")
Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 8
Portal hypertension: an uncommon clinical manifestation of Takayasu arteritis in a 9-year-old child
Authors Herrera CN, Tomala-Haz JE
Received 9 June 2016
Accepted for publication 2 August 2016
Published 15 November 2016 Volume 2016:8 Pages 115—118
DOI https://doi.org/10.2147/OARRR.S114689
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Chuan-Ju Liu
Cristina N Herrera, Javier E Tomala-Haz
Department of Pediatric Rheumatology, Dr Roberto Gilbert Children’s Hospital, Guayaquil, Ecuador
Abstract: Takayasu arteritis (TA) is the third most common childhood vasculitis and its clinical manifestations depend on the arteries involved. We report a case of a 9-year-old boy with multiple aneurysms in carotid and iliac arteries, subclavian and coronary arteries, and abdominal aorta. At the age of 7 years, he presented with recurrent fever and hepatosplenomegaly. An angio-computed tomography scan showed aneurysms in the left subclavian artery, abdominal aorta, and both proximal iliac arteries. He was diagnosed with TA and was treated with corticosteroids, aspirin, and enalapril. One year later, he was admitted to Dr Roberto Gilbert Children’s Hospital because of intracranial hemorrhage. Angiography revealed enlargement of aneurysms enlargement and new aneurysms. He also developed portal hypertension. Treatment with intravenous corticosteroids, azathioprine, and monthly intravenous cyclophosphamide was begun. After 6 months of no improvement, infliximab was begun. The aim of this article was to report the concurrence of coronary involvement and portal vein hypertension in pediatric TA because there were scarce reports on this matter.
Keywords: Takayasu arteritis, coronary aneurysm, children, portal hypertension
Case report
At the age of 6 years, our patient presented with intermittent fever and later on clubbing of fingers and hepatosplenomegaly. A year later, an angio-computed tomography (CT) scan performed at another hospital showed multiple aneurysms on the left subclavian branch, distal abdominal aorta, and proximal iliac arteries (Figure 1A and B). Based on clinical manifestations and CT findings, Takayasu arteritis (TA) was diagnosed, and treatment was started with oral corticosteroids, aspirin, and enalapril. Unfortunately, due to a lack of written medical records from the other hospital, we could not get more precise information regarding the treatment given. At the age of 9 years, the child was admitted to our hospital for the first time and was put on 10 mg/day (0.5 mg/kg/day) of prednisone plus aspirin and enalapril that were continued.
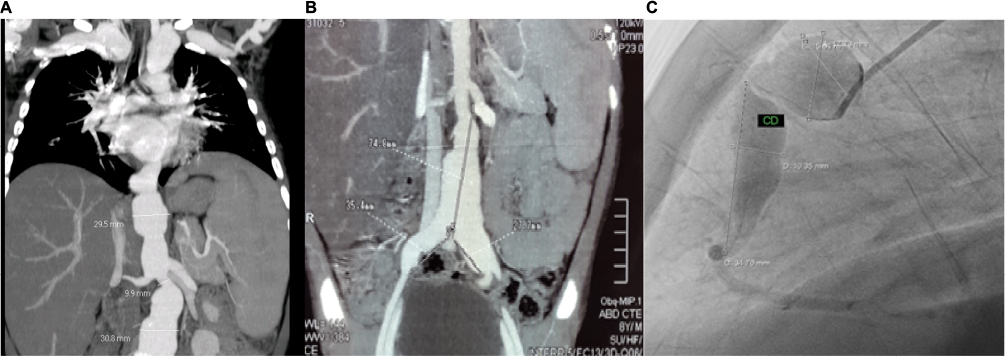 | Figure 1 (A, B) Angio-CT demonstrates subclavian, abdominal aorta, and both proximal iliac arteries aneurysms, (C) angiography shows right coronary aneurysms. Note: Courtesy of Cardiology Department and Radiology Department of Dr Roberto Gilbert Children’s Hospital. Abbreviations: CT, computed tomography; CD, right coronary. |
Owing to a history of 1 month of daily fever and 1 week of headache, the child was hospitalized. A head CT was performed, which revealed a right frontal intracerebral hemorrhage; therefore, aspirin was stopped. An abdominal ultrasonography (showed signs of liver inflammation and also of mild portal hypertension. Only one angio-CT body scan was ever performed on this patient since the diagnosis was made until his admission to our medical facility. A control body angio-CT scan performed at our hospital showed enlargement of aneurysms when compared with the first scan; therefore, pulse therapy with methylprednisolone at a dose of 30 mg/kg/day for 3 consecutive days was begun followed by maintenance prednisone at a dose of 2 mg/kg/day.
