")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Polymeric nanoparticles for targeted treatment in oncology: current insights
Authors Prabhu R, Patravale V, Joshi MD
Received 18 November 2014
Accepted for publication 24 December 2014
Published 2 February 2015 Volume 2015:10(1) Pages 1001—1018
DOI https://doi.org/10.2147/IJN.S56932
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Video abstract presented by Rashmi H Prabhu
Views: 3125
Rashmi H Prabhu,1 Vandana B Patravale,1 Medha D Joshi2
1Department of Pharmaceutical Sciences and Technology, Institute of Chemical Technology, Mumbai, India; 2Department of Pharmaceutical Sciences, Chicago College of Pharmacy, Midwestern University, Downers Grove, IL, USA
Abstract: Chemotherapy, a major strategy for cancer treatment, lacks the specificity to localize the cancer therapeutics in the tumor site, thereby affecting normal healthy tissues and advocating toxic adverse effects. Nanotechnological intervention has greatly revolutionized the therapy of cancer by surmounting the current limitations in conventional chemotherapy, which include undesirable biodistribution, cancer cell drug resistance, and severe systemic side effects. Nanoparticles (NPs) achieve preferential accumulation in the tumor site by virtue of their passive and ligand-based targeting mechanisms. Polymer-based nanomedicine, an arena that entails the use of polymeric NPs, polymer micelles, dendrimers, polymersomes, polyplexes, polymer–lipid hybrid systems, and polymer–drug/protein conjugates for improvement in efficacy of cancer therapeutics, has been widely explored. The broad scope for chemically modifying the polymer into desired construct makes it a versatile delivery system. Several polymer-based therapeutic NPs have been approved for clinical use. This review provides an insight into the advances in polymer-based targeted nanocarriers with focus on therapeutic aspects in the field of oncology.
Keywords: polymeric nanoparticles, cancer, passive delivery, ligand-based delivery
Introduction
Cancer is a disease characterized by the uncontrolled growth and spread of abnormal cells, and is still the second most common cause of death worldwide. Current treatment for cancer includes surgery, radiation, hormone therapy, and chemotherapy. Chemotherapy forms a major strategy for treating the disease. Conventional chemotherapy is highly nonspecific in targeting the drug to cancerous cells, making the normal healthy cells vulnerable to the drug’s undesirable effects. This significantly hampers the maximum allowable dose of the drug. Moreover, rapid elimination and specific distribution into targeted organs and tissues necessitate the administration of large dose of drug, which is not economical and often results in untoward toxicity issues.1,2 Nanoparticles (NPs) are customized drug delivery vectors capable of preferentially targeting large doses of chemotherapeutic agents or therapeutic genes into malignant cells while sparing healthy cells. NPs hold great promise of drastically changing the face of oncology by their ability of targeted delivery, and thereby, overcoming limitations of conventional chemotherapy, which include undesirable biodistribution, cancer cell drug resistance, and severe systemic side effects.3,4
There are numerous NP systems currently being employed for cancer therapeutics. The properties of these systems have been modulated to enhance delivery to the tumor; for instance, hydrophilic surfaces provide the NPs with stealth properties for longer circulation times, and positively charged surfaces can enhance internalization into the cancer cells.1 The types of NPs currently explored for cancer therapeutic applications include dendrimers, liposomes, polymeric NPs, micelles, protein NPs, lipid NPs, ceramic NPs, viral NPs, metallic NPs, and carbon nanotubes.5
The broad scope for chemically modifying the polymeric system facilitates its wide utility for targeted and therapeutic aspects in the field of oncology. Polymeric NPs are defined by their morphology and composition in the core and periphery. The therapeutic agent is either conjugated to the surface of the NP, or encapsulated and protected inside the polymeric core. Polymeric NP platforms are characterized by their unique physicochemical structures, including solid polymeric NP, polymeric micelle, polymer conjugate, dendrimer, polymersome, polyplex, and polymer–lipid hybrid system (Figure 1). The functionalization of the NPs helps to achieve extended blood residence time, reduce nonspecific distribution, and target tissues or specific cell surface antigens with a targeting ligand (peptide, aptamer, antibody/antibody fragment, small molecule).6
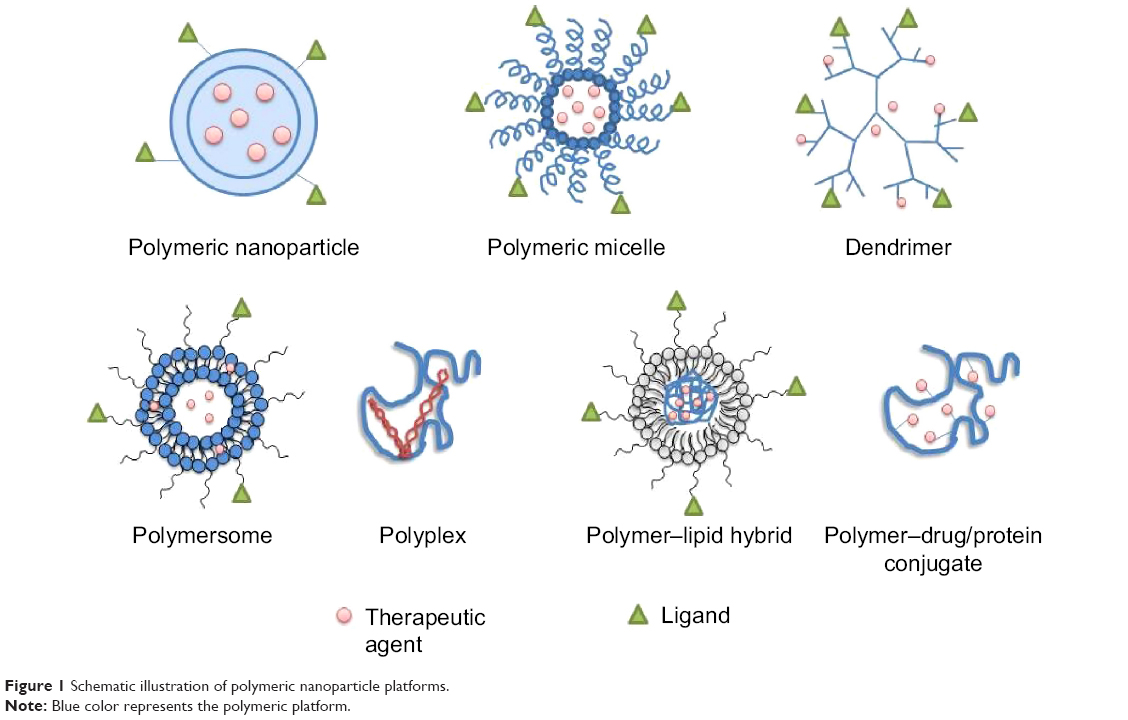 | Figure 1 Schematic illustration of polymeric nanoparticle platforms. |
This review details the targeting aspects and various polymer-based nanocarriers for cancer therapy.
Targeted delivery of NPs
The delivery of an anticancer drug to the target tissue can be achieved by NPs primarily in two ways: passive and ligand-based targeting (Figure 2).
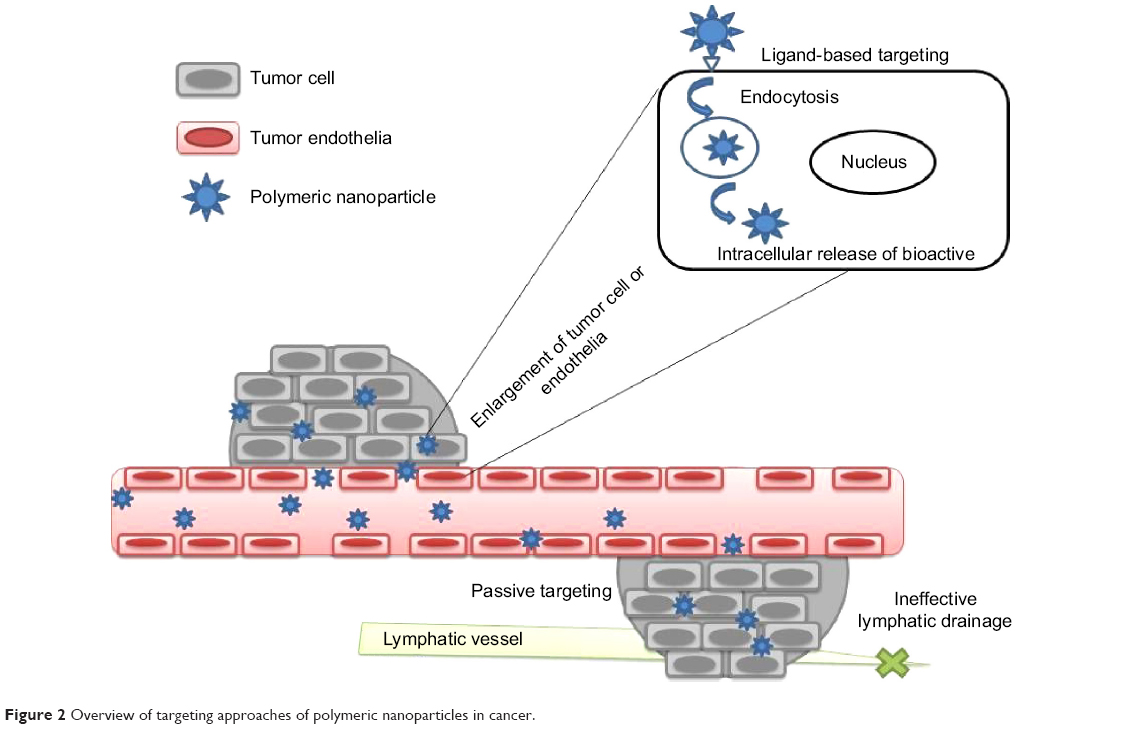 | Figure 2 Overview of targeting approaches of polymeric nanoparticles in cancer. |
Passive targeting
This targeting approach exploits the pathophysiological conditions, such as leaky vasculature, pH, temperature, and surface charge surrounding the tumor for specific delivery of NPs.
