")
Back to Journals » Journal of Pain Research » Volume 15
Post-Inhibitory Rebound Firing of Dorsal Root Ganglia Neurons
Received 12 April 2022
Accepted for publication 2 July 2022
Published 26 July 2022 Volume 2022:15 Pages 2029—2040
DOI https://doi.org/10.2147/JPR.S370335
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qi Fang
Tong Zhu,1,2 Siqi Wei,1 Yuying Wang1,3,4
1Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Xi’an Jiaotong University Health Science Center, Xi’an, Shaanxi, 710061, People’s Republic of China; 2Clinical Experimental Center, Xi’an International Medical Center Hospital, Xi’an, Shaanxi, 710100, People’s Republic of China; 3Institute of Neuroscience, Translational Medicine Institute, Xi’an Jiaotong University Health Science Center, Xi’an, Shaanxi, 710061, People’s Republic of China; 4Key Laboratory of Environment and Genes Related to Diseases (Xi’an Jiaotong University), Ministry of Education, Xi’an, People’s Republic of China
Correspondence: Yuying Wang, Email [email protected]
Background: In the central nervous system, post-inhibitory rebound firing (RF) may mediate overactivity of neurons under pathophysiological condition. RF is also observed in dorsal root ganglion (IRA) neurons. However, the functional significance of RF in primary sensory neurons has remained unknown. After peripheral sensory nerve/neuron injury, DRG neurons exhibit hyperexcitability. Therefore, RF may play a role in neuropathic pain.
Methods: Chronic compression of DRG (CCD) is used as a neuropathic pain model. Rats were divided into 2 groups: Sham and CCD groups. Patch clamp was performed on the whole DRG and cultured DRG neurons to record RF and T-type Ca2+ currents. The blocker of T-type Ca2+ channels, NiCl2, was applied to DRG neurons.
Results: Rebound neurons were more excitable than non-rebound neurons. And they discharged RF with prominent after depolarizing potentials, which were blocked by NiCl2. After DRG injury, the proportion of rebound neurons augmented, and rebound neurons’ excitability increased. Meanwhile, the steady-state activation curve of T-type Ca2+ channels was shifted toward the left.
Conclusion: RF may be related to highly excitable neurons and sensitive to both depolarization and hyperpolarization. T-type Ca2+ channels were critical to RF, potentially enhancing the spontaneous firing of rebound neurons in response to resting membrane potential fluctuations.
Keywords: post-inhibitory rebound firing, dorsal root ganglia, chronic compression, T-type Ca2+ currents
Introduction
Normally, depolarizing inputs may induce neurons to generate action potentials (APs), whereas hyperpolarizing inputs reduce neural excitability. However, rebound neurons exhibit post-inhibitory-rebound firing (RF) following the termination of hyperpolarizing inputs.1 Rebound neurons are widely distributed in the central nervous system (CNS).2–5 RF is associated with the rhythmic activity of central neural networks and is crucial in both onset and offset responses.6–9 T-type Ca2+ channels and hyperpolarization-activated channels contribute to RF,10,11 which has also been associated with neurodegenerative disorders ranging from epilepsy to motor neuron disease.12,13 In neurodegeneration of CNS, hyperexcitability of rebound neuron may be self-corrected against neuronal loss and neurological impairment following neurodegeneration.13 In Parkinson’s disease, the basal ganglia project inhibitory outputs to the thalamus, which may elicit rebound bursts in thalamic neurons. And the rebound firing may account for the unexpected excessive movements (eg, tremors) of patients.10,14–16 Walsh found that nucleus reuniens of a mouse model of Alzheimer’s disease exhibited an increased propensity to rebound burst following hyperpolarizing current stimuli, and this may contribute to hippocampal-thalamo-cortical hyperexcitability.17 In the peripheral nervous system (PNS), a small number of low-threshold medium-sized dorsal root ganglia (DRG) neurons also generate RF.18,19 In neuropathic pain conditions, DRG neurons become hyperexcitable after DRG injury.20–24 To some degree, the hyperexcitability of DRG neurons after injury resembles the hyperexcitability of central rebound neurons in neurodegenerative disorders, suggesting DRG rebound neurons may be hyperexcitable and involved in neuropathic pain. Therefore, RF may play a role in neuropathic pain signal transmission. However, the features of RF and ion channel mechanisms in DRG remain unknown. In this study, we applied chronic compression of DRG (CCD) rats, a neuropathic pain model, to investigate the mechanisms of RF in DRG neurons.
Materials and Methods
Animals
Young male (6–8 weeks old) Sprague-Dawley rats weighing between 100 and 150 g were provided by the Medical Experimental Animal Center of Xi’an Jiaotong University (Shaanxi Province, China). Rats were housed under climate-controlled conditions at a 12-h light/dark cycle with food and water ad libitum. The experimental protocols were approved by the Institutional Animal Care Committee of Xi’an Jiaotong University. The experiments were performed in accordance with the ethical guidelines of the International Association for the Study of Pain.25 All efforts were made to minimize the number of animals used and their suffering.
Chronic-Compression of DRG (CCD) Rat Model
The CCD rat model of neuropathic pain has been reported previously.22,24 Briefly, rats were anesthetized with sodium pentobarbital (45 mg/kg, i.p.). Then, the skin and muscles along the lower lumber vertebra were cut to expose left L4 and L5 intervertebral foramina, and L-shaped rods made of stainless steel (4 * 2 mm in length and 0.6 mm in diameter) were carefully inserted. A transient twitch of the ipsilateral hind leg muscles of the animal was observed when the DRG was touched by the steel rod. After the surgical procedure, antibiotics were administered systemically. Sham animals were also anesthetized and the left L4 and L5 intervertebral foramina were exposed, but no steel rods were inserted. There were 20 rats in CCD group and 16 in sham group.
