")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Pore-Forming Toxins During Bacterial Infection: Molecular Mechanisms and Potential Therapeutic Targets
Received 28 May 2021
Accepted for publication 19 August 2021
Published 7 September 2021 Volume 2021:15 Pages 3773—3781
DOI https://doi.org/10.2147/DDDT.S322393
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Haijie Hu, Min Liu, Shuang Sun
Institute of Biomedical Sciences, Shandong Provincial Key Laboratory of Animal Resistance Biology, Collaborative Innovation Center of Cell Biology in Universities of Shandong, College of Life Sciences, Shandong Normal University, Jinan, 250014, People’s Republic of China
Correspondence: Shuang Sun
Institute of Biomedical Sciences, Shandong Provincial Key Laboratory of Animal Resistance Biology, Collaborative Innovation Center of Cell Biology in Universities of Shandong, College of Life Sciences, Shandong Normal University, Jinan, 250014, People’s Republic of China
Tel/Fax +86-531-8618-2516
Email [email protected]
Abstract: Bacterial infections are predominantly treated with antibiotics, and resistance to antibiotics is becoming an increasing threat to our health. Pore-forming toxins (PFTs) are virulence factors secreted by many pathogenic bacterial strains, both in acute and chronic infections. They are special membrane-targeting proteins that exert toxic effects by forming pores in the cell membrane. Recent studies have elucidated the structure of PFTs and the detailed molecular mechanisms of their pathogenicity. Here, we discuss recent findings that highlight the regulatory mechanisms and important roles of two types of PFTs, α-PFTs and β-PFTs, in mediating the virulence of bacteria, and the therapeutic potential of targeting PFTs for antibacterial treatment. Therapeutic strategies based on PFTs are highly specific and may alleviate the issue of increasing resistance to antibiotics.
Keywords: bacterial infections, pore-forming toxins, virulence factors, disease, treatment
Introduction
Currently, most bacterial infections are treated with one or more antibiotics, but antibiotic resistance has become a significant concern, and has led to the need for new therapy targets and strategies to combat bacterial infections. Pore-forming toxins (PFTs) are produced by a variety of organisms that are widely found in eukaryotes and prokaryotes, including pathogenic bacteria.1,2 They act as crucial virulence factors in the pathogenicity of bacteria by destroying the cell barrier and promoting the survival of pathogenic bacteria under various stimuli and harsh environmental pressures.3 PFTs also can attack the human innate and adaptive immune defense systems, enabling the pathogen to persistently attack the host. Reducing the production of PFTs or interrupting their pathogenesis are therefore important steps to reduce their pathogenicity. PFTs are divided into two categories, α-PFTs and β-PFTs, according to the secondary structure that binds to the cell membrane, namely α-helices or β-barrels.4 Both α-PFTs and β-PFTs are further divided into three families, respectively. The synthesis of various PFTs is regulated via co-ordination of a variety of transcriptional regulators,5,6 but the specific mechanisms of most PFTs are not clear. The PFT structure has recently been elucidated, which has provided the basis for a deeper understanding of its pathogenic mechanism.3,7 When initially secreted, PFTs are in the form of water-soluble monomers, and during the process of invasion, the monomers undergo specific assembly, and oligomerize into a polymer that forms a transmembrane structure, which is the pore.8–11 This oligomerization process may occur before the PFT inserts into the cell membrane, or during interaction of the monomers with membrane receptors.12 It has become necessary to continuously develop new treatments to cope with the toxin-producing pathogens.
In this review, we have focused on Bacillus cereus and Staphylococcus aureus as examples. They are both clinically significant pathogenic bacteria with secreted PFTs as their primary pathogenic factors during infection. We focus on recent studies on the toxicity characteristics of two α-PFTs predominantly produced by B. cereus, namely non-hemolytic enterotoxin (NHE) and hemolysin BL (HBL), and two β-PFTs predominantly produced by S. aureus, namely γ-hemolysin and leukocidin A/B (also named leukocidin G/H) (LukAB). We discuss their synthesis pathways, and corresponding regulatory and pathology mechanisms. With this information we hypothesize potential therapeutic targets to interfere with PFT toxicity.
Synthesis of PFTs
HBL toxin comprises three subunits: HBL-B, HBL-L1, and HBL-L2, encoded by the genes hblA, hblD, and hblC, respectively.13,14 These three genes are located on the operon hblCDAB, and the promoter is located upstream of hblC.15 Alignment of the deduced amino acid sequences of the three proteins indicates significant similarity (20 to 24% identical), and structural analysis of the HBL protein shows that all three components are almost entirely composed of α-helices. HBL-B and HBL-L1 contain predicted transmembrane fragments of 17 and 60 amino acid residues, respectively, at the same position, while HBL-L2 has no predicted transmembrane fragments. These observed similarities indicate that the HBL components are produced by duplication of common genes. Similar to HBL, the coding genes of the three components of NHE, namely NHE-A, NHE-B, and NHE-C, are nheA, nheB, and nheC, respectively, and in an operon containing the three open reading frames (ORFs).16 There is a high degree of homology between NHE and HBL, and similarities between NHE-A and HBL-C, NHE-B and HBL-D, and NHE-C and HBL-A, are the most significant.17,18 The gene encoding γ-hemolysin is present in more than 99.5% of human S. aureus isolates.12 γ-Hemolysin is composed of three subunits: two S components (HlgA and HlgC), and an F subunit (HlgB).19 Unlike hblCDAB and nheABC, hlgACB comprises two transcription units. The first transcription unit comprises a single ORF, hlgB, which encodes an F subunit (HlgB), and the second unit comprises two ORFs, hlgA and hlgC, encoding two S components (HlgA and HlgC), respectively.20 The gene encoding LukAB is a part of the core genome of S. aureus, which exists in most genomes. It is composed of the S component, LukA/H, and the F component, LukB/G, and the sequence homology between the two components is unlike that of other toxins.21
Regulation of PFT Synthesis in Pathogens
HBL and NHE expression is regulated by multiple factors. For example, expression of the primary virulence factors of B. cereus, hblCDAB and nheABC, is regulated by the pleiotropic regulatory factor PlcR, redox regulator Fnr, two-component system ResDE, and the catabolic control protein CcpA, on the promoter regions of the two operons. These transcriptional regulatory factors may regulate toxin expression via a synergistic effect.22–24
PlcR is a small transcriptional activator that participates in the regulation of various virulence factors.25 During the exponential phase of bacterial growth, PlcR promotes specific interactions with PapR quorum sensing peptides.26 PlcR expression reaches its highest level at the beginning of the stationary phase of bacterial growth.25 High levels of PlcR activate most of the virulence factors, including the three components of HBL. PlcR appears to be a key regulator of the effective adaptation of B. cereus to the host environment. The transcription of another virulence factor, cytotoxin K, is also regulated by PlcR.27 PapR is a quorum sensing system signal molecule that is an autoinducing small signal peptide. It is secreted by bacterial cells and reintroduced into cells after processing.28 A previous study showed that a set of synthetic PapR derived peptides could inhibit the activation of many virulence factors. These small artificial peptides interfere with the normal quorum sensing systems and are a potential target as anti-toxin agents.26,29 Thus, novel small molecules that interfere with the quorum sensing systems, or facilitate the quorum quenching systems, may be selected as anti-virulence agents.