Enhanced permeation and retention (EPR) effect
Nanoparticulate systems take advantage of unique pathophysiologic characteristics of tumor vessels for passive targeting. When the tumor volume reaches above 2 mm3, diffusion limitation sets in, which eventually impairs nutrition intake, waste excretion, and oxygen delivery.5 Such rapidly growing cancer cells recruit the generation of new blood vessels, a phenomenon called angiogenesis (or neovascularization). Aberrant tortuosity, abnormalities in the basement membrane, and the lack of pericytes lining endothelial cells are the features of this process, which results in leaky vessels with gap sizes of 100 nm to 2 μm, depending upon the tumor type.7 Moreover, such tumors exhibit poor lymphatic drainage due to the high interstitial pressure at the core of the tumor than at the periphery. This combination of leaky vasculature and poor lymphatic flow results in enhanced permeation and retention (EPR) effect. NPs can preferentially localize in cancerous tissues owing to their size being smaller than blood vessel fenestration and be entrapped in the tumor due to higher retention ability than the normal tissues.5,7,8
Tumor microenvironment
Passive targeting can also be achieved by exploiting the microenvironment surrounding tumor cells, which is distinct from the normal cells. Rapidly dividing cancer cells exhibit a high metabolic rate. Tumor cells utilize glycolysis to maintain adequate supply of nutrients and oxygen, thereby resulting in an acidic environment.9 The pH-sensitive nanoparticulate systems are designed to be stable at a physiologic pH of 7.4 but degraded to release active drug in target tissues in which the pH is less than physiologic values, such as in the acidic environment of tumor cells.10 Hyperthermia is implicated in many pathological areas such as human ovarian carcinoma. Thermo-sensitive polymeric system contains polymer that exhibits a low critical solution temperature (LCST) and that tends to precipitate when the temperature is above LCST in the tumor with concomitant release of payload. Localized hyperthermia in tumors can be induced by physical methods such as ultrasound or photothermal means.11,12 Additionally, cancer cells express and release unique enzymes such as matrix metalloproteinases (MMPs), which are implicated in their movement and survival mechanisms.13 An albumin-bound form of doxorubicin (DOX) incorporating a MMP-2-specific octapeptide sequence between the drug and the carrier was observed to be efficiently and specifically cleaved by MMP-2 in an in vitro study.14
Surface charge
Passive targeting also entails the use of innate feature of the NP such as charge to target the tumor. Tumor cells bear relatively high negative surface charge than normal cells, thereby enabling favored binding by cationic NP systems.15 Targeting of cationic NP system is achieved by electrostatic binding to negatively charged phospholipid headgroups preferentially expressed on tumor endothelial cells.16,17 The cytotoxicity potential of polymeric NPs largely depends on cellular internalization and subcellular localization of the NPs, which is governed by the nature of polymeric surface charge (anionic, cationic, or neutral).18 Cationic NPs have been found to efficiently deliver small interfering RNA (siRNA) to silence target gene in cancer cells and also sensitize the cancer cells to the effect of paclitaxel (PTX) for improved anticancer activity.19,20
Ligand-based targeting
In ligand-based targeting, ligands are conjugated at the periphery of the nanoparticulate system to bind with appropriate receptors at the target tumor site. The targeting ligands can be categorized as proteins (antibody and its fragments), nucleic acids (aptamers), or other ligands (peptides, vitamins, and carbohydrates), which generally bind to the receptor uniquely overexpressed by tumor cells or vasculature.21–23 The targeting ligands play a vital role in enhancing cellular uptake of NPs through the process of endocytosis. Long-circulating NPs enable their efficient delivery to the tumor site by the EPR phenomena, and internalization of the nanosystem results in improved therapeutic effect.24–27 The cellular targets for this strategy have been identified on the tumor cell and endothelium.
Tumor cell targeting
This targeting approach involves targeting of cell surface receptors overexpressed by tumor cells in order to enhance the cellular uptake of the nanocarriers. The ligand-based targeting is more important for the intracellular delivery of macromolecular drugs such as DNA, siRNA, and proteins, whose site of action is located intracellularly. The cellular internalization of nanocarrier increases the antitumoral efficacy of ligand-targeted nanocarriers.5 The ability of the nanocarrier to be internalized post-binding to target cell receptor is requisite for proper selection of targeting ligands.2 The most widely studied targets are transferrin, folate, and epidermal growth factor receptors (EGFRs), and glycoproteins.
Transferrin receptors
Transferrin, a serum non-heme iron-binding glycoprotein, transports iron through the blood and into proliferating cells by attaching to the transferrin receptor. Once the transferrin is internalized, iron is released as a result of endocytosis in the acidic environment of the cell. The transferrin receptor is an important protein responsible for iron homeostasis and regulation of cell growth. Thus, the overexpression of transferrin receptors in metastatic and drug-resistant cancer cells in comparison to the normal cells due to increased requirement of iron makes this receptor a pertinent target for cancer therapy.2,28,29
Folate receptors
The folate receptor is a 38 kDa glycosyl-phosphatidylinositol-conjugated glycoprotein, which is the most widely researched tumor marker. This receptor binds to the vitamin folic acid and folate–drug conjugates or folate-anchored nanocarriers with a high affinity and internalizes into the cells via receptor-mediated endocytosis. Folic acid is necessary for the synthesis of nucleotide bases, viz purines and pyrimidines. Moreover, normal cells transport folic acid only in reduced form such as 5-methyl-tetrahydrofolate and do not transport folate conjugates across their membrane.30 The major route of folate conjugate entry into the cancer cells is mainly via the folate receptors, as these receptors are significantly upregulated on cancer cells compared to normal cells.31 Functional folate receptors are majorly confined to the apical surfaces of polarized epithelia. A wide range of tumors overexpress folate receptors, including ovary, lung, brain, head and neck, renal cell, and breast cancers. The great utility of these folate ligands stems from the fact that they are inexpensive, nontoxic, and non-immunogenic. They also have high binding affinity, stability on storage and in circulation, and are easily conjugated to nanocarriers.30–32
Epidermal growth factor receptors
The EGFRs belonging to a family of tyrosine kinase receptors are highly upregulated on tumor cell surfaces. EGFR binds to six known endogenous ligands: EGF, transforming growth factor-α, amphiregulin, betacellulin, heparin-binding EGF, and epiregulin.33 Activation of EGFR by one of these ligands stimulates intracellular signaling processes involved in tumor growth and progression that include proliferation, angiogenesis, invasion, and metastasis.34,35 The EGFR is overexpressed in breast, lung, colorectal, and brain cancers.36
Glycoproteins
Lectins are proteins that can identify and attach specifically to the carbohydrate entity of glycoproteins expressed on tumor cell surface. Glycoproteins expressed on tumor cells are different from that of normal cells. Lectin targeting can be characterized as direct lectin targeting (lectins included in nanosystems as ligand to target cell surface glycoprotein) and reverse lectin targeting (conjugating nanosystem with carbohydrate moiety to target lectins). The lectin-based targeting has been applied majorly in targeting colon.37
Tumor endothelium targeting
The growth of solid tumors can be inhibited by preventing angiogenesis, which is the production of new blood vessels for adequate blood supply mainly in the tumor core to provide oxygen and essential nutrients. Thus, designing of nanocarriers that actively target angiogenesis can prove to be very useful for regulating cancer growth and associated metastatic potential.38 Targeting the tumor endothelium has following merits: (i) there is no need for the nanocarriers to cross endothelial barriers to reach their target site; (ii) nanocarriers have the ease of accessibility to bind to endothelial receptors post-intravenous injection; (iii) endothelial cells are less prone to the risk of developing resistance to treatment than tumor cells because of high genetic stability; and (iv) this approach can be applied to all types of tumor as most of the markers are expressed on endothelial cells.39,40
VEGF receptor
The vascular endothelial growth factors (VEGFs) induce tumor angiogenesis and neovascularization by virtue of their ability to bind and activate the VEGF receptor (VEGFR) signaling cascade.41 These receptors seem to be promising strategy for angiogenesis-associated targeting of NP systems. Oncogenes and tumor hypoxia augment VEGF levels in the tumor cells, which leads to an overexpression of two types of VEGFRs, viz VEGFR-1 (fms-like tyrosine kinase) and VEGFR-2 (fetal liver kinase-1), on tumor endothelial cells. VEGFR-2 is the most widely explored among the VEGF class of receptors. Angiogenesis can be inhibited either by targeting VEGF to prevent ligand binding to VEGFR-2 or by targeting VEGFR-2 to reduce VEGF binding and activate an endocytic pathway.5,42
αvβ3 integrin
The αvβ3 integrin, an endothelial cell receptor for extracellular matrix proteins, includes von Willebrand factor, fibrinogen (fibrin), vibronectin, thrombospondin, osteopontin, and fibronectin. These proteins share a common structural feature of the presence of three-amino acid sequence (ie, arginine-glycine-aspartic acid, RGD). The αvβ3 integrin is highly upregulated on neovascular endothelial and tumor cells than on resting endothelial cells and other normal organs. It also plays a vital role in the calcium-dependent signaling pathway, thereby causing migration of endothelial cell. Derivatives of RGD (Arg-Gly-Asp) oligopeptides can bind and block the endothelial αvβ3 integrins. αvβ3 integrin is also related intrinsically to the VEGFR-2 signaling. Blocking αvβ3 integrin receptor binding is found to be associated with downregulation of VEGF, thereby inhibiting the tumor angiogenesis synergistically.43
Vascular cell adhesion molecule-1
Vascular cell adhesion molecule-1 (VCAM-1), a transmembrane glycoprotein, is expressed exclusively on the surface of endothelial tumor cells. VCAM-1 promotes cell-to-cell adhesion during tumor angiogenesis. Increased expression of VCAM-1 is usually found in leukemia, breast and lung cancer, renal cell carcinoma, melanoma, gastric cancer, and nephroblastoma.44,45
Matrix metalloproteinases
The matrix metalloproteinases (MMPs) belong to a class of structurally related zinc-dependent endopeptidases. MMPs are known to be an essential physiologic component involved in tissue repair, morphogenesis, and angiogenesis. Membrane type 1 matrix metalloproteinase (MT1-MMP) is expressed on angiogenic endothelial tumor cells, including colon, cervical, and gastric carcinomas, and gliomas, melanomas, and malignancies of the lung.46,47 MT1-MMP functions i) by degrading the extracellular matrix, ii) by playing a role in angiogenesis, metastasis, endothelial cell invasion, and migration, iii) in the formation of capillary tubes, and iv) in recruiting accessory cells.47 It also activates MMP-2 that hydrolyzes Type IV collagen, a cementing component of basement membrane. In addition, targeting the MT1-MMP limits the ligand binding to αvβ3 integrin, thereby suggesting it to be a valuable target.46
Polymer-based nanocarriers for targeted cancer therapy
An arsenal of polymeric nanocarriers, viz polymeric NPs, polymeric micelles, dendrimers, polymersomes, polyplexes, polymer hybrid systems, and polymer conjugates, has been explored for targeted delivery of therapeutic moiety in cancer. The polymers employed for fabrication of these nanocarriers may be either of natural or of synthetic origin. These polymeric NPs are capable of ferrying wide range of drugs for a sustained period of time in a controlled manner at target sites to provide enhanced antitumor efficacy with minimal systemic side effects. Also, these nanosystems protect drugs from their rapid metabolism during systemic circulation, and clearance by the liver, kidney, and reticuloendothelial system, which further improves drug’s stability and target specificity.3,48 Several polymer-based NPs have been approved clinically.