Preparation of DRG Neurons
The DRG neurons were prepared from the CCD rats or sham rats on the day 3 ˜ 6 post operation, as described previously.26 Briefly, the rats were deeply anesthetized with sodium pentobarbital (55 mg/kg, i.p.) and then decapitated, left L4 and L5 DRGs were removed and placed in a tube containing protease (0.4 mg/mL; Sigma) and collagenase (1 mg/mL; Sigma type 1). The tube was bathed for 30–40 min at 35℃. Later, the ganglia were rinsed three times with Hank’s balanced salt solution (HBSS) to remove enzymes. They were then kept in oxygenated artificial cerebral spinal fluid (ACSF) (95% O2 and 5% CO2) at 24℃ for 1 h. The ACSF contained (in mM): 125 NaCl, 3.8 KCl, 1.2 KH2PO4, 1.0 MgCl2, 2.5 CaCl2, 25 NaHCO3, 10 glucose. The ganglia were then fixed in a recording chamber with a grid of nylon threads glued to a “U”-shaped platinum frame. The chamber was mounted on the stage of microscope (BX51WI; Olympus, Tokyo, Japan). During recording, ganglia were perfused with oxygenated ACSF.16
In order to record T-type Ca2+ currents, left L4 and L5 DRG were cut into sections and placed into a tube containing trypsin (1 mg/mL; Sigma) and collagenase (2.0 mg/mL; Sigma, type 1) at 37 ℃ for 45 min. Then, the tube was centrifuged, the supernatant was removed and the cell pellet was resuspended. The cell suspensions were later plated onto 35 mm dishes. The cells were grown in Dulbecco’s modified Eagle’s Medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin (100 U/mL) at 37℃ with 5% CO2. The recordings were performed on DRG neurons grown in vitro for 2–6 h.
Cellular membrane capacitance (Cm) was measured in patch clamp recording, which was related to neuron size. The unit of Cm is picofarad (1pF is 10−12 farad).
Electrophysiological Recordings
Whole-cell patch-clamp recordings were performed at room temperature using a Multiclamp 700B amplifier (Axon Instruments, Sunnyvale, CA, US). A dish with DRG neurons was mounted on an inverted microscope (Olympus 1 × 71). Patch pipettes were pulled from thick-wall borosilicate glass pipettes using a horizontal puller (Model P-97, Sutter Instruments). To record RF and hyperpolarization-activated currents, a recording electrode with a resistance of 1 ˜ 3 MΩ was filled with physiological solution (140 mM KCl, 5 mM NaCl, 5 mM Mg-ATP, 1 mM MgCl2, 5 mM ethylene glycol tetraacetic acid (EGTA), 2 mM CaCl2 and 15 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH was adjusted to 7.3 by adding KOH). The bath solution contained the following: 144 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM HEPES and 10 mM glucose with pH adjusted to 7.3 by adding NaOH. Na+ free external solution contained the following: 125 mM N-Methyl-D-glucamine (NMDG), 2.5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM HEPES and 10 mM glucose with pH adjusted to 7.3 by adding KOH.26
Ca2+ currents were recorded using the whole-cell configuration. The pipette solution contained the following: 135 mM tetramethylammonium hydroxide (TMA-OH), 2 mM MgCl2, 10 mM EGTA, 40 mM HEPES and 1 mM Mg-ATP with pH titrated to 7.2 using hydrofluoric acid (HF), F− was used to facilitate high voltage activated (HVA) Ca2+ current rundown. The external solution consisted of 135 mM tetraethylammonium chloride (TEA-Cl), 1 mM MgCl2, 10 mM BaCl2, 10 mM HEPES, and 11 mM glucose, with pH adjusted to 7.3 using TEA-OH. The osmolarities of all pipette solutions and external solutions were approximately adjusted to 295˜305 mOsm/L. On the recording day, nifedipine (10 μM) and tetrodotoxin (TTX, 500 nM) were added to the bath solution.27
The rheobase was determined to be the minimum current able to elicit an AP with incremental depolarizing current injection of 10 pA for 500 ms. The RF was recorded similarly, with a hyperpolarizing current provided for 500 ms with −10 pA increments.
T-type Ca2+ currents were recorded in voltage clamp mode, in step sweeps of 250 ms duration beginning at −90 mV and increasing in 10 mV intervals to 50 mV, or ramp sweep of 500 ms duration ranging from −90 to 30 mV. Current amplitudes were normalized to Cm to determine the current density (pA/pF). Activation curves were plotted according to the step voltage clamp recording described above. In the inactivation analysis, T-type Ca2+ currents were evoked by a 50 mV test-pulse of 200 ms duration after a pre-pulse of 3-s duration ranging from −120 to −50 mV in 10 mV increments. The voltage dependencies of steady-state activation and inactivation were described with single Boltzmann distributions of the following formulas:
Activation: G(V) = Gmax/(1 + exp[-(V − V50)/k])
Inactivation: I(V) = Imax/(1 + exp[(V − V50)/k])
where Gmax is the maximal conductance, Imax is the maximal amplitude of current, V50 is the voltage at which half the current is activated or inactivated, and k represents the voltage dependence (slope) of the distribution.
The data were analyzed using Origin Pro 8.0 (OriginLab Corporation, Northampton, MA, USA).
For electrophysiological recordings, the resting membrane potential (RMP) was more negative than −50 mV. Membrane properties were monitored with the pCLAMP membrane test. Data acquisition was filtered at 10 kHz and sampled at 20 kHz with the Clampex 9.0 software system (Axon Instruments) through the Digidata 1322A (Axon Instruments). The series resistance was <10 megohms. The data were analyzed using Clampfit 10.4 (Axon Instruments) and Origin Pro 8.0 (OriginLab Corporation, Northampton, MA, US).
Drugs
All reagents were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA).
Statistical Analysis
All data are reported as the mean ± standard error of the mean (SEM). Statistical comparisons were performed using independent t-test, chi-squared test and one-way ANOVA. Statistical analysis was performed using Origin Pro 8.0 (OriginLab Corporation, Northampton, MA, US).
Results
Patch-clamp recordings were performed on 70 injured and 70 sham DRG neurons. Among them, 45 out of 70 injured neurons and 20 out of 70 sham neurons were rebound neurons exhibiting RF (CCD group vs sham group: 45/70 vs 20/70, P < 0.0001). The rebound neurons were small (Cm < 35 pF) or medium-sized (Cm ≥ 35 and ≤ 70 pF). An overview of the electrophysiological properties of the neuronal subgroups is shown in Table 1. Because there were only 2 small rebound neurons in both the CCD and sham rats, the parameters of small rebound neurons were not compared between them. Regarding RMP, there was no significant difference in RMP between the CCD and sham rats in the same size category. The rheobase was significantly lower and the input resistance was significantly higher in rebound neurons than in non-rebound neurons, implying that excitability was higher in rebound neurons than in non-rebound neurons.