Cardiology and Neurosurgery Department performed a body pan-angiography that showed extensive damage and enlargement of aneurysms in both subclavian arteries, left internal carotid, distal abdominal aorta, both iliac arteries, and right coronary (middle and proximal segments) and left coronary arteries (trunk and anterior descending) (Figure 1C). An incomplete right bundle branch block was detected on electocardiogram. Neither murmurs nor arterial hypertension was found. The laboratory findings were as follows: white blood cells 4×100 cells/mm3 (neutrophils 65%, lymphocytes 29%), hemoglobin 10 mg/dL, hematocrit 30.6%, aspartate transferase (AST) 256 U/L, alanine transaminase (ALT) 235 U/L, alkaline phosphatase 760 U/L, albumin 2.7 g/dL, and globulin 4.0 g/dL. Prothrombin time/partial thromboplastin time, renal function, and C-reactive protein were normal. The Mantoux test was negative, and the pediatric vasculitis damage index (PDVI) was 3/72.
Twelve days after receiving monthly intravenous cyclophosphamide (increased from 500 mg/m2/month until 750 mg/m2/month), azathioprine (2 mg/kg/day), and subcutaneous enoxaparin, he developed headache and diplopia. Head CT demonstrated a posterior cranial fossa hemorrhage with hydrocephalus and supratentorial hemorrhage. Enoxaparin was discontinued and an external ventriculostomy was performed.
During treatment with cyclophosphamide and azathioprine, steroids were weaned off 20% of the dose, every 3 weeks until reaching 0.5 mg/Kg/day. During the first 5 months of treatment, the child was afebrile with no clinical worsening.
Treatment with infliximab was delayed because of the patient’s concurrent infection with Candida in oral cavity and urine. Azathioprine was temporarily discontinued because of lymphopenia (700 cells/mm3) and 15-fold increase in the levels of liver enzymes (AST/ALT). Once treatment with infliximab began, azathioprine was discontinued. Infliximab was administered at a dose of 8 mg/kg following the 0-, 2-, 6-, and 8-week regimen. During treatment with antitumor necrosis factor-alpha (TNF-α), the patient developed several infections such as complicated pneumonia with pleural effusion, a second episode of oral candidiasis, dengue fever, and brain abscess. In order to rule out concomitant Mycobacterium tuberculosis infection in this patient, a polymerase chain reaction test was performed on pleural fluid and cerebrospinal fluid which turned out to be negative.
After 5 months of treatment with infliximab, it was interrupted because of infections and no signs of improvement. On the last control body angio-CT scan, abdominal aneurysm had grown 1 cm in diameter and PDVI increased to 4. Our patient died 16 months after his first admission to our hospital. Written informed consent was obtained from the patient’s parent to publish this paper and accompanying images.