ResDE and Fnr are both regulators of redox regulatory pathways, and Fnr is a member of the iron-sulfur protein Crp/Fnr superfamily.30 ResDE is a typical two-component system (TCS) consisting of a membrane-bound histidine receptor kinase (ResE) and a cytoplasmic response regulator (ResD). ResE senses signals inside and outside the cell, such as oxygen limitation and NO, and undergoes autophosphorylation of the conserved histidine residues.31 ResD phosphorylation is achieved by transphosphorylation with ResE as a phosphate donor. The phosphorylated ResD acts as a transcription activator for the transcription of hblCDAB and nheABC. Phosphorylated ResD also interacts with Fnr to form a transcriptionally active complex.30,31 Fnr plays an important role in the fermentation and metabolism of B. cereus and the transcriptional regulation of the enterotoxins, HBL and NHE.22,32,33 CcpA is a member of the LacI family of transcriptional regulators. CcpA binds to a histidine-containing phosphocarrier protein to enable it to function as an effector. CcpA acts as a repressor to inhibit transcription in the catabolic regulation of HBL and NHE. The expression of NHE and HBL in CcpA-deficient strains is higher than that of the wild-type.22,31
The expression of γ-hemolysin and LukAB are strictly regulated by a highly complex, and multifaceted, regulatory network. An important regulator, accessory gene regulator (AGR) and several TCSs, such as S. aureus exoprotein expression RS (SaeRS), autolysis-related locus ArlRS, and staphylococcal respiratory response AB (SrrAB), are all involved in the regulatory systems.34–37 AGR acts as an activator of toxin production. At the population level, AGR is an auto-inducing peptide, which produces an effector RNA molecule, called RNAIII, in the process of activating its own synthesis. RNAIII acts as a direct or indirect regulator of many virulence genes, either up- or down-regulating their transcription and protein production.7 The SaeRS system is a key switch that regulates the production of leukocidins.34 It plays an important role in protecting microbial cells from exposure to the cytotoxic concentration of intracellular heme.38 In the ArlRS TCS, ArlS is a membrane-anchored sensor protein, and ArlR is an intracellular response regulator.37 The ArlRS two-component system inhibits the transcription of the Staphylococcus AGR operon via interaction with the AGR promoter. SrrAB regulates the pathogenic factors of S. aureus by directly binding the corresponding promoter region, including TSST-1, RNAIII, and protein A. SrrAB TCS is the main regulator of the respiratory growth and virulence of S. aureus.36
Pathogenicity of PFTs
Hemolysin HBL has been identified as a three-component toxin. Recent studies have demonstrated that the formation of pores in macrophages by HBL leads to the outflow of potassium ions, which triggers rapid cell death, and the pyrin domain-containing protein, NLRP3 inflammasome.39 HBL also has a variety of toxic effects, such as enteric toxicity, hemolysis, cytotoxicity, vascular permeability, and skin necrosis.40–42 It is a heat-sensitive protein comprising binding component B and two soluble components, L1 and L2, which assemble on the cell membrane in a specific order; B-L1-L2.43,44 HBL-B is the component believed to bind to the cell. The binding of HBL-B to target cells is a key step in the formation of the membrane pore, and pores will not form if there is insufficient B binding to the cells, or if there are excess quantities of L2 or L1 (Figure 1). Optimal pore-forming activity occurs when the B:L1:L2 ratio is 10:10:1.45 Of the three components, only HBL-B binds to Chinese hamster CHO cells in vitro, and one study showed that only HBL-L1, and not HBL-L2, binds to HBL-B.45 The three components of HBL are essential for its cytotoxicity and pore formation. The combination of any two components will not produce cytotoxicity, and maximum activity can only be exerted when all three components are present at the same time, and in the correct ratio.46
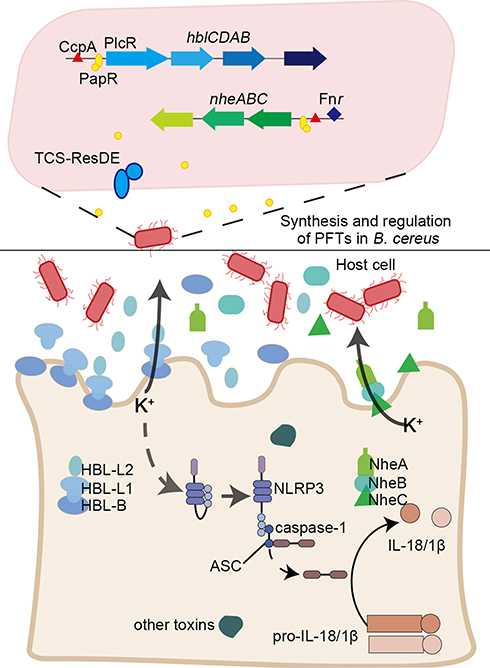 |
Figure 1 A brief summary of the regulation and pathogenic mechanisms of two typical α-PFTs, non-hemolytic enterotoxin (NHE) and hemolysin BL (HBL) toxins. The promoter regions of hblCDAB and nheABC are regulated by pleiotropic regulatory factor PlcR (along with the quorum sensing system signal molecule PapR), redox regulator Fnr, catabolic control protein CcpA, and two-component system ResDE. The three components of HBL and NHE were secreted and assemble on the cell membrane to form the pore in a specific order; B-L1-L2 and C-B-A. NHE and HBL cooperate to influence the secretion of K+ and activate an inflammatory response. |
The non-hemolytic enterotoxin, NHE, is a homolog of HBL, and another important toxin secreted by B. cereus. Single, or combinations of two NHE components cannot induce hemolysis; maximum activity is only achieved when all three subunits (NHE-A, NHE-B, and NHE-C) interact in a molar ratio of 10:10:1.44 This was confirmed in a eukaryotic, cell-free protein synthesis system using the three purified proteins, which showed that the toxicity of NHE was diminished when NHE-C was present at higher concentrations. NHE-B and NHE-C form a complex that was shown to directly bind to Vero kidney epithelial cells; however, only NHE-C was required to induce cytotoxicity.44 NHE-A is dispensable for initial cell binding, but necessary for cytotoxic effect.47,48 NHE was recently reported to induce rapid cell death via the NLRP3 inflammasome, and NHE and HBL cooperate to activate an inflammatory response.49 Compared with the wild-type, the cytotoxicity of the Δnhe mutant was shown to be reduced, and the B. cereus Δhbl and Δnhe mutant strain failed to activate the NLRP3 inflammasome.39,41,49 Based on this feature, screening small molecules to target one of these components, in particular HBL-B or NHE-C, may be an effective strategy to inhibit the cytotoxicity of HBL and NHE.