Polymeric NPs
Polymeric NPs are solid colloidal systems in which the therapeutic agent is dissolved, entrapped, encapsulated, or adsorbed onto the constituent polymer matrix. Depending upon the process of formation of NPs, the structure of resulting polymeric NPs may vary from nanospheres (matrix systems in which the drug is dispersed throughout the particles) to nanocapsules (vesicular reservoir systems in which the drug is confined to an aqueous or oily cavity surrounded by a single polymeric membrane).49,50 Several polymers such as poly(lactide-co-glycolide) (PLGA), polylactide (PLA), polyglycolide, polycaprolactone (PCL), poly(d,l-lactide), chitosan, and PLGA–polyethylene glycol (PEG) have been developed for passive and ligand-targeted delivery of therapeutic moieties exemplified in Tables 1 and 2.51–83 PEGylated PLGA NPs were employed as carrier for PTX to improve its therapeutic index. PTX-loaded NPs were found to be three times more cytotoxic on HeLa cells than Taxol. In vivo in transplantable liver tumor-bearing mice, PTX-loaded NPs showed noticeable tumor growth inhibition and enhanced survival rate (14 days) in comparison to Taxol (11 days). This resulted due to EPR phenomena of PTX-loaded NPs and their ability to sustain the drug levels in blood for a longer time.51 Biodegradable polyethylene oxide (PEO)–PCL NPs loaded with PTX and tamoxifen (TMX) were found to be efficient in overcoming multidrug resistance in ovarian adenocarcinoma. The cytotoxicity assay demonstrated that such NPs led to IC50 tenfold and twofold lower in sensitive SKOV3 and resistant SKOV3 cell lines in comparison to drug solution, respectively. Upon intravenous administration of PTX–TMX combination in PEO–PCL NP formulations, significant enhancement in antitumor efficacy and negligible treatment-associated toxicity were observed.71 Chittasupho et al formulated PLGA NPs targeting the immunologically active receptor, intercellular adhesion molecule-1, by attaching the Cyclo-(1,12)-PenITDGEATDSGC (cLABL) peptide to the NP surface.74 DOX-loaded cLABL peptide-conjugated PLGA NPs showed more rapid cellular uptake by A549 lung epithelial cancer cells compared to NPs without peptide. The cytotoxicity assessment of cLABL-NPs compared to free drug showed similar IC50 values implying that activity of the drug released from NPs was retained. Cheng et al reported A10 aptamer-functionalized PLGA–PEG NPs against prostate-specific membrane antigen (PSMA)-overexpressing LNCaP cancer cells.76 PLGA–PEG NPs functionalized with aptamer ligand have shown 3.77-fold increased delivery of NPs to tumors at 24 hours as compared to equivalent NPs lacking this aptamer.PLGA–PEG copolymer-based NPs have been investigated as an active delivery system for DOX by conjugating a novel heptapeptide that targets EGFR. The IC50 of DOX-loaded peptide-conjugated PLGA–PEG NPs in SKOV3 cells was lower by 62.4-fold, and cellular uptake efficiency was higher by 3.3-fold than that of peptide-free PLGA–PEG NPs. Biodistribution study in mice highlighted the fact that the accumulation of peptide-conjugated NPs was 30 times more in tumor tissue in comparison with free DOX.81
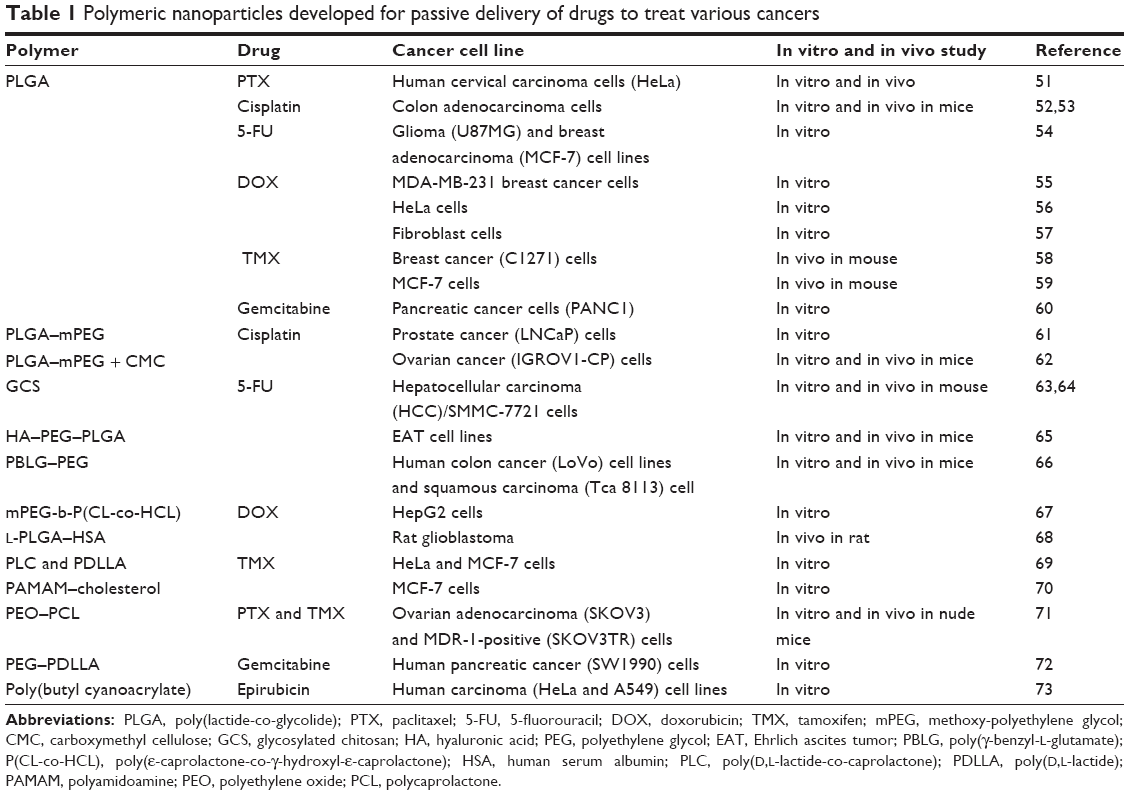 | Table 1 Polymeric nanoparticles developed for passive delivery of drugs to treat various cancers |
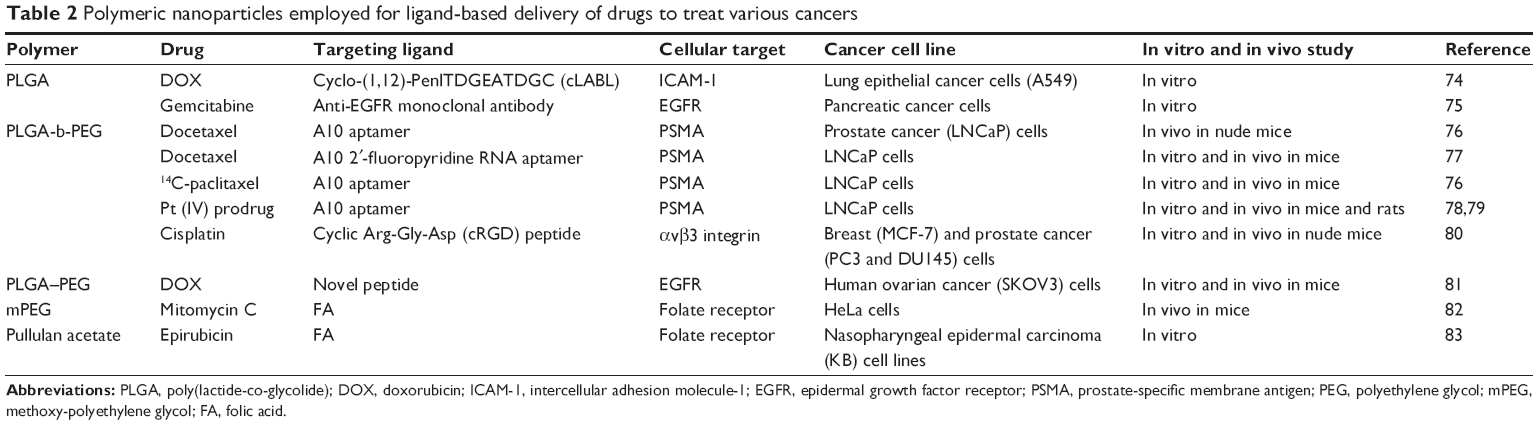 | Table 2 Polymeric nanoparticles employed for ligand-based delivery of drugs to treat various cancers |
Polymeric micelles
The ability of amphiphilic di- or tri-block copolymers to self-assemble into spherical nanosized core/shell structure in aqueous media forms polymeric micelles. The hydrophobic part of the copolymer, which forms the core, allows for the encapsulation of anticancer drugs, whereas the hydrophilic portion of the copolymer forming the shell of the micelles provides stealth property to the micellar system. This property prevents its uptake by reticuloendothelial system, and thereby, prolongs its circulation time in bloodstream. Polymer-derived micelles exhibit greater stability and lower critical micellar concentration value (in the order of 10–6 M) in comparison to surfactant-based micelles.84,85 Recent clinical study data on few micellar-based preparations of anticancer drugs have highlighted their utility as potential drug carrier in oncotherapy.85
Bisht et al synthesized polymeric micelles of cross-linked and random copolymers of N-isopropylacrylamide, with N-vinyl-2-pyrrolidone and PEG monoacrylate to encapsulate curcumin.86 These micelles demonstrated in vitro anticancer efficacy comparable to free curcumin against human pancreatic cancer cell lines and were found to retain the mechanistic activity specific to curcumin. Jin et al explored the utility of PTX-loaded N-octyl-O-sulfate chitosan micelles for treating multidrug-resistant (MDR) cancer.87 These micelles exhibited high cellular uptake about twofold more than Taxol, and the low efflux of PTX resulted in the optimal cytotoxicity in both human hepatocellular liver carcinoma (HepG2) cells and the MDR HepG2 (HepG2-P) cells. Intravenous injection of PTX-loaded micelles into the tumor-bearing mice demonstrated high tumor inhibition rate of 75.5% than that of Taxol (45.3%). These micelles also prolonged survival time of mice, thereby expressing greater therapeutic efficacy than Taxol. Synergetic effect of micelles on drug delivery and permeability glycoprotein inhibition enabled the delivery of PTX in intact form at the tumor site. Polymeric micelles composed of dextran and poly(d,l-lactide-co-glycolide) block copolymer was investigated for delivery of DOX. The in vitro anticancer effects of the polymeric micelles were investigated in DOX-resistant human cholangiocarcinoma (HuCC-T1) cells and compared with free DOX. The polymeric micelles showed about fourfold higher cytotoxicity to