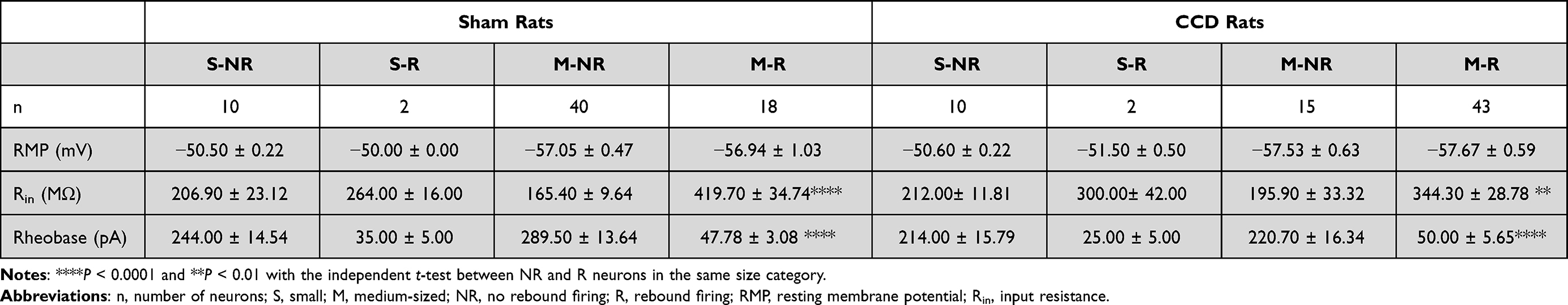 |
Table 1 The Electrophysiological Parameters of DRG Neurons in the Sham and CCD Rats |
Rebound Neurons
Rebound neurons (40 out of 45 in the CCD rats and 20 out of 20 in the sham rats) generated RF with prominent after-depolarizing potentials (ADP) (Figure 1A-a1, a2, a4). It is known that T-type Ca2+ currents contribute to ADP.28,29 In order to reveal the role of T-type Ca2+ channels in RF, we classified rebound neurons in 3 groups according to the occurrence of ADP: group 1 (Figure 1A-a1) and group 2 (Figure 1A-a2) generating RF with ADP, and group 3 generating RF without ADP (Figure 1A-a3). More specifically, group 1 generated AP with ADP, while group 2 and 3 generated AP without ADP.
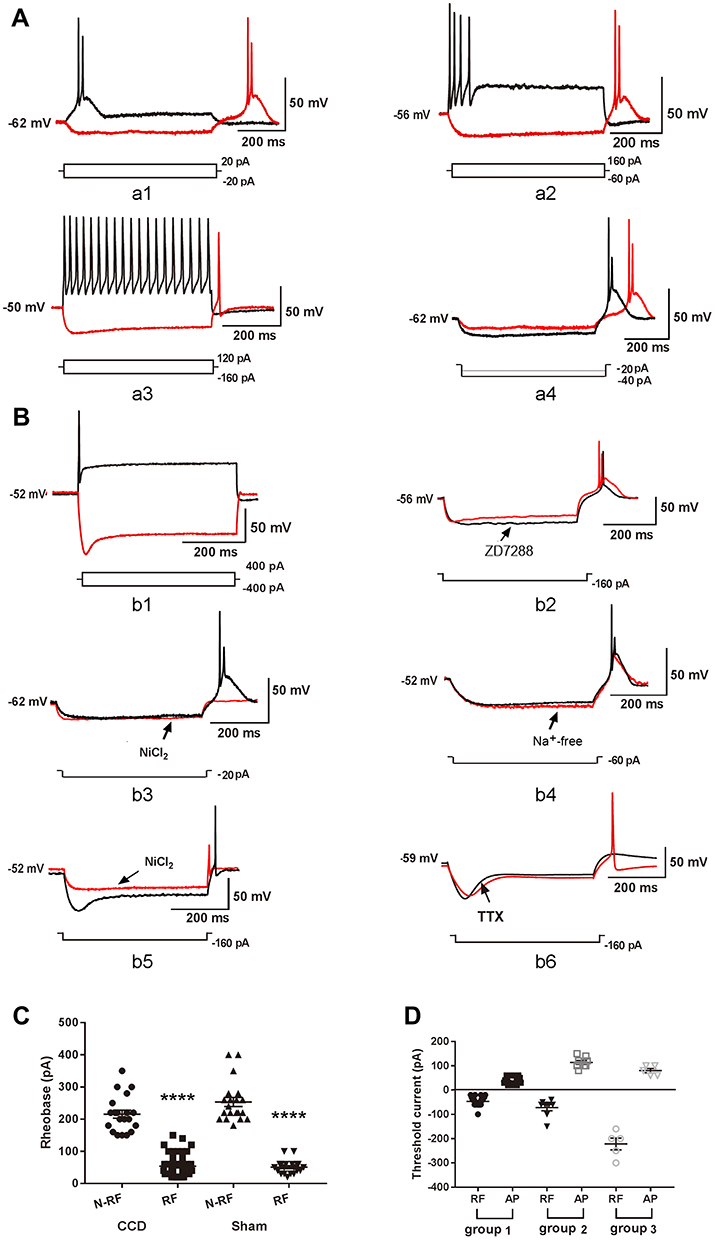 |
Figure 1 The post-inhibitory rebound firing of DRG neurons. (A) Representation of RF and AP. a1: Group 1 rebound neurons discharged RF and AP with prominent ADP. The arrow indicates ADP. ADP: after-depolarizing potential. a2: Group 2 rebound neurons discharged RF with ADP and AP without ADP. A3: Group 3 rebound neurons discharged RF and AP without ADP. a4: The latency of RF reduced in response to increasing hyperpolarization. (B) Underlying ion mechanism of RF. b1: Non-rebound neurons discharged AP with sag but without RF. b2: ZD7288 slightly inhibited ADP in RF. b3: NiCl2 inhibited ADP and RF. b4: Neurons only generated large-amplitude rebound depolarizations in Na+ free bath. b5: high concentration of NiCl2 reduced the amplitude of RF in group 3 rebound neurons. b6: Administration of TTX fully inhibited RF. (C) The rheobase was compared between RF and N-RF neurons in the CCD and sham rats. RF: rebound firing, N-RF: non-rebound firing. ****P < 0.0001. (D) The threshold current was compared between RF and AP in group 1, group 2 and group 3 rebound neurons. |
The latency of RF decreased in response to increasing hyperpolarization, suggesting that the latency of RF is related to the hyperpolarization level (Figure 1A-a4). It is known that hyperpolarization-activated current (Ih)11 and T-type Ca2+ currents contribute to RF of CNS rebound neurons. Hyperpolarization-activated cyclic nucleotide-gated cation channels opened in response to hyperpolarization and then induced a sag on membrane potential. In response to hyperpolarization pulse, although some rebound neurons exhibited sag (Figure 1A-a3), some non-rebound neurons also exhibited sag (Figure 1B-b1), whereas some rebound neurons did not exhibit any sag (Figure 1A-a1, a4). We applied ZD7288 (10 μM), a blocker of hyperpolarization-activated cyclic nucleotide-gated cation channels,20 to rebound neurons. ZD7288 did not inhibit RF (Figure 1B-b2), suggesting that Ih was not essential for the generation of RF. ZD7288 slightly inhibited the amplitude of ADP (n = 3, from 23.67 ± 4.98 mV to 15.67 ± 4.70 mV).