The pore-forming γ-hemolysin and LukAB secreted by S. aureus are members of the β‑PFT family and consist of two different monomeric protein components.50 According to their chromatographic elution characteristics, these are named the S and F subunits, where S and F represent slow elution and fast elution, respectively.51 The S component is responsible for recognizing and binding to the cell membrane receptors, and recruiting the F component to dimerize through conformational changes. These proteins bind to the host leukocyte membrane and form beta-barrel pores that span the phospholipid bilayer (Figure 2).52 γ-Hemolysin exhibits lytic activity on human and rabbit cells and recognizes and binds to the red blood cell membrane as an octamer through direct interaction with host lipids and glycolipid derivatives.19 γ-Hemolysin HlgCB targets the human complement membrane receptors, C5a anaphylatoxin chemotactic receptor 1 and C5a anaphylatoxin chemotactic receptor 2, to mediate interaction with the cell membrane.53 γ-Hemolysin HlgAB also induces human phagocyte cell death by interacting with the chemokine receptors, chemokines CXC chemokine receptor 1 (CXCR1), CXCR2 and CC-chemokine receptor 2 (CCR2).12 This interaction appears to be species specific as, in a mouse model, HlgAB did not bind to the mouse neutrophil CCR2 receptor. In addition to the formation of induction pores, HlgAB and HlgCB can also induce pro-inflammatory reactions, which have been shown to be related to the pathogenesis of septic arthritis.12,19,54 Studies show that the S. aureus USA300 strain, lacking hlgABC, has less ability to form pores in human neutrophils than wild-type strains, and the mutant strain can improve the survival rate in a mouse bacteremia model.55
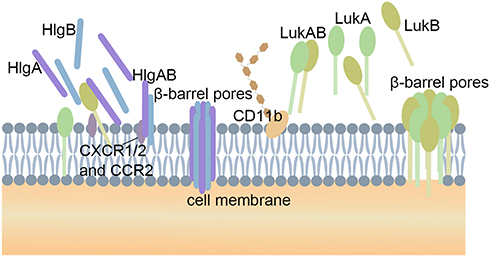 |
Figure 2 The pore-forming process of two β-PFTs. The water-soluble monomers S subunits (HlgA and HlgC) and one F subunit (HlgB) of γ-hemolysin target the receptors C5aR and C5L2 on the cell membrane. These proteins form beta-barrel pores that span the phospholipid bilayer. LukAB are pre-assembled into heterodimers in solution. Four LukAB heterodimers form an octamer on the cell membrane through direct interact with the receptor CD11b. The potential therapeutic targets are highlight by pentagram. |
LukAB, with a molecular weight of 75KDa, is a newly discovered member of the two-component interleukin family.56 LukAB fundamentally differs from the other two-component toxins in its mechanism of toxicity. The monomers are pre-assembled into heterodimers in solution, and these insert into the cell membrane and recruit other heterodimers to finally form an octamer β-barrel pore spanning the phospholipid bilayer (Figure 2).50 The individual subunits are not cytotoxic.57 Compared with other leukotoxins, this unique assembly method eliminates the pore formation process, and seems to increase the toxicity of LukAB. The ability of LukAB to cause disease in the body is largely due to its precise targeted killing of specific immune cells, including neutrophils, monocytes, macrophages, and lymphocytes, to avoid the hosts natural immunity.57,58 LukAB promotes bacterial survival by facilitating their escape from the neutrophils after phagocytosis.19,59 LukAB interacts directly with CD11b via the binding site in the I domain.60,61 As LukA and LukB are secreted as monomers and form heterodimers before recognizing and binding to receptors on the cell membrane, it would seem unlikely to be able to inhibit the toxic effect by inhibiting the dimerization of LukA and LukB. A better strategy may be to interfere with the binding of the LukAB complex to the cell, or with dimer oligomerization into the pore complexes. The I domain of mouse CD11b differs from human CD11b, which results in a weaker affinity of LukAB with mouse CD11b. Thus, even when mouse leukocytes express high levels of CD11b they have a greater tolerance to high doses of toxins.19
HlgAB, HlgCB, and LukAB can also activate NLRP3 inflammasomes in macrophages and monocytes, and promote their lysis and pro-inflammatory activity.62 These two types of pore-forming toxins mediate cell death by inserting into the cell membrane to “dissolve” the cell membrane.7 The formation of pores leads to a K+ efflux, considered to be the key factor for activation of the NLRP3 inflammasome. The NLRP3 inflammasome then initiates the release of caspase-1-dependent cytokines, IL-1β and IL-18, and induces cell necrosis.63
Potential PFT-Derived Strategies for the Treatment of Bacterial Infections
Antibiotics are, and have always been, the primary treatment for bacterial infections. However, with the continuous mutation of drug-resistant strains, and the overuse of antibiotics, the antibiotic-based treatment strategy needs to be challenged.64 It is worth noting that with the continuous progress of research, a variety of treatment methods have been explored and innovated. PFTs are a powerful tool in the pathogen’s armor to attack and resist the host organism. Potential treatment strategies based on PFTs can be divided into two parts: targeting the PFT-producing strains, and targeting the host cells after infection.