DOX-resistant HuCC-T1 cells than treatment with free DOX, suggesting that the polymeric micelles were effectively taken up by tumor cells by overcoming drug resistance, while free DOX hardly gained access into tumor cell.88 PEG–polyglutamic acid block copolymer micelles loaded with cisplatin demonstrated remarkably prolonged blood circulation and accumulation in solid tumors (Lewis lung carcinoma cells) about 20-fold higher than free cisplatin. The micellar system was found to confer both sufficient stability to ensure prolonged circulation in the bloodstream and sustained drug release kinetics upon accumulation at the delivery site via the EPR effect. Treatment with micelles led to complete tumor regression with no significant body weight loss, whereas free drug treatment resulted in tumor survivals and approximately 20% of body weight loss at the equivalent dose.89
Vega et al have synthesized immunopolymeric micelle by coupling antibody C225 against EGFR to the poly(L-glutamic acid)-co-PEG block copolymer for targeted delivery of DOX.90 When assessed on human vulvar squamous carcinoma A431 cells that overexpress EGFR, this antibody conjugate exhibited an IC50 of 1.7 μM which was significantly lower than free DOX having an IC50 >10 μM. Thus, the antibody conjugate proved to be more potent than free DOX in inhibiting the growth of A431 cells, owing to selective binding to EGFR and receptor-mediated uptake of the micellar system. Polymeric micelles composed of PEG–phosphatidylethanolamine were attached to antitumor mAb 2C5 having nucleosome-restricted specificity for different cancer cells for target-specific delivery of PTX. These immunomicelles effectively recognized and bound to various cancer cells (murine Lewis lung carcinoma and EL4 T cell lymphoma and human BT20 breast adenocarcinoma cell lines) in vitro. When administered intravenously into experimental mice bearing Lewis lung carcinoma, tumor-specific 2C5 immunomicelles loaded with PTX showed increased accumulation of PTX in the tumor and enhanced tumor growth inhibition by almost 2.5 times as compared with free PTX or Taxol® in non-targeted micelles.91 cRGD-labeled poly(ε-caprolactone)–PEG micelles encapsulating DOX were found to greatly enhance internalization of micelles in tumor endothelial cells (human Kaposi’s sarcoma) that overexpress αvβ3 integrins through receptor-mediated endocytosis than non-functionalized micelles.92 Yoo and Park exploited the folate receptor by functionalizing folic acid onto DOX-loaded PEG–PLGA micelles by covalently coupling the ligand via its γ-carboxyl group. In vitro cytotoxicity study of the folate–micelles demonstrated enhancement in cell uptake and cytotoxicity against KB cells (human nasopharyngeal epidermal carcinoma cell line) over non-targeted micelles.93 Marked improvement in in vivo antitumor efficacy with two times decrease in the tumor growth rate was demonstrated by folate-conjugated micelles compared to non-targeted micelles.
Park et al fabricated folate receptor-targeted PEG–PCL micelles entrapping PTX, which demonstrated higher cell viability of over 80% when tested against normal fibroblast cells than PTX (around 65%) suggesting the role of active targeting ligand folic acid in site-specific delivery of nanocarriers.94 Folate targeting was also explored by Han et al.95 They prepared folate-conjugated polymer micelles assembled from mixture of folate–polyethylene glycol–distearoylphosphatidylethanolamine (FA–PEG–DSPE) and methoxy-polyethylene glycol–distearoylphosphatidylethanolamine (mPEG–DSPE) to encapsulate anticancer agent 9-nitro-camptothecin. The molar ratio 1:100 of FA–PEG–DSPE and mPEG–DSPE was found to avoid macrophages and express high-selective targeting ability. The folate-conjugated micelles showed a greater ability to actively target the tumor cells (pancreatic cancer cell line, human uterine cervix cancer cell line, and human gastric cancer cell line) with overexpressed folate receptors on cell surface in comparison with folate-free micelles or free anticancer agents.Folate-anchored pluronic P105 and L101 were investigated as micellar carriers for the delivery of PTX for overcoming multidrug resistance in human breast cancer MCF-7 and MDR cell sublines, MCF-7/ADR. Pluronic micellar PTX significantly reduced IC50 of PTX in MDR cells compared to free PTX, indicating the susceptibility of MDR cells to the cytotoxic effects of pluronic micellar PTX than the non-resistant cell lines. Increased internalization of folate-anchored micelles was observed due to enhanced uptake by folate receptors. The authors have suggested that the synergistic action of pluronics-based micelles to overcome MDR and folate-mediated uptake would prove beneficial for treating MDR solid tumors.96
Jeong et al designed galactose-conjugated PEG-co-poly(γ-benzyl-l-glutamate) block copolymer loaded with PTX for targeting asialoglycoprotein receptors (ASGPRs) overexpressed in hepatocellular carcinoma.97 Cytotoxicity of these micelles was more pronounced in HepG2 cells (ASGPR-expressing cancer cell line) than SK-Hep 01 cells (non-ASGPR-expressing cell line) due to active delivery of PTX to HepG2 cells through receptor-mediated mechanism.Farokhzad et al utilized an RNA aptamer for the PSMA to target docetaxel-loaded PLA-block-PEG copolymer micelles to prostate tumors.77,98 The uptake of aptamer bioconjugates was found to be specific for the cells that express PSMA than the control group cells. Aptamer-encoded micelles demonstrated lower cell viability of around 48% over non-targeted counterparts with 30% cell viability when assessed in LNCaP prostate cancer cells. Intra-tumoral injection of the micelle NPs into LNCaP xenograft mouse model exhibited significant anticancer efficacy with complete tumor reduction in tested mice as compared with non-targeted NPs.