- type Ca2+ channels opened and then induced a depolarization potential, which may be ADP. NiCl2 is a non-specific blocker of T-type Ca2+ channels.10 In group 1 and group 2 rebound neurons, the delivery of NiCl2 (50–100 μM) inhibited RF and ADP (Figure 1B-b3). The Na+ free bath blocked RF but not the depolarization potential (Figure 1B-b4), suggesting that T-type Ca2+ channel was critical to the generation of RF through the rebound depolarization potential. In group 3 rebound neurons, a high concentration of NiCl2 (500 μM) did not fully inhibit RF (Figure 1B-b5). The amplitude of RF was reduced (n = 3, from 91.67 ± 4.41 mV to 48.33 ± 1.67 mV), while the latency was shortened from 18.33 ± 2.40 ms to 7.67 ± 1.45 ms (n = 3). The application of TTX (300 nM) fully inhibited the RF (Figure 1B-b6).
RF of DRG Neurons
The features of RF were compared among group 1, group 2 and group 3 rebound neurons, including the rheobase, the hyperpolarizing current threshold, the latency and the amplitude of ADP. The rheobase of classical AP was 35.31 ± 2.54 pA (n = 32) in the CCD rats and 46.5 ± 2.93 pA (n = 20) in the sham rats (Figure 1C). In group 1 rebound neurons, the hyperpolarizing current threshold evoking RF was −42.50 ± 2.69 pA (n = 32) in the CCD rats and −50.00 ± 3.70 pA (n = 20) in the sham rats (Figure 1D). There was no difference between the CCD and sham rats on hyperpolarizing current threshold evoking RF and the rheobase. In group 2 rebound neurons, the hyperpolarizing current threshold evoking RF was −65.00 ± 9.06 pA (n = 8), which was higher than in group 1 rebound neurons (Figure 1D, −65.00 ± 9.06 pA vs −42.50 ± 2.69 pA, ** P < 0.01). Similarly, the rheobase was higher in group 2 rebound neurons (n = 8) than in group 1 rebound neurons (102.5 ± 13.86 pA vs 35.31 ± 2.54 pA, P < 0.0001) (Figure 1C). In group 3 rebound neurons (n = 5), the hyperpolarizing current threshold evoking RF was −212.00 ± 23.32 pA and the rheobase was 80.00 ± 8.94 pA (Figure 1D). The hyperpolarizing current threshold evoking RF was the highest among 3 groups (P < 0.0001), and the rheobase in group 3 rebound neurons was not significantly lower than in group 2 rebound neurons (80.00 ± 8.94 pA vs 102.5 ± 13.86 pA, P > 0.05) (Figure 1C). The amplitude of ADP and the latency were measured for RF in response to the threshold hyperpolarizing current evoking RF (Table 2). Concerning RF, the amplitude of ADP was larger in group 1 rebound neurons than in group 2 rebound neurons. The latency of RF was longer in group 1 rebound neurons than in group 2 rebound neurons and 3 rebound neurons.
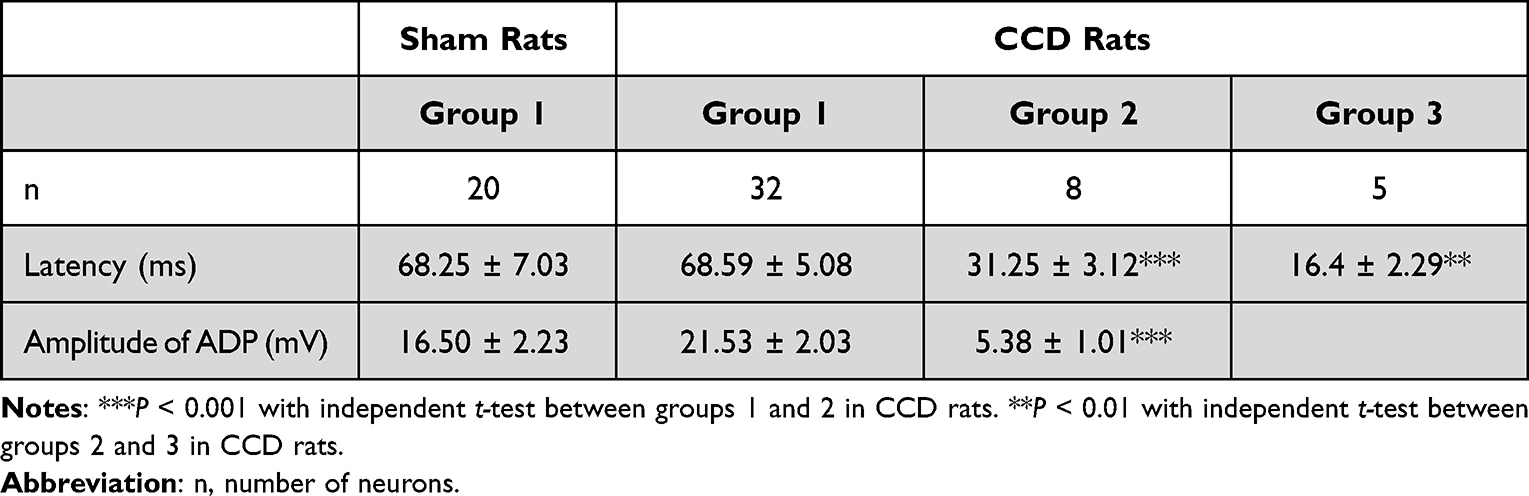 |
Table 2 The Latency of RF and ADP Amplitude of RF in the 3 Groups of Rebound Neurons |
Spontaneous Firing of Rebound Neurons in the CCD Rats
We evaluated the excitability of neurons collected for the present study and other 60 chronic compressed neurons from other studies that were not recorded for RF. The spontaneous firing patterns in these neurons were similar to RF, indicating that the excitability of rebound neurons after injury may increase. Among them, four injured neurons discharged spontaneous ADP-firing (Figure 2A). Two injured neurons discharged spontaneous burst firing (Figure 2B). The other two injured neurons discharged spontaneous tonic firing. After a slight hyperpolarization, the spontaneous tonic firing changed to a spontaneous repetitive burst firing (Figure 2C), implying hyperpolarization modulated neuron’s excitability and firing pattern.