Strategies for Targeting PFT-Producing Strains
We put forward three possible avenues to reduce the virulence of PFTs-producing strains: toxin synthesis, toxin secretion, and the process of invasion. The coding genes of most PFTs are in an operon. siRNAs or microRNAs can be used to directly decrease the expression level of the PFTs. siRNAs are currently used in the treatment of cancer and present as a promising treatment for diseases caused by bacterial infections. However, it is a challenge to accurately deliver the siRNA into the PFTs producing strains, while avoiding the degradation and removal of siRNAs in the host cells. Therefore, the development of safe and effective delivery systems is crucial to the application of siRNAs as a treatment strategy to reduce toxin synthesis. Currently, various of siRNA delivery systems have been proposed, such as chemical modification of siRNA, lipid-based siRNA delivery systems, polymer-based siRNA delivery systems, and conjugate siRNA delivery systems.65 The conjugate siRNA delivery system involves covalently linking the siRNA with small molecules and antibodies to improve targeting to specific cells.66 Antibodies to bacterial cell wall components are the preferred choice to link to siRNAs, enabling far more accurate delivery of the siRNA. Research into effective delivery systems has room for innovation.
Many PFTs, such as HBL and NHE, contain Sec-type signal peptides that are necessary for secretion through the Sec pathway.67 SecA, the ATPase motor of the Sec pathway, is unique to bacteria, and is thus an ideal target for the development of novel antibacterial agents.68 Some in vitro studies have confirmed that certain antibacterial effects can be achieved using SecA inhibitors, and a variety of SecA inhibitors have been discovered and designed, such as azide compounds, imino-containing molecules, pannomycinan, 5-cyano-6-aryl-2-thiouracils derivatives, thiazolo [4,5-d] pyrimidine derivatives, and indole derivatives.69–71 However, whether these inhibitors can achieve the conditions and standards used for in vivo treatment need to be further explored.71 The Sec system is widely used in various strains and inhibiting this system may disrupt the normal metabolism and balance of beneficial gut microbiota. Thus, therapeutics that target PFTs themselves are a more effective alternative to inhibitors and antibiotics for the treatment of pathogenic infections.72,73 Thirdly, PFTs are cell-specific and recognize lipid, sugar, and protein receptors on the cell membrane. Surface binding to target cells leads to an increase in the concentration of PFTs and promotes the formation of oligomers. This is a key step in the formation of the pores in the cell membrane that destroy the epithelial barrier and cellular structure to promote bacterial growth. Interfering with the formation of these pores in an appropriate ratio may be an effective therapeutic strategy. In S. aureus some compounds, including Oroxylin A, Oroxylin A 7-O-glucuronide (OLG), Oroxin A (ORA), and Oroxin B (ORB), have been shown to bind to α-hemolysin and inhibit the self-assembly process, thus making it impossible to form pores in the membrane.74,75
Strategies for Targeting the Host Cell After Infection
Previous studies have shown that monoclonal antibodies (mAbs) are effective in the treatment of experimental models of infection by PFTs.76 For example, two cross-neutralizing leukocidin mAbs, SAN177 and SAN481 can cross-neutralize a variety of toxins, including HlgAB and HlgBC, by binding to the F subunit of the toxin.77 The human LukAB antibody can be combined with toxin monomers or dimers to neutralize cytotoxicity.78 Similarly, the use of monoclonal antibodies to neutralize α-PFTs is worthy of further investigation. Recent studies have also shown that nanoparticles and toxins coated with a biofilm can effectively neutralize toxins, and the development of this kind of nanotoxoid vaccine is expected to slow down the threat of many types of toxins.79,80 Furthermore, single-stranded DNA or RNA oligonucleotides, named aptamers, are screened against α-toxin-induced cell death. These aptamers inhibit toxin-mediated activation of the transcription factors TNF-α and IL-17. However, development of agents based on these aptamers for the treatment of infections need further exploration.74
Specific receptors on the cell surface are the key to the toxic effects of β-PFTs, and some receptors are shared by multiple toxins. Therefore, the inhibitors of specific receptors may be used to simultaneously slow the effects multiple toxins. Current studies have shown that the C5a receptor is the co-receptor of HlgCB and another β-PFT, Panton-Valentine Leukocidin (PVL), on the surface of human immune cells, and a variety of C5a receptor inhibitors have been demonstrated in in vitro experiments. This inhibition has a significant effect on neutralizing the PVL toxin, but the effect on the HlgCB toxin is not obvious.53 Therefore, it is feasible that an inhibitor may block the interaction between the toxin and the C5aR without interfering with the normal C5aR immune function. Further research to better understand the mechanism involved here is required.