Dendrimers
Dendrimers are synthetic, repeatedly branched polymeric macromolecules having numerous extensions from central core, resulting in a tree-like structure. The structure of dendrimers and modifiable surface functionality allow for either encapsulation/conjugation of therapeutic agent, in the core or on the surface, making them attractive carriers for anticancer therapeutics.99
Poly(glycerol-succinic acid) dendrimers were explored as potential carriers for camptothecin.100 The anticancer activity of the camptothecin-encapsulated dendrimer formulation was examined using human breast adenocarcinoma (MCF-7), colorectal adenocarcinoma (HT-29), non-small-cell lung carcinoma (NCI-H460), and glioblastoma (SF-268). Increased cytotoxicity of delivered camptothecin was observed due to the dendrimer carrier, which lowered the IC50s in two- to sevenfold range in various cancer cells when compared with camptothecin dissolved in dimethyl sulfoxide. Cell uptake of the dendrimer carrier increased by 16-fold than free drug when assessed in MCF-7 cells.101,102 Star amphiphilic block copolymer containing poly(ε-caprolactone) and PEG was evaluated as carrier of hydrophobic anticancer drug etoposide. Etoposide-encapsulated dendrimers showed comparable toxicity than free etoposide when tested on porcine kidney epithelial cells.103 Dendrimers based on melamine were found to reduce the organ toxicity of anticancer agents, methotrexate (MTX) and 6-mercaptopurine, which are hepatotoxic. Treatment of C3H mice with subchronic doses of drug-encapsulated dendrimers leads to significant reduction in hepatotoxicity as evaluated by alanine transaminase (ALT) levels. ALT levels were reduced to 27% for MTX-encapsulated dendrimers and 36% for the 6-mercaptopurine dendrimers than ones treated with non-encapsulated drugs.104
Padilla De Jesús et al explored the use of a 2,2-bis(hydroxymethyl) propanoic acid-based dendrimer as carrier for DOX in vitro and in vivo.105 DOX was covalently attached to dendrimer hybrid through a hydrazone linkage. The DOX–polymer conjugate was found to be cytotoxic and less potent than free drug when tested on murine melanoma cell line (B16F10) and breast cancer cell lines (MDA-MB-231 and MDA-MB-435), which is indicative of release of drug from the dendrimer conjugate. The biodistribution study showed no significant accumulation of the DOX–polymer conjugate in vital organs, and prolonged half-life of DOX in conjugate form (72 minutes) than free drug (8 minutes). The authors have suggested that drug attached to the appropriate high-molecular weight system could further extend the half-life, which is requisite for efficiently exploiting the EPR phenomenon.105 A polyamidoamine (PAMAM) dendrimer generation 3.5 was conjugated to cisplatin (Pt) through sodium carboxylate surface synthesizing a dendrimer-platinate, which released platinum slowly in vitro. On intraperitoneal administration, dendrimer-Pt was eightfold less toxic than free drug and showed superior activity against B16F10 melanoma-bearing mice. Moreover, the dendrimer-Pt exhibited anticancer activity, whereas cisplatin was found to be inactive after intravenous administration to treat melanoma. High maximum tolerated dose (15 mg/kg) of cisplatin in the form of dendrimer formulation was indicative of its selective accumulation in solid tumor tissue by EPR effect when compared with cisplatin (1 mg/kg).106 Folic acid conjugated at the surface of generation 5 PAMAM dendrimers was investigated for the targeted delivery of MTX by comparing the two aspects of drug loading into the dendrimer system, viz encapsulated and covalently bound drug. The drug release from encapsulated MTX was more than 70% as compared to slow release of covalently bound drug which was less than 5% in 2.5 hours, thereby suggesting that the covalently bound drug does not release the drug prematurely in biological conditions. Also, the diffusion characteristic of encapsulated drug was similar to that of free drug. The cytotoxicity study revealed that MTX as the dendrimer inclusion complex retained anticancer activity similar to the free drug in in vitro conditions of free and blocked receptors of folic acid. Folic acid-targeted MTX conjugates demonstrated high specificity and antiproliferative activity for KB cells, which overexpress folic acid receptors. However, when the folic acid receptors are blocked, these conjugates lose their antiproliferative effect, indicating intracellular delivery of the drug through receptor-mediated endocytosis.107 A similar study was performed with folic acid, fluorescein, and PTX conjugated to partially acetylated PAMAM dendrimers. Internationalization of the conjugate occurred by selective targeting to folate receptors and preferentially delivering PTX-conjugated dendrimers to KB cells.108
Polymersomes
Polymersomes are self-assembled polymer vesicles of synthetic amphiphilic block copolymers consisting of discrete hydrophilic and hydrophobic blocks. Although they exhibit an architecture similar to that of liposomes (vesicles derived from phospholipids), polymersomes possess greater stability, storage capability, and prolonged circulation time.109
Polymer vesicles can efficiently encapsulate DOX in their aqueous center. The therapeutic potential of DOX-loaded PEO-block-PCL polymersomes was assessed in xenotransplanted (T6-17 cells) tumor-bearing mice, and their capability to retard tumor growth was examined. DOX-loaded polymersomes were able to retard tumor growth as comparable to commercially available agent DOXIL® (a clinically administered liposomal formulation of DOX).109 Polymersomes based on polyphosphazene were investigated as delivery systems of hydrophilic DOX hydrochloride (DOX·HCl) or hydrophobic DOX base (DOX) for breast cancer therapy. Strong interaction with polymersomes enabled higher encapsulation of DOX·HCl or DOX. In vivo administration of these polymersomes in nude mice bearing MCF-7 xenograft tumors demonstrated similar tumor growth inhibition but enhanced life safety, especially for DOX·HCl-loaded polymersomes in comparison with free DOX·HCl.110 Li et al evaluated the ability of poly(butadiene)-b-PEO polymersomes for delivery of PTX by embedding the drug in its hydrophobic bilayer.111 The PTX-loaded polymersome formulation showed comparable activity against PTX alone to inhibit proliferation of MCF-7 human breast cancer cells, thereby maintaining the cytotoxic property of the drug.Polymersomes have also been used to co-encapsulate PTX (in hydrophobic bilayer) and DOX (in hydrophilic core) for efficient passive delivery to MDA-MB231 human breast tumor-bearing mice. Such dual drug-loaded PEG–polyester-based polymer vesicles exhibited increased synergistic anticancer effect, a higher maximum tolerated dose, as well as increased suppression of tumor in comparison to free drugs.112,113
Petersen et al have developed bioresorbable polymersomes for efficient and site-specific delivery of cisplatin to human colon cancer cells overexpressing α(5)β(1) integrin.114 PEO-block-poly(γ-methyl-ε-caprolactone) polymersomes were functionalized with α(5)β(1) integrin-specific targeting peptide named PR_b. This allowed for specific binding and enhanced uptake into α(5)β(1)-overexpressing cancer cells in comparison to conventionally used RGD-targeting peptides which bind to a variety of integrins. Cisplatin-loaded PR_b-functionalized polymersomes demonstrated enhanced cytotoxicity toward DLD-1 colon cancer cells than non-targeted polymersomes. Targeted polymersomes were found to be less toxic to CACO-2 model human epithelial cells which express low α(5)β(1) integrin levels, signifying that targeting was specific to α(5)β(1)-overexpressing cells.The in vivo antitumor efficacy of DOX-loaded poly(γ-benzyl-l-glutamate)-block-hyaluronan-based polymersomes was evaluated in Ehrlich ascites tumor-bearing mice. Biodistribution study in mice revealed that the polymersomes selectively accumulated in the tumor by virtue of passive accumulation and active targeting (CD44-mediated endocytosis) due to the presence of hyaluronic acid on the surface of polymersomes. This site-specific delivery of drug leads to prolongation in tumor doubling time and increased survival of mice.115 Polymersomes self-assembled from PEO-b-poly(butadiene) diblock copolymers were functionalized with PR_b for targeted delivery of therapeutic protein named tumor necrosis factor-α (TNF-α) to prostate cancer cells. Efficient internalization of PR_b-functionalized polymersomes was achieved by specifically attaching to α5β1 integrins expressed on prostate cancer cells. Increased cytotoxic potential of delivered TNF-α was seen with targeted polymersomes than non-targeted polymersomes.116
Polyplexes
Polyplexes are polymeric systems in which gene or siRNA is condensed and/or complexed through electrostatic interactions between the cationic groups of the polymer and the negatively charged nucleic acids. The polyplexes protect the nucleic acids from enzymatic degradation and prevent the release of cargo at off-target sites. Also, polyplexes with excess positive charge may preferentially enhance the transfection by interaction with negatively charged cell surfaces. Specific delivery of therapeutic nucleic acids to tumor sites is a promising approach in anticancer strategies.117