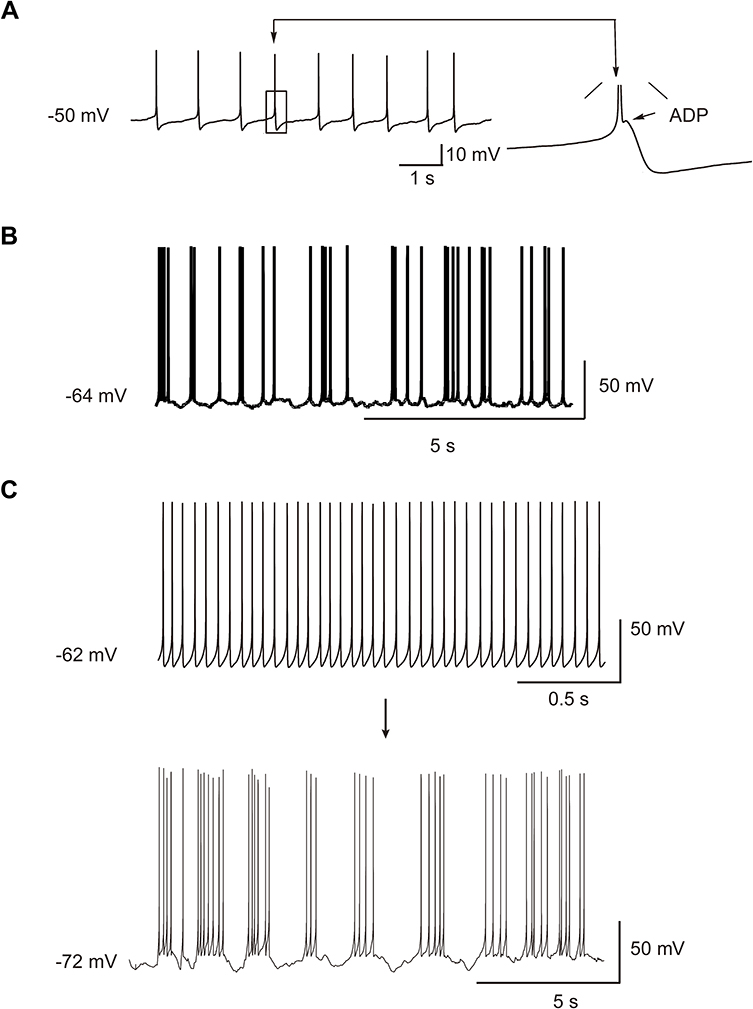 |
Figure 2 Spontaneous firing of DRG neurons. (A) Spontaneous ADP firing. The arrow indicates a single, enlarged ADP. ADP: after-depolarizing potential. (B) Spontaneous burst firing. (C) Spontaneous tonic firing turned to a spontaneous burst firing after a slight hyperpolarization. |
T-Type Ca2+ Currents in DRG Neurons
The T-type Ca2+ currents were recorded according to a voltage protocol shown in Figure 3A-a1 (inset). A total of 18/32 injured DRG neurons and 18/60 sham DRG neurons exhibited T-type calcium currents (56.25% vs 30.00%, P < 0.05). Low voltage activated (LVA) T-type Ca2+ currents and high voltage activated (HVA) Ca2+ currents were recorded with ramp voltage command from −90 mV to 30 mV (Figure 3A-a1, a2). The LVA T-type Ca2+ currents peak was around −40 mV, while the HVA Ca2+ currents peak was around 0 mV (Figure 3A-a1, a2). The voltage at which the currents approached peak (Vpeak) was different between the CCD and sham rats (n = 18, −41.39 ± 1.40 mV vs −48.22 ± 1.88 mV, P < 0.01) (Figure 3A-a3).
