clinical trials, is needed to ensure patients and clinicians, as well as manufacturers and regulators, have a comprehensive understanding of efficacy and safety for higher-risk medical devices.
Acknowledgments
The authors acknowledge the suggestions on study design and comments on an early draft of the manuscript received from Danica Marinac-Dabic, MD, PhD, and Benjamin C Eloff, PhD, both from the US Food and Drug Administration, Bethesda, MD, USA. In addition, the authors acknowledge the suggestions on study design received from Richard E Kuntz, MD, MSc from Medtronic Inc.; and Dale Wahlstrom, MS from LifeScience Alley. This study was supported by research grants from the US Food and Drug Administration (1U01FD004585-01) and Medtronic, Inc.
Author contributions
JSR and KLB drafted the manuscript and conducted the statistical analysis. JSR and HMK were responsible for the conception and design of this work. JDR and KLB were responsible for acquisition of data. All authors participated in the analysis and interpretation of the data and critically revised the manuscript for important intellectual content.
Disclosure
All authors receive research support from Janssen, the Pharmaceutical Companies of Johnson & Johnson, to develop methods to promote data sharing. JSR and HMK receive research support from the Centers for Medicare and Medicaid Services (CMS) to develop and maintain hospital performance measures that are used for public reporting. JSR is a member of a scientific advisory board for FAIR Health, Inc. HMK chairs a scientific advisory board for UnitedHealthcare. The authors have no other conflicts of interest in this work.
References
US Food and Drug Administration. Medical Devices: Post-Market Requirements (Devices). Available from: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Postmarket Requirements/. Accessed April 17, 2015. | |
Institute of Medicine. Medical Devices and the Public’s Health: The FDA 510(k) Clearance Process at 35 Years. Washington, DC: National Academy Press; 2011. | |
US Food and Drug Administration, Center for Devices and Radiological Health. Strengthening Our National System for Medical Device Postmarket Surveillance. September 2012; Available from: http://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProducts andTobacco/CDRH/CDRHReports/UCM301924.pdf. Accessed April 17, 2015. | |
US Food and Drug Administration, Center for Devices and Radiological Health. Strengthening Our National System for Medical Device Postmarket Surveillance: Update and Next Steps. April 2013; Available from: http://www.fda.gov/downloads/MedicalDevices/Safety/CDRH PostmarketSurveillance/UCM348845.pdf. Accessed April 17, 2015. | |
California Healthcare Institute. Available from: http://www.chi.org/. Accessed April 17, 2015. | |
Indiana Medical Device Manufacturers Council. Available from: http://www.imdmc.org/. Accessed April 17, 2015. | |
LifeScience Alley. Available from: https://www.lifesciencealley.org/. Accessed April 17, 2015. | |
Massachusetts Medical Device Industry Council. Available from: http://www.massmedic.com/. Accessed April 17, 2015. | |
US Food and Drug Administration. Devices@FDA. Available from: http://www.accessdata.fda.gov/scripts/cdrh/devicesatfda/. Accessed April 17, 2015. | |
Seapine Software. State of Medical Device Development Survey. 2013; Available from: http://www.seapine.com/2013-medical-device-development-survey-report.html. Accessed April 17, 2015. | |
Makower J, Meer A, Denend L. FDA Impact on US Medical Technology Innovation. November 2010; Available from: http://www.nvca.org/index.php?option=com_docman&task=doc_download&gid=668&Itemid=93. Accessed April 17, 2015. | |
Chen CE, Dhruva SS, Redberg RF. Inclusion of comparative effectiveness data in high-risk cardiovascular device studies at the time of premarket approval. JAMA. 2012;308(17):1740–1742. | |
Dhruva SS, Bero LA, Redberg RF. Strength of study evidence examined by the FDA in premarket approval of cardiovascular devices. JAMA. 2009;302(24):2679–2685. | |
Gartenberg AJ, Peleg A, Dhruva SS, Redberg RF. Presumed safe no more: lessons from the Wingspan saga on regulation of devices. BMJ. 2014;348:g93. | |
Kesselheim AS, Avorn J. New and unproved medical devices. BMJ. 2013;347:f7413. | |
Prasad V, Rho J, Cifu A. The inferior vena cava filter: how could a medical device be so well accepted without any evidence of efficacy? JAMA Intern Med. 2013;173(7):493–495; discussion 495. | |
US Food and Drug Administration. MAUDE – Manufacturer and User Facility Device Experience. Available from: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfMAUDE/search.CFM. Accessed April 17, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.