Poly-l-lysine-based vector was explored by Zhao et al for cancer-specific gene therapy.118 The polymer was modified with histidine group to impart endosome escape property, and cationic peptide moiety to aid polyplex formation with pDNA and act as substrate for protein kinase Cα (PKCα) which is specifically activated in cancer cells. The polyplexes demonstrated PKCα-responsive gene expression immediately after their application into cancer cells, and the gene expression was found to continue for 24 hours.Phosphorylcholine-modified polyethyleneimine (PEI) was employed as an effective strategy for delivery of DNA in cancer therapy. These polyplexes were shown to be selectively uptaken by liver cancer HepG2 cells compared to PEGylated polyplexes and also exhibit sixfold more gene expression in liver cancer cells than normal cells.119
Nie et al developed synthetic dual-ligand-targeted polyplex system based on plasmid DNA condensation by PEI.120 The peptide B6 targeting transferrin receptor and RGD-containing peptide for simultaneous integrin targeting were evaluated in the context of PEGylated PEI-based polyplexes. Cellular association and cellular uptake studies demonstrated specificity of both ligands for each targeted receptor in two prostate cancer cell lines (DU145 and PC-3). Increased transfection efficiency by fourfold and targeting synergism were evident for dual targeting over the combination of single-targeted polyplexes in the ratio of 1:1.van Steenis et al prepared PEGylated poly(dimethylaminomethyl methacrylate)-based polyplexes containing folate as targeting ligand at their surface.121 Higher cytotoxicity of the folate-containing polyplex by 2.5 times was observed due to increased cellular association of the folate-targeted complex than non-folate polyplexes. Transfection of human ovarian cancer cell line (OVCAR-3) in vitro was distinctly increased compared to untargeted PEGylated polyplexes, implying targeted gene delivery. Monoclonal antibodies targeting EGFR conjugated with PEI-grafted-α,β-poly(N-3-hydroxypropyl)-dl-aspartamide were complexed with pDNA for targeted therapy of hepatocellular carcinoma. Enhanced transfection efficiency in liver cells overexpressing EGFR in vitro compared to non-targeted polyplexes was observed.122
Galactose-modified trimethyl chitosan-cysteine-based polymeric vectors were explored for its ability to deliver siRNA. These polyplexes resulted in efficient and persistent gene knockdown when tested in human liver cancer (QGY-7703) cells and human lung cancer (A549) cells. Remarkable antitumor efficacy with respect to the tumor growth retardation, gene knockdown, angiogenesis inhibition, and apoptosis induction was achieved in QGY-7703 tumor-bearing mice.123 Cationic (oligoethanamino)amide-based polymers were conjugated with folic acid for targeted delivery of siRNA in human cervix carcinoma cells. These polyplexes achieved folate receptor-specific cell targeting, and silencing of the EG5 gene in receptor-positive tumors. In vivo administration of these polyplexes resulted in silencing of reporter gene and the absence of accumulation in non-target tissues such as the liver, lung, and spleen.124 A polymeric system was devised for delivery of prostate cancer cell-specific VEGF siRNA. Prostate cancer cell-targeting peptide was conjugated with PEI via a PEG linker. This polymeric conjugate could efficiently condense siRNA to form stable polyplexes. These polyplexes exhibited significantly higher gene silencing than unmodified polymeric carriers (PEI–PEG or PEI) due to targeting peptide-mediated specific internalization in human prostate carcinoma cells (PC-3 cells).125
Polymer hybrid systems
Polymer–lipid hybrid system
Polymer–lipid hybrid system is a combination of polymeric NPs and liposomes. This hybrid system has the following components: i) a biodegradable hydrophobic polymeric core encapsulating poorly water-soluble anticancer drugs to provide sustained release, ii) a hydrophilic shell providing stealth property to evade identification by the immune system and prolong the systemic circulation, and iii) a lipidic monolayer separating hydrophobic core and hydrophilic shell to prevent diffusion of encapsulated agent and decrease water penetration rate into the NPs. This hybrid system combines the unique features of both polymeric NPs and liposomes that include high drug encapsulation, desirable sustained drug release prolife, and good serum stability, and allows for surface functionalization to achieve cancer cell targeting.126
A polymer–lipid hybrid nanoparticulate (PLN) system containing DOX was developed by complexing the cationic DOX with anionic soybean-oil-based polymer and dispersing this complex with lipid (stearic acid) in water. Effective delivery of DOX and enhanced cytotoxicity by eightfold against P-gp-overexpressing human breast cancer cell line were observed from PLN system but no difference on a wild-type cell line when compared to DOX solution.127 It was revealed by endocytosis inhibition studies that phagocytosis is the important pathway for improved cellular uptake of PLN system. Physical association of DOX with the PLN system enables it to bypass the membrane-bound P-gp, thereby resulting in enhanced DOX uptake and retention in P-gp-overexpressing cells than free drug.128 DOX–PLN possesses significant in vivo cytotoxic activity against solid tumors when administered in mice intratumorally with minimal systemic toxicity.129 Hu et al have synthesized a targeted delivery system by conjugating anti-carcinoembryonic antigen (CEA) half-antibody to lipid–polymer hybrid NPs to target CEA overexpressed in pancreatic cancer cells.130 These hybrid NPs comprise polymeric core made of poly(d,l-lactic-co-glycolic acid), a monolayer of phospholipids, and an outer corona layer made oPEG. In vitro cell uptake study demonstrated specificity of targeted NPs toward CEA-presenting pancreatic cancer (BxPC-3) cells than CEA-negative (XPA-3) cells. PTX-loaded targeted NPs exhibited enhanced cellular cytotoxicity against BxPC-3 when compared with non-targeted NPs.
Folic acid-conjugated NPs of mixed lipid monolayer shell and biodegradable polymer (PLGA) core were fabricated for controlled and targeted delivery of docetaxel. Functional components of mixed lipid shell were 1,2-dilauroylphosphatidylocholine (for stabilization of NPs in aqueous phase), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (to impart stealth property), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[folate(polyethylene glycol)-5000] (functionalization with folic acid for targeted delivery). Folic acid-conjugated NPs demonstrated higher cellular uptake cytotoxicity than non-conjugated formulation at the same drug concentration and exposure time.131 Zhang et al fabricated PLN by self-assembly of PLGA and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine–PEG conjugate for targeted delivery of docetaxel.126 They functionalized PLN with A10 aptamer which binds to PSMA overexpressed by prostate cancer cells. Selective uptake of aptamer-functionalized PLN was evident in PSMA-expressing cancer cells than non-expressing cells.
Polymer–surfactant hybrid system
Chavanpatil et al have fabricated novel polymer–surfactant NP system for encapsulation and sustained release of water-soluble drugs.132 This hybrid system constitutes polymer (sodium alginate) and anionic surfactant (dioctylsodium sulfosuccinate; AOT). AOT–alginate NPs enhanced the cytotoxicity of DOX significantly due to increased cellular uptake and drug accumulation in drug-resistant MCF-7 cells in comparison to DOX solution.
Polymer–cyclodextrin hybrid system
Bellocq et al developed a transferrin-modified, cyclodextrin polymer-based system for delivery of siRNA.133 This hybrid system comprises an NP assembly formed by condensation of a cyclodextrin polycation with nucleic acid, PEG at the surface for increasing stability in biological fluids, and transferrin for targeting of cancer cells that express transferrin receptor. The transferrin–PEG–adamantane conjugate self-assembles with the NPs by adamantane (host) and particle surface cyclodextrin (guest) inclusion complex formation and also retains high receptor binding. This system was found to transfect K562 leukemia cells with a fourfold enhancement over non-targeted NPs.
Polymer conjugates
Water-soluble polymers conjugated to anticancer drugs or proteins are referred as polymer conjugates. These have a pharmacokinetic prolife different from that of the parent drug, and hence are considered as new chemical entities. Polymer conjugation to proteins reduces immunogenicity, extends plasma half-life, and enhances protein stability, whereas polymer–drug conjugation promotes tumor targeting through the EPR effect and enables endocytic capture at cellular level, resulting in lysosomotropic drug delivery. Linear polymers such as N-(2-hydroxypropyl)methacrylamide copolymers, polyglutamic acid, PEG, and polysaccharides (dextran) with drugs (DOX, PTX, camptothecin, and platinate) have been widely developed for the fabrication of polymer–drug conjugates. Polymer–drug/protein conjugates represent the most widely tested polymeric therapeutic in clinical setting.134,135 Numerous polymer conjugates successfully employed in oncotherapy are reviewed in Tables 3 and 4.