PFTs form pores in the cell membrane and change the permeability of the cell, which results in increased Ca2+ inflows, and the stimulation of Ca2+-dependent repair mechanisms. Compared with the protein pores caused by mechanical damage of the cell membrane, the pores formed by PFTs are proteolipid pores which have certain boundaries. These proteolipid pores are considered to stimulate two main repair mechanisms; shedding and endocytosis,81 both of which remove damaged membranes through vesicles or endocytosis.81,82 Recent studies have shown that Ca2+-activated lipid scramblase TMEM16F can enhance the fluidity and plasticity of the membrane, and the release of damaged membrane bubbles, to promote the repair of the plasma membrane after pore formation.83 To treat cellular inflammation caused by Ca2+ outflow mediated by PFTs, some studies have proposed the use of DNA aptamers to inhibit toxin-mediated cytolysis and activation of transcription factors, but this protective mechanism needs further exploration.84
Conclusion
Pathogenic bacteria secrete various virulence factors to attack host cells; PFTs being one of the most complex of these. This review has highlighted the synthesis of two types of PFTs, α-PFTs and β-PFTs. The regulatory systems especially transcriptional regulatory factors have been concluded which provided several potential targets for therapeutic. The invasive mechanism of these two types PFTs are also discussed. Though PFTs are not very widely spread, these various toxins are mostly participated in destroying the host immune system in different ways to mediate different cellular responses, which leading to high levels of resistance to the innate and adaptive immune defenses.3,7 These effects lower the immunity of the host and trigger more infections. Though deletion of PFTs will not completely protect from disease in animal models, it will be helpful in enhancing the immunity. Therefore, an in-depth exploration of the structure of toxins secreted by these pathogens, and their pathogenic mechanisms have enabled us to understand the heterogeneity of pore formation more clearly.
Overall, we propose several potential strategies based on our understanding of PFTs, with particular emphasis on their biosynthesis and pathogenic features. The possible strategies are focused mainly on the synthesis, secretion, and invasion process of PFTs. Such as SecA inhibitors and some compound like Oroxylin A and its derivatives which can bind to α-hemolysin to inhibit the pore-forming process. Furthermore, targeting the host cell after infection are also important strategies that can decrease pathogenicity or facilitate recovery of the host cells. Here, we discussed the application of monoclonal antibodies, nanoparticles and DNA aptamers. Further studies are warranted for the application of these strategies to treat PFT-induced diseases. A better understanding of the mechanisms of action of PFTs may aid in the design of novel strategies for the prevention and treatment of bacterial PFT-induced diseases.
Acknowledgments
This work was supported by grants from the Natural Science Foundation of Shandong Province (ZR2018BC002) and the National Natural Science Foundation of China (32000490).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bräuning B, Bertosin E, Praetorius F, et al. Structure and mechanism of the two-component α-helical pore-forming toxin YaxAB. Nat Commun. 2018;9(1):1806. doi:10.1038/s41467-018-04139-2
2. Kathuria R, Chattopadhyay K. Vibrio cholerae cytolysin: multiple facets of the membrane interaction mechanism of a beta-barrel pore-forming toxin. IUBMB Life. 2018;70(4):260–266. doi:10.1002/iub.1725
3. Los FC, Randis TM, Aroian RV, Ratner AJ. Role of pore-forming toxins in bacterial infectious diseases. Microbiol Mol Biol Rev. 2013;77(2):173–207. doi:10.1128/MMBR.00052-12
4. Brauning B, Groll M. Structural and mechanistic features of ClyA-like α-pore-forming toxins. Toxins. 2018;10(9):343. doi:10.3390/toxins10090343
5. Stenfors Arnesen LP, Fagerlund A, Granum PE. From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol Rev. 2008;32(4):579–606. doi:10.1111/j.1574-6976.2008.00112.x
6. Jenul C, Horswill AR. Regulation of Staphylococcus aureus virulence. Microbiol Spectr. 2019;7(2):7. doi:10.1128/microbiolspec.GPP3-0031-2018
7. Dal Peraro M, van der Goot FG. Pore-forming toxins: ancient, but never really out of fashion. Nat Rev Microbiol. 2016;14(2):77–92. doi:10.1038/nrmicro.2015.3
8. Nguyen VT, Kamio Y, Higuchi H. Single-molecule imaging of cooperative assembly of gamma-hemolysin on erythrocyte membranes. EMBO J. 2003;22(19):4968–4979. doi:10.1093/emboj/cdg498
9. Roderer D, Glockshuber R. Assembly mechanism of the α-pore–forming toxin cytolysin A from Escherichia coli. Philos Trans R Soc Lond B Biol Sci. 2017;372(1726):20160211. doi:10.1098/rstb.2016.0211
10. Benke S, Roderer D, Wunderlich B, Nettels D, Glockshuber R, Schuler B. The assembly dynamics of the cytolytic pore toxin ClyA. Nat Commun. 2015;6:6198. doi:10.1038/ncomms7198
11. Gilbert RJ, Dalla Serra M, Froelich CJ, Wallace MI, Anderluh G. Membrane pore formation at protein-lipid interfaces. Trends Biochem Sci. 2014;39(11):510–516. doi:10.1016/j.tibs.2014.09.002
12. Spaan AN, Vrieling M, Wallet P, et al. The staphylococcal toxins gamma-haemolysin AB and CB differentially target phagocytes by employing specific chemokine receptors. Nat Commun. 2014;5:5438. doi:10.1038/ncomms6438
13. Bohm ME, Huptas C, Krey VM, Scherer S. Massive horizontal gene transfer, strictly vertical inheritance and ancient duplications differentially shape the evolution of Bacillus cereus enterotoxin operons hbl, cytK and nhe. BMC Evol Biol. 2015;15:246. doi:10.1186/s12862-015-0529-4
14. Worthy HL, Williamson LJ, Auhim HS, et al. The crystal structure of Bacillus cereus HblL1. Toxins. 2021;13(4):253. doi:10.3390/toxins13040253
15. Bohm ME, Krey VM, Jessberger N, Frenzel E, Scherer S. Comparative bioinformatics and experimental analysis of the intergenic regulatory regions of Bacillus cereus hbl and nhe enterotoxin operons and the impact of CodY on virulence heterogeneity. Front Microbiol. 2016;7:768. doi:10.3389/fmicb.2016.00768
16. López AC, Alippi AM. Enterotoxigenic gene profiles of Bacillus cereus and Bacillus megaterium isolates recovered from honey. Rev Argent Microbiol. 2010;42(3):216–225.