Discussion
TA is a rare chronic idiopathic granulomatous vasculitis that affects mainly the aorta and its main branches. Vessel inflammation begins from the adventitia and progresses to the intima producing segmental stenosis, occlusion, dilatation, and/or aneurysms.1–4
Clinical manifestations can vary depending on the arteries involved. Patients can be asymptomatic and incidental findings may lead to diagnosis (eg, decreased peripheral artery pulses, blood pressure difference between arms and legs, bruits and hypertension) or may present dramatically (eg, congestive heart failure, cerebrovascular event, or aortic aneurysm disruption).1–4
TA is rare in childhood, but up to 20% of patients are diagnosed under the age of 19 years.5 Although there is a lack of data on childhood-onset TA (c-TA) worldwide, a few series of cases of c-TA have been reported. Diagnosis delay observed in these cases was 17 months and in our patient it was 24 months.1–3,6 He had neither murmurs nor hypertension at the beginning, but one remarkable sign was daily fever. Hepatosplenomegaly is not a frequent sign of TA, but in this case led to perform a body angio-CT which helped to make the diagnosis.
Our patient had portal hypertension confirmed by Doppler ultrasonography with no evidence of portal thrombosis. Because of the severity of the disease, a liver biopsy was not performed. In the literature, we found three cases of TA with liver involvement, in which two had hepatic sinusoidal dilatation and the other had hepatosplenomegaly even before the peripheral pulse difference stage.7,8
The body angiogram performed in our patient revealed coronary involvement. Coronary aneurysms have been described in childhood vasculitis, mainly in Kawasaki disease. We found seven case reports of coronary involvement in cTA, of which two of them were identified postmortem.9–13 Endo et al reported an adult cohort of 130 patients with TA; among them, 81 underwent selective coronary angiography, and 31 of these patients (38%) (four males and 27 females, mean age 41.1±13.2 years) had reported abnormal coronary angiographic findings.14 We believe it is important to make a coronary evaluation in all of the patients with TA even in the absence of symptoms.
Our patient had two intracranial hemorrhages, one associated with the use of aspirin and the other with the use of enoxaparin. The use of an anticlotting agent is controversial, and there is no agreement on this matter yet.6,15,16 After the second intracranial hemorrhage, our patient needed an external ventriculostomy, and according to the patient’s risk–benefit ratio neither aspirin nor anticoagulation was continued.
Morbidity and mortality were higher in children than in adults due to a quick development of hypertension and heart failure.3,4 On follow-up, a mortality of 10%–35% had been reported.2–4. Treatment was based on high-dose corticosteroids and immunosuppressive drugs (eg, methotrexate, azathioprine, and cyclophosphamide).2,5,17
Considering the fact that proinflammatory cytokines like TNF-α and interleukin-6 (IL-6) are implicated in blood vessel inflammation and injury, inhibitory molecules such as infliximab (anti-TNF-α) and tocilizumab (anti-IL-6) have been used in cases of corticosteroid resistance or relapses.18,19 A large series of cases with a long-term follow-up from the Mayo Clinic in the US reported better survival rates than the past (97% at 10 years and 86% at 15 years), and Serra et al5 showed a decrease in the mortality rate by 2.8% during the 2000–2010 follow-up period. The use of disease-modifying antirheumatic drugs and biological agents seems to be associated with a decreased rate of mortality.2 However, due to the absence of randomized trials comparing different immunosuppressive agents, no immunosuppressive agent can be considered more effective than others.5 Other interventional or surgical options are available (balloon dilatation, stents placement, and bypass grafting).6,15,16.
Eleftheriou et al2 found permanent damage items on PVDI scoring in the follow-up (score ≥3) were associated with mortality (P=0.024). Other factors such as the time to diagnosis >12 months, cardiac involvement, or need for biological therapy were not significantly associated.2. We finally conclude that the severity of the disease (PVDI was always >3) led to a poor prognosis in our patient.
Disclosure
The authors report no conflicts of interest in this work.