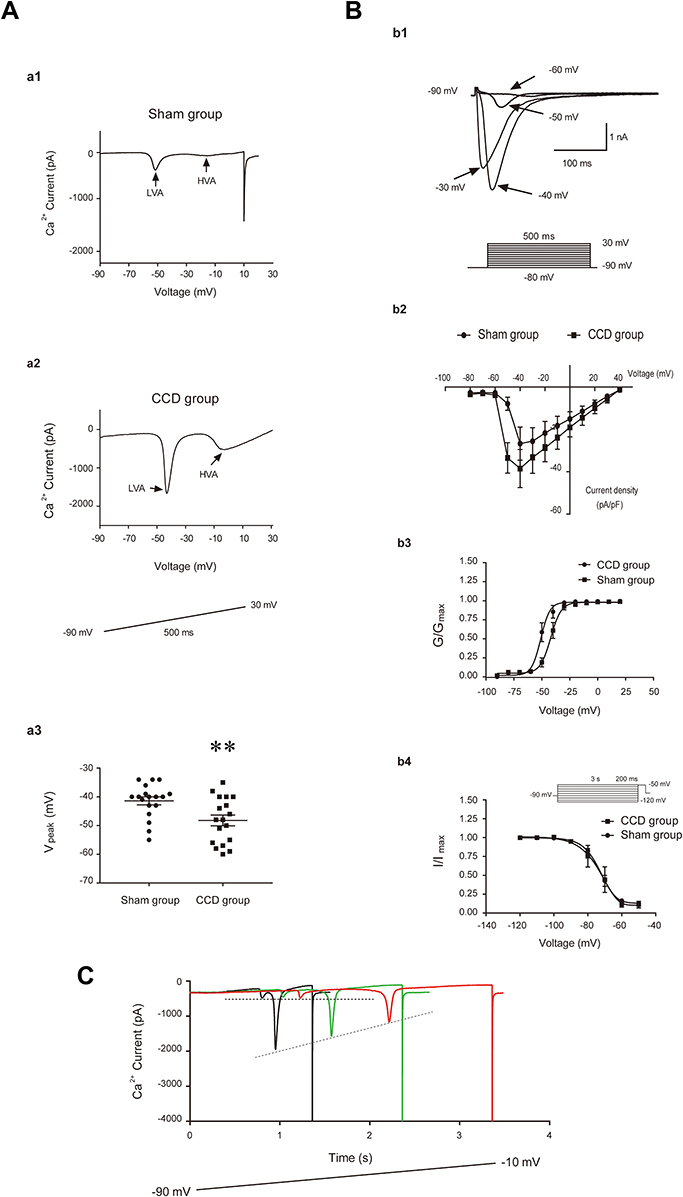 |
Figure 3 The T-type Ca2+ currents of DRG neurons. (A) T-type Ca2+ currents in ramp voltage recording and Vpeak of T-type Ca2+ currents. a1 and a2: T-type Ca2+ currents recorded in the sham and CCD rats. LVA: low voltage-activated currents. HVA: high voltage-activated currents. Inset: the protocol of ramp voltage clamp. The arrow and gray rectangle indicate LVA and HVA, respectively. a3: The Vpeak of T-type Ca2+ currents in the CCD and sham rats, **P < 0.01. (B) T-type Ca2+ currents in step voltage recording and activation, inactivation of T-type Ca2+ currents. b1: T-type Ca2+ currents in step voltage recording. Inset: the protocol of step voltage clamp. b2: I–V plots of T-type Ca2+ currents in sham and CCD group. b3 and b4: steady-state activation and inactivation curves of T-type Ca2+ currents in the CCD and sham rats. After injury, the steady-state activation curve shifted toward left. G is conductance and Gmax is the maximal conductance, I is current and Imax is the maximal amplitude of current. (C) the LVA Ca2+ currents were recorded with increasing duration of the ramp voltage command. Two dashed lines indicate the first peak and the second peak of current, respectively. The second peak gradually decreased with increasing duration of the ramp voltage command. |
The step voltage protocol to analyze activation and inactivation is shown in Figure 3B-b1 (inset). At −40 mV, the T-type Ca2+ currents were higher in the CCD rats (n = 9) than in the sham rats (n = 10), but the result was not statistically significant (44.04 ± 8.71 vs 25.61 ± 8.11 pA/pF, P > 0.05) (Figure 3B-b2). After CCD injury, the steady state activation curve of T type Ca2+ channel was shifted toward the left (CCD rats: n = 9, V50 = −50.94 ± 0.79 mV, K = 4.68 ± 0.70; sham rats: n = 10, V50 = −42.02 ± 0.79 mV, K = 4.09 ± 0.80; P < 0.0001 for V50 and P > 0.05 for K) (Figure 3B-b3), indicating the activation of T type Ca2+ channels in injured DRG neurons was enhanced. However, there was no difference regarding the steady-state inactivation between the CCD and sham rats (CCD rats: n = 8, V50 = −73.39 ± 0.80 mV, K = 5.37 ± 0.40; sham rats: n = 8, V50 = −74.87 ± 1.56 mV, K = 5.08 ± 0.67; P > 0.05 for both V50 and K) (Figure 3B-b4).
The ramp voltage command was applied to record Ca2+ currents. In response to a long duration between 1 and 3 s, the LVA Ca2+ currents were divided into two peaks (Figure 3C). There were 5 neurons from the CCD rats and 3 neurons from the sham rats exhibiting two peaks of LVA Ca2+ currents. The first peak was around −60 mV, the second peak was around −40 mV. In response to an increasing duration of ramp voltage command, the amplitude of the first current peak did not change, while the amplitude of the second peak gradually reduced (Figure 3C), suggesting that the first peak was not influenced by the ramp slope and may be spontaneously activated around RMP and then contribute to spontaneous firing. The current peak amplitudes around −60 mV were 5.31 ± 0.45 pA/pF in the sham rats and 3.62 ± 0.51 pA/pF in the CCD rats (Figure 3C).
Discussion and conclusions
The ability to generate classical AP in response to depolarizing input signifies cells’ excitability. However, some neurons have the ability to generate RF in response to hyperpolarizing input. In the present study, we investigated the features of RF of DRG neurons.
Unlike an AP, RF is generated after the termination of hyperpolarization. Hyperpolarization makes more Na+ and Ca2+ channels in a state of non-inactivated, which in turn open and then depolarize MP to reach AP threshold after the termination of hyperpolarization. Therefore, theoretically rebound neurons should be low threshold neurons. Consistently, the rheobase is lower in rebound neurons than in non-rebound neurons. In this study, we classified rebound neurons according to the occurrence of ADPs in AP and RF. In group 1 rebound neurons, both classical AP and RF coexisted with prominent large-amplitude ADPs. The value of rheobase was similar to the hyperpolarizing current threshold evoking RF. In group 2 rebound neurons, the classical AP did not coexist with ADPs, but RF coexisted with prominent small-amplitude ADPs. The value of rheobase was higher than the hyperpolarizing current threshold evoking RF. Compared with group 1 rebound neurons, the hyperpolarizing current threshold evoking RF was higher. The above results suggest that neurons’ ability to generate RF may be related to the amplitude of ADP in the 2 groups of neurons. In group 3 rebound neurons, both classical AP and RF did not coexist with ADPs. The value of rheobase was not significantly different from that in group 2, but the hyperpolarizing current threshold evoking RF was the highest among 3 groups of rebound neurons. Collectively, the ability to generate RF was low in neurons without ADP in RF and the excitability was low in neurons without ADP in AP, suggesting that both the hyperpolarizing current threshold evoking RF and rheobase may be associated with ADP. It is known that T-type Ca2+ currents contribute to the ADP.28,29 Considering the occurrence of ADP in RF and the effect of Ni2+ on ADP, we proposed that T-type Ca2+ channels contribute mostly to the generation of RF in group 1 and 2 rebound neurons. In group 3 rebound neurons, TTX-sensitive Na+ currents were mainly responsible for the generation of RF. In all rebound neurons, Ih was not essential for the generation of RF, but it might enhance the generation of RF. Group 1 and group 2 rebound neurons were observed in both the CCD and sham rats, but group 3 rebound neurons were only exhibited in the CCD rats, reinforcing that neurons’ excitability increased after DRG injury.