17. Granum PE, O’Sullivan K, Lund T. The sequence of the non-haemolytic enterotoxin operon from Bacillus cereus. FEMS Microbiol Lett. 1999;177(2):225–229. doi:10.1111/j.1574-6968.1999.tb13736.x
18. Cui Y, Martlbauer E, Dietrich R, Luo H, Ding S, Zhu K. Multifaceted toxin profile, an approach toward a better understanding of probiotic Bacillus cereus. Crit Rev Toxicol. 2019;49(4):342–356. doi:10.1080/10408444.2019.1609410
19. Alonzo F, Torres VJ. The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol Mol Biol Rev. 2014;78(2):199–230. doi:10.1128/mmbr.00055-13
20. Kaneko J, Kamio Y. Bacterial two-component and hetero-heptameric pore-forming cytolytic toxins: structures, pore-forming mechanism, and organization of the genes. Biosci Biotechnol Biochem. 2004;68(5):981–1003. doi:10.1271/bbb.68.981
21. Badarau A, Trstenjak N, Nagy E. Structure and function of the two-component cytotoxins of Staphylococcus aureus - learnings for designing novel therapeutics. Adv Exp Med Biol. 2017;966:15–35. doi:10.1007/5584_2016_200
22. Esbelin J, Jouanneau Y, Armengaud J, Duport C. ApoFnr binds as a monomer to promoters regulating the expression of enterotoxin genes of Bacillus cereus. J Bacteriol. 2008;190(12):4242–4251. doi:10.1128/JB.00336-08
23. Heinrichs JH, Beecher DJ, MacMillan JD, Zilinskas BA. Molecular cloning and characterization of the hblA gene encoding the B component of hemolysin BL from Bacillus cereus. J Bacteriol. 1993;175(21):6760–6766. doi:10.1128/jb.175.21.6760-6766.1993
24. van der Voort M, Kuipers OP, Buist G, de Vos WM, Abee T. Assessment of CcpA-mediated catabolite control of gene expression in Bacillus cereus ATCC 14579. BMC Microbiol. 2008;8:62. doi:10.1186/1471-2180-8-62
25. Gohar M, Økstad OA, Gilois N, Sanchis V, Kolstø AB, Lereclus D. Two-dimensional electrophoresis analysis of the extracellular proteome of Bacillus cereus reveals the importance of the PlcR regulon. Proteomics. 2002;2(6):784–791. doi:10.1002/1615-9861(200206)2:6<784::Aid-prot784>3.0.Co;2-r
26. Slamti L, Lereclus D. Specificity and polymorphism of the PlcR-PapR quorum-sensing system in the Bacillus cereus group. J Bacteriol. 2005;187(3):1182–1187. doi:10.1128/jb.187.3.1182-1187.2005
27. Brillard J, Lereclus D. Comparison of cytotoxin cytK promoters from Bacillus cereus strain ATCC 14579 and from a B. cereus food-poisoning strain. Microbiology. 2004;150(Pt8):2699–2705. doi:10.1099/mic.0.27069-0
28. Pomerantsev AP, Pomerantseva OM, Camp AS, Mukkamala R, Goldman S, Leppla SH. PapR peptide maturation: role of the NprB protease in Bacillus cereus 569 PlcR/PapR global gene regulation. FEMS Immunol Med Microbiol. 2009;55(3):361–377. doi:10.1111/j.1574-695X.2008.00521.x
29. Yehuda A, Slamti L, Malach E, Lereclus D, Hayouka Z. Elucidating the hot spot residues of quorum sensing peptidic autoinducer PapR by multiple amino acid replacements. Front Microbiol. 2019;10:1246. doi:10.3389/fmicb.2019.01246
30. Esbelin J, Jouanneau Y, Duport C. Bacillus cereus Fnr binds a [4Fe-4S] cluster and forms a ternary complex with ResD and PlcR. BMC Microbiol. 2012;12:125. doi:10.1186/1471-2180-12-125
31. Esbelin J, Armengaud J, Zigha A, Duport C. ResDE-dependent regulation of enterotoxin gene expression in Bacillus cereus: evidence for multiple modes of binding for ResD and interaction with Fnr. J Bacteriol. 2009;191(13):4419–4426. doi:10.1128/JB.00321-09
32. Zigha A, Rosenfeld E, Schmitt P, Duport C. The redox regulator Fnr is required for fermentative growth and enterotoxin synthesis in Bacillus cereus F4430/73. J Bacteriol. 2007;189(7):2813–2824. doi:10.1128/JB.01701-06
33. Duport C, Zigha A, Rosenfeld E, Schmitt P. Control of enterotoxin gene expression in Bacillus cereus F4430/73 involves the redox-sensitive ResDE signal transduction system. J Bacteriol. 2006;188(18):6640–6651. doi:10.1128/JB.00702-06
34. Venkatasubramaniam A, Kanipakala T, Ganjbaksh N, et al. A critical role for HlgA in Staphylococcus aureus pathogenesis revealed by a switch in the SaeRS two-component regulatory system. Toxins. 2018;10(9):377. doi:10.3390/toxins10090377
35. Schmitt J, Joost I, Skaar EP, Herrmann M, Bischoff M. Haemin represses the haemolytic activity of Staphylococcus aureus in an SAE-dependent manner. Microbiology. 2012;158(Pt10):2619–2631. doi:10.1099/mic.0.060129-0
36. Tiwari N, Lopez-Redondo M, Miguel-Romero L, et al. The SrrAB two-component system regulates Staphylococcus aureus pathogenicity through redox sensitive cysteines. Proc Natl Acad Sci U S A. 2020;117(20):10989–10999. doi:10.1073/pnas.1921307117
37. Yan H, Wang Q, Teng M, Li LX. The DNA-binding mechanism of the TCS response regulator ArlR from Staphylococcus aureus. J Struct Biol. 2019;208(3):107388. doi:10.1016/j.jsb.2019.09.005
38. Guo H, Hall JW, Yang J, Ji Y. The SaeRS two-component system controls survival of Staphylococcus aureus in human blood through regulation of coagulase. Front Cell Infect Microbiol. 2017;7:204. doi:10.3389/fcimb.2017.00204