References
Szugye HS, Zeft AS, Spalding SJ. Takayasu Arteritis in the pediatric population: a contemporary United States-based single center cohort. Pediatr Rheumatol Online J. 2014;12:21. | ||
Eleftheriou D, Varnier G, Dolezalova P, McMahon AM, Al-Obaidi M, Brogan PA. Takayasu arteritis in childhood: retrospective experience from a tertiary referral centre in the United Kingdom. Arthritis Res Ther. 2015;17:36. | ||
Katsicas M, Pompozib L, Russo R. Arteritis de Takayasu en pediatría [Takayasu arteritis in pediatrics]. Arch Argent Pediatr. 2012;110(3):251–255. Spanish. | ||
Vaideeswar P, Deshpande JR. Pathology of Takayasu arteritis: a brief review. Ann Pediatr Cardiol. 2013;6(1):52–58. | ||
Serra R, Butrico L, Fugetto F, et al. Update in pathophysiology, diagnosis and management of Takayasu arteritis. Ann Vasc Surg. Epub 2016 May 27. | ||
Mathew AJ, Goel R, Kumar S, Danda D. Childhood-onset Takayasu arteritis: an update. Int J Rheum Dis. 2016;19(2):116–126. | ||
Durant C, Martin J, Hervier B, Gournay J, Hamidou M. Takayasu arteritis associated with hepatic sinusoidal dilatation. Ann Hepatol. 2011;10(4):559–561. | ||
Yotsuyanagi H, Chikatsu N, Kaneko Y, Kurokawa K. Takayasu’s arteritis in prepulseless stage manifesting lymph node swelling and hepatosplenomegaly. Intern Med. 1995;34(5):455–459. | ||
Baruteau AE, Martins RP, Boulmier D, et al. Acquired left ventricular submitral aneurysms in the course of Takayasu arteritis in a child. Congenit Heart Dis. 2012;7(1):76–79. | ||
Nakao M, Ong KK, Lim YP, Wong KY. Aortic intimal fold in Takayasu arteritis causing obstruction of left coronary ostium. Pediatr Cardiol. 2011;32 (7):990–992. | ||
Moortgat S, Tuerlinckx D, Bodart E, Khoury G, Moniotte S. Severe ostial stenosis of the left coronary artery in a 12-year-old girl as the first manifestation of Takayasu’s arteritis. Acta Cardiol. 2009;64(6):825–829. | ||
Limsuwan A, Khowsathit P, Pienvichit P. Left main coronary occlusion from Takayasu arteritis in an 8-year-old child. Pediatr Cardiol. 2007;28(3):234–237. | ||
Hlavaty L, Diaz F, Sung L. Takayasu arteritis of the coronary arteries presenting as sudden death in a white teenager. Am J Forensic Med Pathol. 2015;36(3):221–223. | ||
Endo M, Tomizawa Y, Nishida H, et al. Angiographic findings and surgical treatments of coronary artery involvement in Takayasu arteritis. J Thorac Cardiovasc Surg. 2003;125(3):570–577. | ||
Muranjan MN, Bavdekar SB, More V, Deshmukh H, Tripathi M, VaswaniR. Study of Takayasu’s arteritis in children: clinical profile and management. J Postgrad Med. 2000;46(1):3–8. | ||
Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis. 2009;68(3):318–323. | ||
Kötter I, Henes JC, Wagner AD, Loock J, Gross WL. Does glucocorticosteroid-resistant large-vessel vasculitis (giant cell arteritis and Takayasu arteritis) exist and how can remission be achieved? A critical review of the literature. Clin Exp Rheumatol. 2012;30(1 Suppl 70):S114–S129. | ||
Abisror N, Mekinian A, Lavigne C, Vandenhende MA, Soussan M, Fain O; Club Rhumatismes et Inflammation and SNFMI. Tocilizumab in refractory Takayasu arteritis: a case series and updated literature review. Autoimmun Rev. 2013;12(12):1143–1149. | ||
Loricera J, Blanco R, Hernández JL, Pina T, González-Vela MC, González-Gay MA. Biologic therapy in ANCA-negative vasculitis. Int Immunopharmacol. 2015;27(2):213–219. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.