After DRG injury, the proportion of rebound neurons significantly increased. As previously described, DRG neurons become highly excitable.22 Consistently, more injured neurons displayed T-type Ca2+ currents and the Vpeak shifted to a more negative value, suggesting that the generation of RF and rebound neurons’ excitability might be enhanced. Thus, rebound neurons can be sensitive to both hyperpolarization and depolarization. Highly excitable rebound neurons might be able to generate AP/RF in response to a weak depolarization/hyperpolarization around RMP. Injured DRG neurons also exhibited Ca2+ currents with peak at −60 mV which were activated by ramp voltage with long duration and not influenced by ramp voltage slope. Accordingly, injured neurons fired spontaneously. The spontaneous firing patterns were similar to rebound firing patterns, including ADP-firing, non-ADP burst firing and tonic firing. In all, the above results suggest that injured rebound neurons may contribute to tactile allodynia and spontaneous neuropathic pain through low threshold firing and spontaneous firing.
Collectively, RF is one of the intrinsic properties of neurons. In the central nervous system, the inhibitory synapse transmission may induce excitatory signals via RF.10,14–16,30,31 Although RF was elicited in vitro14,30,31 or in vivo from isoflurane-anaesthetized adult mice,32 however, Alviña et al found that intact mouse or rat deep cerebellar nuclei neurons rarely show RF under physiological conditions in vivo,33 implying RF may not be observed in awake behaving intact animals but is most likely to contribute to neural hyperexcitability under pathophysiological condition. In PNS, different from central neuron, DRG neurons do not receive synapse transmission, but there are a variety of receptors on DRG neurons. Inhibitory receptors’ activation induces hyperpolarization of DRG neurons34 and then induces more activity of DRG neurons via RF. As a result, inhibitory receptors of DRG neurons may not be an effective therapeutic target. It is known that some DRG neurons exhibit membrane potential fluctuations.35 Injured rebound neurons potentially generate spontaneous AP and RF in response to small membrane potential fluctuations, contributing to neuropathic pain.
Conclusions
After DRG injury, DRG neurons exhibited three types of RF: both RF and AP with ADP, RF with ADP and AP without ADP, both RF and AP without ADP. Intact DRG neurons exhibited one type of RF: both RF and AP with ADP. Rebound neurons are presumably low threshold neurons sensitive to both excitatory and inhibitory stimuli. The T-type Ca2+ channels are critical for rebound firing. After injury, more DRG neurons become hyper-excitable and exhibit RF or even discharge spontaneously, potentially being responsible for neuropathic pain and allodynia.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant No. 81300972), Natural Science Foundation of Shaanxi province (grant No. 2020JM-075) and Scientific Research Foundation for Returned Scholars of Ministry of Education (CN).
Disclosure
The authors report no conflicts of interest.
References
1. Rajaram E, Kaltenbach C, Fischl MJ, et al. Slow NMDA-mediated excitation accelerates offset-response latencies generated via a post-inhibitory rebound mechanism. eNeuro. 2019;6. doi:10.1523/ENEURO.0106-19.2019
2. Ferrante M, Shay CF, Tsuno Y, et al. Post-inhibitory rebound spikes in rat medial entorhinal layer II/III principal cells: in vivo, in vitro, and computational modeling characterization. Cereb Cortex. 2017;27(3):2111–2125. doi:10.1093/cercor/bhw058
3. Hansel C. Reading the clock: how Purkinje cells decode the phase of olivary oscillations. Neuron. 2009;62(3):308–309. doi:10.1016/j.neuron.2009.04.020
4. Mesnage B, Gaillard S, Godin AG, et al. Morphological and functional characterization of cholinergic interneurons in the dorsal horn of the mouse spinal cord. J Comp Neurol. 2011;519(16):3139–3158. doi:10.1002/cne.22668
5. Sangrey T, Jaeger D. Analysis of distinct short and prolonged components in rebound spiking of deep cerebellar nucleus neurons. Eur J Neurosci. 2010;32(10):1646–1657. doi:10.1111/j.1460-9568.2010.07408.