39. Mathur A, Feng S, Hayward JA, et al. A multicomponent toxin from Bacillus cereus incites inflammation and shapes host outcome via the NLRP3 inflammasome. Nat Microbiol. 2019;4(2):362–374. doi:10.1038/s41564-018-0318-0
40. Jessberger N, Dietrich R, Bock S, Didier A, Martlbauer E. Bacillus cereus enterotoxins act as major virulence factors and exhibit distinct cytotoxicity to different human cell lines. Toxicon. 2014;77:49–57. doi:10.1016/j.toxicon.2013.10.028
41. Tausch F, Dietrich R, Schauer K, et al. Evidence for complex formation of the Bacillus cereus haemolysin BL components in solution. Toxins. 2017;9(9):288. doi:10.3390/toxins9090288
42. Senesi S, Ghelardi E. Production, secretion and biological activity of Bacillus cereus enterotoxins. Toxins. 2010;2(7):1690–1703. doi:10.3390/toxins2071690
43. Madegowda M, Eswaramoorthy S, Burley SK, Swaminathan S. X-ray crystal structure of the B component of hemolysin BL from Bacillus cereus. Proteins. 2008;71(2):534–540. doi:10.1002/prot.21888
44. Lindback T, Hardy SP, Dietrich R, et al. Cytotoxicity of the Bacillus cereus Nhe enterotoxin requires specific binding order of its three exoprotein components. Infect Immun. 2010;78(9):3813–3821. doi:10.1128/IAI.00247-10
45. Jessberger N, Dietrich R, Schwemmer S, et al. Binding to the target cell surface is the crucial step in pore formation of hemolysin BL from Bacillus cereus. Toxins. 2019;11(5):281. doi:10.3390/toxins11050281
46. Sastalla I, Fattah R, Coppage N, et al. The Bacillus cereus Hbl and Nhe tripartite enterotoxin components assemble sequentially on the surface of target cells and are not interchangeable. PLoS One. 2013;8(10):e76955. doi:10.1371/journal.pone.0076955
47. Ramarao N, Lereclus D. Adhesion and cytotoxicity of Bacillus cereus and Bacillus thuringiensis to epithelial cells are FlhA and PlcR dependent, respectively. Microbes Infect. 2006;8(6):1483–1491. doi:10.1016/j.micinf.2006.01.005
48. Heilkenbrinker U, Dietrich R, Didier A, et al. Complex formation between NheB and NheC is necessary to induce cytotoxic activity by the three-component Bacillus cereus Nhe enterotoxin. PLoS One. 2013;8(4):e63104. doi:10.1371/journal.pone.0063104
49. Fox D, Mathur A, Xue Y, et al. Bacillus cereus non-haemolytic enterotoxin activates the NLRP3 inflammasome. Nat Commun. 2020;11(1):760. doi:10.1038/s41467-020-14534-3
50. Oliveira D, Borges A, Simoes M. Staphylococcus aureus toxins and their molecular activity in infectious diseases. Toxins. 2018;10(6):252. doi:10.3390/toxins10060252
51. Seilie ES, Bubeck Wardenburg J. Staphylococcus aureus pore-forming toxins: the interface of pathogen and host complexity. Semin Cell Dev Biol. 2017;72:101–116. doi:10.1016/j.semcdb.2017.04.003
52. Yamashita K, Kawai Y, Tanaka Y, et al. Crystal structure of the octameric pore of staphylococcal gamma-hemolysin reveals the beta-barrel pore formation mechanism by two components. Proc Natl Acad Sci U S A. 2011;108(42):17314–17319. doi:10.1073/pnas.1110402108
53. Spaan AN, Schiepers A, de Haas CJ, et al. Differential interaction of the staphylococcal toxins panton-valentine leukocidin and gamma-hemolysin CB with human C5a receptors. J Immunol. 2015;195(3):1034–1043. doi:10.4049/jimmunol.1500604
54. Tam K, Torres VJ. Staphylococcus aureus secreted toxins and extracellular enzymes. Microbiol Spectr. 2019;7(2):7. doi:10.1128/microbiolspec.GPP3-0039-2018
55. Malachowa N, Whitney AR, Kobayashi SD, et al. Global changes in Staphylococcus aureus gene expression in human blood. PLoS One. 2011;6(4):e18617. doi:10.1371/journal.pone.0018617
56. Ventura CL, Malachowa N, Hammer CH, et al. Identification of a novel Staphylococcus aureus two-component leukotoxin using cell surface proteomics. PLoS One. 2010;5(7):e11634. doi:10.1371/journal.pone.0011634
57. Dumont AL, Nygaard TK, Watkins RL, et al. Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol Microbiol. 2011;79(3):814–825. doi:10.1111/j.1365-2958.2010.07490.x
58. Rouha H, Weber S, Janesch P, et al. Disarming Staphylococcus aureus from destroying human cells by simultaneously neutralizing six cytotoxins with two human monoclonal antibodies. Virulence. 2018;9(1):231–247. doi:10.1080/21505594.2017.1391447
59. DuMont AL, Yoong P, Surewaard BG, et al. Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect Immun. 2013;81(5):1830–1841. doi:10.1128/IAI.00095-13
60. DuMont AL, Yoong P, Day CJ, et al. Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc Natl Acad Sci U S A. 2013;110(26):10794–10799. doi:10.1073/pnas.1305121110
61. Trstenjak N, Milic D, Graewert MA, et al. Molecular mechanism of leukocidin GH-integrin CD11b/CD18 recognition and species specificity. Proc Natl Acad Sci U S A. 2020;117(1):317–327. doi:10.1073/pnas.1913690116
62. Perret M, Badiou C, Lina G, et al. Cross-talk between Staphylococcus aureus leukocidins-intoxicated macrophages and lung epithelial cells triggers chemokine secretion in an inflammasome-dependent manner. Cell Microbiol. 2012;14(7):1019–1036. doi:10.1111/j.1462-5822.2012.01772.x
63. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–1153. doi:10.1016/j.immuni.2013.05.016