6. Astori S, Wimmer RD, Prosser HM, et al. The Ca(V)3.3 calcium channel is the major sleep spindle pacemaker in thalamus. Proc Natl Acad Sci U S A. 2011;108:13823–13828. doi:10.1073/pnas.1105115108
7. Blitz DM. Circuit feedback increases activity level of a circuit input through interactions with intrinsic properties. J Neurophysiol. 2017;118(2):949–963. doi:10.1152/jn.00772.2016
8. Park YG, Park HY, Lee CJ, et al. Ca(V)3.1 is a tremor rhythm pacemaker in the inferior olive. Proc Natl Acad Sci U S A. 2010;107:10731–10736. doi:10.1073/pnas.1002995107
9. Pressler RT, Rozman PA, Strowbridge BW. Voltage-dependent intrinsic bursting in olfactory bulb Golgi cells. Learn Mem. 2013;20(9):459–466. doi:10.1101/lm.031856.113
10. Kim J, Kim Y, Nakajima R, et al. Inhibitory Basal Ganglia inputs induce excitatory motor signals in the thalamus. Neuron. 2017;95(1181–1196):e1188. doi:10.1016/j.neuron.2017.08.028
11. Picton LD, Sillar KT, Zhang HY. Control of xenopus tadpole locomotion via selective expression of ih in excitatory interneurons. Curr Biol. 2018;28(3911–3923):e3912. doi:10.1016/j.cub.2018.10.048
12. Li Y, Davey RA, Sivaramakrishnan S, Lynch WP. Postinhibitory rebound neurons and networks are disrupted in retrovirus-induced spongiform neurodegeneration. J Neurophysiol. 2014;112(3):683–704. doi:10.1152/jn.00227.2014
13. Sivaramakrishnan S, Lynch WP. Rebound from inhibition: self-correction against neurodegeneration? J Clin Cell Immunol. 2017;8. doi:10.4172/2155-9899.1000492
14. Edgerton JR, Jaeger D. Optogenetic activation of nigral inhibitory inputs to motor thalamus in the mouse reveals classic inhibition with little potential for rebound activation. Front Cell Neurosci. 2014;8:36. doi:10.3389/fncel.2014.00036
15. Goldberg JH, Farries MA, Fee MS. Basal ganglia output to the thalamus: still a paradox. Trends Neurosci. 2013;36(12):695–705. doi:10.1016/j.tins.2013.09.001
16. Kim J, Kim D. Rebound excitability mediates motor abnormalities in Parkinson’s disease. BMB Rep. 2018;51:3–4. doi:10.5483/BMBRep.2018.51.1.004
17. Walsh DA, Brown JT, Randall AD. Neurophysiological alterations in the nucleus reuniens of a mouse model of Alzheimer’s disease. Neurobiol Aging. 2020;88:1–10. doi:10.1016/j.neurobiolaging.2019.12.006
18. Cardenas CG, Del Mar LP, Scroggs RS. Variation in serotonergic inhibition of calcium channel currents in four types of rat sensory neurons differentiated by membrane properties. J Neurophysiol. 1995;74(5):1870–1879. doi:10.1152/jn.1995.74.5.1870
19. Kanda H, Clodfelder-Miller BJ, Gu JG, Ness TJ, DeBerry JJ. Electrophysiological properties of lumbosacral primary afferent neurons innervating urothelial and non-urothelial layers of mouse urinary bladder. Brain Res. 2016;1648:81–89. doi:10.1016/j.brainres.2016.06.042
20. Liu DL, Wang X, Chu WG, et al. Chronic cervical radiculopathic pain is associated with increased excitability and hyperpolarization-activated current (Ih) in large-diameter dorsal root ganglion neurons. Mol Pain. 2017;13:1744806917707127. doi:10.1177/1744806917707127
21. Ma C, LaMotte RH. Enhanced excitability of dissociated primary sensory neurons after chronic compression of the dorsal root ganglion in the rat. Pain. 2005;113(1):106–112. doi:10.1016/j.pain.2004.10.001
22. Song XJ, Hu SJ, Greenquist KW, Zhang JM, LaMotte RH. Mechanical and thermal hyperalgesia and ectopic neuronal discharge after chronic compression of dorsal root ganglia. J Neurophysiol. 1999;82:3347–3358. doi:10.1152/jn.1999.82.6.3347
23. Yang J, Xie MX, Hu L, et al. Upregulation of N-type calcium channels in the soma of uninjured dorsal root ganglion neurons contributes to neuropathic pain by increasing neuronal excitability following peripheral nerve injury. Brain Behav Immun. 2018;71:52–65. doi:10.1016/j.bbi.2018.04.016
24. Zhang JM, Song XJ, LaMotte RH. Enhanced excitability of sensory neurons in rats with cutaneous hyperalgesia produced by chronic compression of the dorsal root ganglion. J Neurophysiol. 1999;82:3359–3366. doi:10.1152/jn.1999.82.6.3359
25. Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16(2):109–110. doi:10.1016/0304-3959(83)90201-4
26. Wang Y, Huo F. Inhibition of sympathetic sprouting in CCD rats by lacosamide. Eur J Pain. 2018;22(9):1641–1650. doi:10.1002/ejp.1246
27. Liu QY, Chen W, Cui S, et al. Upregulation of Cav3.2 T-type calcium channels in adjacent intact L4 dorsal root ganglion neurons in neuropathic pain rats with L5 spinal nerve ligation. Neurosci Res. 2019;142:30–37. doi:10.1016/j.neures.2018.04.002
28. Cain SM, Snutch TP. Contributions of T-type calcium channel isoforms to neuronal firing. Channels. 2010;4(6):475–482. doi:10.4161/chan.4.6.14106
29. Dubreuil AS, Boukhaddaoui H, Desmadryl G, et al. Role of T-type calcium current in identified D-hair mechanoreceptor neurons studied in vitro. J Neurosci. 2004;24:8480–8484. doi:10.1523/JNEUROSCI.1598-04.2004
30. Nejad MM, Rotter S, Schmidt R. Basal ganglia and cortical control of thalamic rebound spikes. Eur J Neurosci. 2021;54(1):4295–4313. doi:10.1111/ejn.15258
31. Zheng N, Raman IM. Prolonged post inhibitory rebound firing in the cerebellar nuclei mediated by group I metabotropic glutamate receptor potentiation of L-type calcium currents. J Neurosci. 2011;31(28):10283–10292. doi:10.1523/JNEUROSCI.1834-11.2011
32. Otomo K, Perkins J, Kulkarni A, Stojanovic S, Roeper J, Paladini CA. In vivo patch-clamp recordings reveal distinct subthreshold signatures and threshold dynamics of midbrain dopamine neurons. Nat Commun. 2020;11(1):6286. doi:10.1038/s41467-020-20041-2
33. Alvina K, Walter JT, Kohn A, Ellis-Davies G, Khodakhah K. Questioning the role of rebound firing in the cerebellum. Nat Neurosci. 2008;11(11):1256–1258. doi:10.1038/nn.2195
34. Gong L, Gao F, Li J, et al. Oxytocin-induced membrane hyperpolarization in pain-sensitive dorsal root ganglia neurons mediated by Ca(2+)/nNOS/NO/KATP pathway. Neuroscience. 2015;289:417–428. doi:10.1016/j.neuroscience.2014.12.058
35. Liu CN, Michaelis M, Amir R, Devor M. Spinal nerve injury enhances subthreshold membrane potential oscillations in DRG neurons: relation to neuropathic pain. J Neurophysiol. 2000;84:205–215. doi:10.1152/jn.2000.84.1.205
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.