64. Ventola CL. The antibiotic resistance crisis: part 1: causes and threats. P T. 2015;40(4):277–283.
65. Singh A, Trivedi P, Jain NK. Advances in siRNA delivery in cancer therapy. Artif Cells Nanomed Biotechnol. 2018;46(2):274–283. doi:10.1080/21691401.2017.1307210
66. Dong Y, Siegwart DJ, Anderson DG. Strategies, design, and chemistry in siRNA delivery systems. Adv Drug Deliv Rev. 2019;144:133–147. doi:10.1016/j.addr.2019.05.004
67. Fagerlund A, Lindback T, Granum PE. Bacillus cereus cytotoxins Hbl, Nhe and CytK are secreted via the Sec translocation pathway. BMC Microbiol. 2010;10:304. doi:10.1186/1471-2180-10-304
68. Jin J, Hsieh YH, Chaudhary AS, et al. SecA inhibitors as potential antimicrobial agents: differential actions on SecA-only and SecA-SecYEG protein-conducting channels. FEMS Microbiol Lett. 2018;365(15). doi:10.1093/femsle/fny145
69. Cranford-Smith T, Jamshad M, Jeeves M, et al. Iron is a ligand of SecA-like metal-binding domains in vivo. J Biol Chem. 2020;295(21):7516–7528. doi:10.1074/jbc.RA120.012611
70. Jang MY, De Jonghe S, Segers K, Anne J, Herdewijn P. Synthesis of novel 5-amino-thiazolo[4,5-d]pyrimidines as E. coli and S. aureus SecA inhibitors. Bioorg Med Chem. 2011;19(1):702–714. doi:10.1016/j.bmc.2010.10.027
71. Chaudhary AS, Chen W, Jin J, Tai PC, Wang B. SecA: a potential antimicrobial target. Future Med Chem. 2015;7(8):989–1007. doi:10.4155/fmc.15.42
72. Subramanian K, Iovino F, Tsikourkitoudi V, et al. Mannose receptor-derived peptides neutralize pore-forming toxins and reduce inflammation and development of pneumococcal disease. EMBO Mol Med. 2020;12(11):e12695. doi:10.15252/emmm.202012695
73. Escajadillo T, Nizet V. Pharmacological targeting of pore-forming toxins as adjunctive therapy for invasive bacterial infection. Toxins. 2018;10(12):Dec. doi:10.3390/toxins10120542
74. Dong J, Qiu J, Zhang Y, et al. Oroxylin A inhibits hemolysis via hindering the self-assembly of alpha-hemolysin heptameric transmembrane pore. PLoS Comput Biol. 2013;9(1):e1002869. doi:10.1371/journal.pcbi.1002869
75. Qiu J, Wang D, Zhang Y, Dong J, Wang J, Niu X. Molecular modeling reveals the novel inhibition mechanism and binding mode of three natural compounds to Staphylococcal α-hemolysin. PLoS One. 2013;8(11):e80197. doi:10.1371/journal.pone.0080197
76. Ortines RV, Wang Y, Liu H, et al. Efficacy of a multimechanistic monoclonal antibody combination against Staphylococcus aureus surgical site infections in mice. Antimicrob Agents Chemother. 2019;63(8):e00346–19. doi:10.1128/AAC.00346-19
77. Vu TTT, Nguyen NTQ, Tran VG, et al. Protective efficacy of monoclonal antibodies neutralizing alpha-hemolysin and bicomponent leukocidins in a rabbit model of Staphylococcus aureus necrotizing pneumonia. Antimicrob Agents Chemother. 2020;64(3):e02220–19. doi:10.1128/AAC.02220-19
78. Thomsen IP, Sapparapu G, James DBA, et al. Monoclonal antibodies against the Staphylococcus aureus bicomponent leukotoxin AB isolated following invasive human infection reveal diverse binding and modes of action. J Infect Dis. 2017;215(7):1124–1131. doi:10.1093/infdis/jix071
79. Hu CM, Fang RH, Luk BT, Zhang L. Nanoparticle-detained toxins for safe and effective vaccination. Nat Nanotechnol. 2013;8(12):933–938. doi:10.1038/nnano.2013.254
80. Hu CM, Zhang L. Nanotoxoid vaccines. Nano Today. 2014;9(4):401–404. doi:10.1016/j.nantod.2014.06.001
81. Brito C, Cabanes D, Sarmento Mesquita F, Sousa S. Mechanisms protecting host cells against bacterial pore-forming toxins. Cell Mol Life Sci. 2018;76(7):1319–1339. doi:10.1007/s00018-018-2992-8
82. Etxaniz A, Gonzalez-Bullon D, Martin C, Ostolaza H. Membrane repair mechanisms against permeabilization by pore-forming toxins. Toxins. 2018;10(6):234. doi:10.3390/toxins10060234
83. Wu N, Cernysiov V, Davidson D, et al. Critical role of lipid scramblase TMEM16F in phosphatidylserine exposure and repair of plasma membrane after pore formation. Cell Rep. 2020;30(4):1129–1140e5. doi:10.1016/j.celrep.2019.12.066
84. Vivekananda J, Salgado C, Millenbaugh NJ. DNA aptamers as a novel approach to neutralize Staphylococcus aureus alpha-toxin. Biochem Biophys Res Commun. 2014;444(3):433–438. doi:10.1016/j.bbrc.2014.01.076
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.