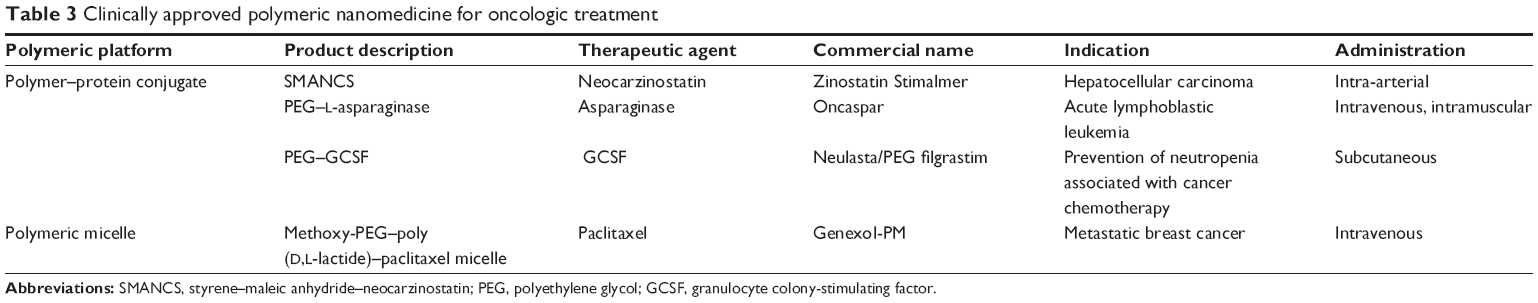 | Table 3 Clinically approved polymeric nanomedicine for oncologic treatment |
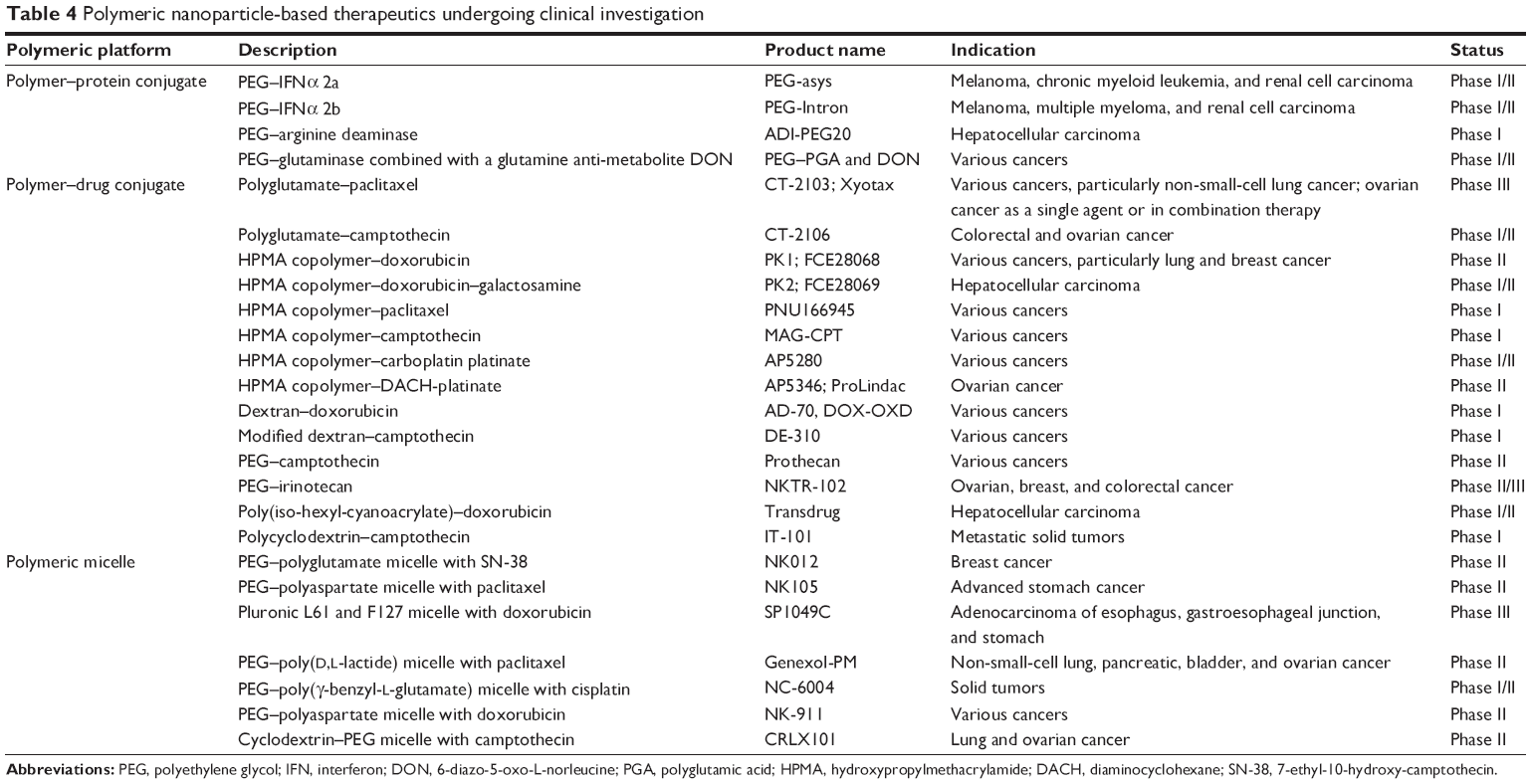 | Table 4 Polymeric nanoparticle-based therapeutics undergoing clinical investigation |
Clinical status of polymeric nanomedicine
Advances in the field of polymeric nanomedicine have rapidly paved its way into clinical trials. Majority of the NP-based therapeutic systems being investigated in clinical and preclinical study level belong to polymeric type (Tables 3 and 4).22,38,135–139 The advantage of ligand-based targeted NPs seems to be widely established; this strategy has resulted in two clinically validated polymeric nanoproducts. CALAA-01 was the first tumor-targeted polymeric nanoformulation to reach clinical development for siRNA delivery. This nanosystem consists of transferrin-functionalized cyclodextrin-based PEGylated NPs containing siRNA for reduction in expression of the M2 subunit of ribonucleotide reductase. CALAA-01 was evaluated in a Phase I clinical trial by intravenous administration to patients with solid tumors refractory to standard treatment.140 Another clinically tested tumor-targeted NP was BIND-014, which comprises biodegradable copolymeric core (PLA, PLGA, and PEG), a pseudo-mimetic dipeptide as a PSMA-targeting ligand, and docetaxel as the anticancer drug. PSMA is a tumor antigen expressed on prostate cancer cells and on the neovasculature of most non-prostate solid tumors. This formulation has entered Phase II clinical trial and is indicated for treatment of solid tumors.139,141
Conclusion
Nanocarriers have emerged as an important treatment modality for therapeutic oncology. Polymer-based nanocarriers have established excellent therapeutic potential at both preclinical and clinical development stages. The fact that polymer-based nanosystems are already in clinical use further validates the efficiency of polymeric platforms for delivery of anticancer agents. The wide scope provided by polymeric platform for functionalization with targeting ligand needs to be validated for its successful application in clinic, although such targeted systems have proven their efficacy in preclinical development. Safety of polymeric nanocarriers is an important consideration, which needs to be assessed before proceeding to clinical study.
Versatility of polymer chemistry enables synthesis of novel polymers with desired properties. The investigation for new molecular targets will advance the ability to improve delivery at the tumor level while reducing toxicity to normal tissues. The field of theranosis is rapidly progressing, and polymer-based carrier system is finding its place in this field for the targeted and image-guided therapy of cancer. This allows for monitoring drug delivery and therapeutic response. Blend of polymers is currently being explored to modulate the properties of the polymeric matrix to achieve high therapeutic load and release-control ability with resultant strong implication on cancer treatment.
Acknowledgment
The authors are thankful to the INSPIRE Program, Department of Science and Technology, Government of India for providing research fellowship.
Disclosure
The authors report no conflicts of interest in this work.
References
Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol. 2008;26(1):57–64. | ||
Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14(5):1310–1316. | ||
Sinha R, Kim GJ, Nie S, Shin DM. Nanotechnology in cancer therapeutics: bioconjugated nanoparticles for drug delivery. Mol Cancer Ther. 2006;5(8):1909–1917. | ||
Gu FX, Karnik R, Wang AZ, et al. Targeted nanoparticles for cancer therapy. Nano Today. 2007;2(3):14–21. | ||
Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60(15):1615–1626. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. | ||
Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249–257. | ||
Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25(34):4633–4646. | ||
Yatvin MB, Kreutz W, Horwitz BA, Shinitzky M. pH-sensitive liposomes: possible clinical implications. Science. 1980;210(4475):1253–1255. | ||
Brewer E, Coleman J, Lowman A. Emerging technologies of polymeric nanoparticles in cancer drug delivery. J Nanomater. 2011;2011:1–10. | ||
Cheng FY, Su CH, Wu PC, Yeh CS. Multifunctional polymeric nanoparticles for combined chemotherapeutic and near-infrared photothermal cancer therapy in vitro and in vivo. Chem Commun (Camb). 2010;46(18):3167–3169. | ||
Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25(1):9–34. | ||
Mansour AM, Drevs J, Esser N, et al. A new approach for the treatment of malignant melanoma: enhanced antitumor efficacy of an albumin-binding doxorubicin prodrug that is cleaved by matrix metalloproteinase 2. Cancer Res. 2003;63(14):4062–4066. | ||
James AM, Ambrose EJ, Lowick JH. Differences between the electrical charge carried by normal and homologous tumour cells. Nature. 1956;177(4508):576–577. | ||
Ran S, Downes A, Thorpe PE. Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2002;62(21):6132–6140. | ||
Krasnici S, Werner A, Eichhorn ME, et al. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int J Cancer. 2003;105(4):561–567. | ||
Asati A, Santra S, Kaittanis C, Perez JM. Surface-charge-dependent cell localization and cytotoxicity of cerium oxide nanoparticles. ACS Nano. 2010;4(9):5321–5331. | ||
Boyer C, Teo J, Phillips P, et al. Effective delivery of siRNA into cancer cells and tumors using well-defined biodegradable cationic star polymers. Mol Pharm. 2013;10(6):2435–2444. | ||
Beh CW, Seow WY, Wang Y, et al. Efficient delivery of Bcl-2-targeted siRNA using cationic polymer nanoparticles: downregulating mRNA expression level and sensitizing cancer cells to anticancer drug. Biomacromolecules. 2009;10(1):41–48. | ||
Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148(2):135–146. | ||
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. | ||
Kirpotin DB, Drummond DC, Shao Y, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66(13):6732–6740. | ||
Drummond DC, Hong K, Park JW, Benz CC, Kirpotin DB. Liposome targeting to tumors using vitamin and growth factor receptors. Vitam Horm. 2000;60:285–332. | ||
Iinuma H, Maruyama K, Okinaga K, et al. Intracellular targeting therapy of cisplatin-encapsulated transferrin-polyethylene glycol liposome on peritoneal dissemination of gastric cancer. Int J Cancer. 2002;99(1):130–137. | ||
Kobayashi T, Ishida T, Okada Y, Ise S, Harashima H, Kiwada H. Effect of transferrin receptor-targeted liposomal doxorubicin in P-glycoprotein-mediated drug resistant tumor cells. Int J Pharm. 2007;329(1–2):94–102. | ||
Lopes de Menezes DE, Pilarski LM, Allen TM. In vitro and in vivo targeting of immunoliposomal doxorubicin to human B-cell lymphoma. Cancer Res. 1998;58(15):3320–3330. | ||
Pastorino F, Brignole C, Di Paolo D, et al. Targeting liposomal chemotherapy via both tumor cell-specific and tumor vasculature-specific ligands potentiates therapeutic efficacy. Cancer Res. 2006;66(20):10073–10082. | ||
Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin Immunol. 2006;121(2):159–176. | ||
Low PS, Kularatne SA. Folate-targeted therapeutic and imaging agents for cancer. Curr Opin Chem Biol. 2009;13(3):256–262. | ||
Sudimack J, Lee RJ. Targeted drug delivery via the folate receptor. Adv Drug Deliv Rev. 2000;41(2):147–162. | ||
Low PS, Antony AC. Folate receptor-targeted drugs for cancer and inflammatory diseases. Adv Drug Deliv Rev. 2004;56(8):1055–1058. | ||
Laskin JJ, Sandler AB. Epidermal growth factor receptor: a promising target in solid tumours. Cancer Treat Rev. 2004;30(1):1–17. | ||
Acharya S, Dilnawaz F, Sahoo SK. Targeted epidermal growth factor receptor nanoparticle bioconjugates for breast cancer therapy. Biomaterials. 2009;30(29):5737–5750. | ||
Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res. 2006;12(18):5268–5272. | ||
Lurje G, Lenz HJ. EGFR signaling and drug discovery. Oncology. 2009;77(6):400–410. | ||
Minko T. Drug targeting to the colon with lectins and neoglycoconjugates. Adv Drug Deliv Rev. 2004;56(4):491–509. | ||
Lammers T, Hennink WE, Storm G. Tumour-targeted nanomedicines: principles and practice. Br J Cancer. 2008;99(3):392–397. | ||
Davis DW, Herbst R, Abbruzzese JL. Antiangiogenic Cancer Therapy. Boca Raton: CRC Press; 2008. | ||
Kumar S, Li C. Targeting of vasculature in cancer and other angiogenic diseases. Trends Immunol. 2001;22(3):129. | ||
Shadidi M, Sioud M. Selective targeting of cancer cells using synthetic peptides. Drug Resist Updat. 2003;6(6):363–371. | ||
Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(suppl 3):4–10. | ||
Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9–22. | ||
Dienst A, Grunow A, Unruh M, et al. Specific occlusion of murine and human tumor vasculature by VCAM-1-targeted recombinant fusion proteins. J Natl Cancer Inst. 2005;97(10):733–747. | ||
Osborn L, Hession C, Tizard R, et al. Direct expression cloning of vascular cell adhesion molecule 1, a cytokine induced endothelial protein that binds to lymphocytes. Cell. 1989;59(6):1203–1211. | ||
Genís L, Gálvez BG, Gonzalo P, Arroyo AG. MT1-MMP: universal or particular player in angiogenesis? Cancer Metastasis Rev. 2006;25(1):77–86. | ||
Vihinen P, Ala-aho R, Kähäri VM. Matrix metalloproteinases as therapeutic targets in cancer. Curr Cancer Drug Targets. 2005;5(3):203–220. | ||
Chan JM, Valencia PM, Zhang L, Langer R, Farokhzad OC. Polymeric nanoparticles for drug delivery. Methods Mol Biol. 2010;624:163–175. | ||
Parveen S, Sahoo SK. Polymeric nanoparticles for cancer therapy. J Drug Target. 2008;16(2):108–123. | ||
Letchford K, Burt H. A review of the formation and classification of amphiphilic block copolymer nanoparticulate structures: micelles, nanospheres, nanocapsules and polymersomes. Eur J Pharm Biopharm. 2007;65(3):259–269. | ||
Danhier F, Lecouturier N, Vroman B, et al. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluation. J Control Release. 2009;133(1):11–17. | ||
Moreno D, de Ilarduya CT, Bandrés E, et al. Characterization of cisplatin cytotoxicity delivered from PLGA-systems. Eur J Pharm Biopharm. 2008;68(3):503–512. | ||
Moreno D, Zalba S, Navarro I, Tros de Ilarduya C, Garrido MJ. Pharmacodynamics of cisplatin-loaded PLGA nanoparticles administered to tumor-bearing mice. Eur J Pharm Biopharm. 2010;74(2):265–274. | ||
Nair KL, Jagadeeshan S, Nair SA, Kumar GS. Biological evaluation of 5-fluorouracil nanoparticles for cancer chemotherapy and its dependence on the carrier, PLGA. Int J Nanomed. 2011;6:1685–1697. | ||
Betancourt T, Brown B, Brannon-Peppas L. Doxorubicin-loaded PLGA nanoparticles by nanoprecipitation: preparation, characterization and in vitro evaluation. Nanomedicine (Lond). 2007;2(2):219–232. | ||
Park H, Yang J, Lee J, Haam S, Choi IH, Yoo KH. Multifunctional nanoparticles for combined doxorubicin and photothermal treatments. ACS Nano. 2009;3(10):2919–2926. | ||
Amjadi I, Rabiee M, Hosseini MS, Mozafari M. Synthesis and characterization of doxorubicin-loaded poly(lactide-co-glycolide) nanoparticles as a sustained release anticancer drug delivery system. Appl Biochem Biotechnol. 2012;168(6):1434–1447. | ||
Jain AK, Swarnakar NK, Godugu C, Singh RP, Jain S. The effect of the oral administration of polymeric nanoparticles on the efficacy and toxicity of tamoxifen. Biomaterials. 2011;32(2):503–515. | ||
Renoir JM, Stella B, Ameller T, Connault E, Opolon P, Marsaud V. Improved anti-tumoral capacity of mixed and pure anti-oestrogens in breast cancer cell xenografts after their administration by entrapment in colloidal nanosystems. J Steroid Biochem Mol Biol. 2006;102(1–5):114–127. | ||
Papa AL, Basu S, Sengupta P, Banerjee D, Sengupta S, Harfouche R. Mechanistic studies of gemcitabine-loaded nanoplatforms in resistant pancreatic cancer cells. BMC Cancer. 2012;12:419. | ||
Gryparis EC, Hatziapostolou M, Papadimitriou E, Avgoustakis K. Anticancer activity of cisplatin-loaded PLGA-mPEG nanoparticles on LNCaP prostate cancer cells. Eur J Pharm Biopharm. 2007;67(1):1–8. | ||
Cheng L, Jin C, Lv W, Ding Q, Han X. Developing a highly stable PLGA-mPEG nanoparticle loaded with cisplatin for chemotherapy of ovarian cancer. PLoS One. 2011;6(9):e25433. | ||
Cheng MR, Li Q, Wan T, et al. Galactosylated chitosan/5-fluorouracil nanoparticles inhibit mouse hepatic cancer growth and its side effects. World J Gastroenterol. 2012;18(42):6076–6087. | ||
Cheng M, He B, Wan T, et al. 5-Fluorouracil nanoparticles inhibit hepatocellular carcinoma via activation of the p53 pathway in the orthotopic transplant mouse model. PLoS One. 2012;7(10):e47115. | ||
Yadav AK, Agarwal A, Rai G, et al. Development and characterization of hyaluronic acid decorated PLGA nanoparticles for delivery of 5-fluorouracil. Drug Deliv. 2010;17(8):561–572. | ||
Li S, Wang A, Jiang W, Guan Z. Pharmacokinetic characteristics and anticancer effects of 5-fluorouracil loaded nanoparticles. BMC Cancer. 2008;8:103. | ||
Chang L, Deng L, Wang W, et al. Poly(ethyleneglycol)-b-poly(ε-caprolactone-co-γ-hydroxyl- ε -caprolactone) bearing pendant hydroxyl groups as nanocarriers for doxorubicin delivery. Biomacromolecules. 2012;13(10):3301–3310. | ||
Wohlfart S, Khalansky AS, Gelperina S, et al. Efficient chemotherapy of rat glioblastoma using doxorubicin loaded PLGA nanoparticles with different stabilizers. PLoS One. 2011;6(5):e19121. | ||
Pérez E, Benito M, Teijón C, Olmo R, Teijón JM, Blanco MD. Tamoxifen-loaded nanoparticles based on a novel mixture of biodegradable polyesters: characterization and in vitro evaluation as sustained release systems. J Microencapsul. 2012;29(4):309–322. | ||
Cavalli R, Bisazza A, Bussano R, et al. Poly(amidoamine)-cholesterol conjugate nanoparticles obtained by electrospraying as novel tamoxifen delivery system. J Drug Deliv. 2011;2011:587604. | ||
Devalapally H, Duan Z, Seiden MV, Amiji MM. Modulation of drug resistance in ovarian adenocarcinoma by enhancing intracellular ceramide using tamoxifen-loaded biodegradable polymeric nanoparticles. Clin Cancer Res. 2008;14(10):3193–3203. | ||
Jia L, Zheng JJ, Jiang SM, Huang KH. Preparation, physicochemical characterization and cytotoxicity in vitro of gemcitabine-loaded PEG-PDLLA nanovesicles. World J Gastroenterol. 2010;16(8):1008–1013. | ||
Yordanov G, Skrobanska R, Evangelatov A. Entrapment of epirubicin in poly(butyl cyanoacrylate) colloidal nanospheres by nanoprecipitation: formulation development and in vitro studies on cancer cell lines. Colloids Surf B Biointerfaces. 2012;92:98–105. | ||
Chittasupho C, Xie SX, Baoum A, Yakovleva T, Siahaan TJ, Berkland CJ. ICAM-1 targeting of doxorubicin-loaded PLGA nanoparticles to lung epithelial cells. Eur J Pharm Sci. 2009;37(2):141–150. | ||
Aggarwal S, Yadav S, Gupta S. EGFR targeted PLGA nanoparticles using gemcitabine for treatment of pancreatic cancer. J Biomed Nanotechnol. 2011;7(1):137–138. | ||
Cheng J, Teply BA, Sherifi I, et al. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28(5):869–876. | ||
Farokhzad OC, Cheng J, Teply BA, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103(16):6315–6320. | ||
Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A. 2008;105(45):17356–17361. | ||
Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc Natl Acad Sci U S A. 2011;108(5):1850–1855. | ||
Graf N, Bielenberg DR, Kolishetti N, et al. α(V)β(3) integrin-targeted PLGA-PEG nanoparticles for enhanced anti-tumor efficacy of a Pt(IV) prodrug. ACS Nano. 2012;6(5):4530–4539. | ||
Liu CW, Lin WJ. Polymeric nanoparticles conjugate a novel heptapeptide as an epidermal growth factor receptor-active targeting ligand for doxorubicin. Int J Nanomedicine. 2012;7:4749–4767. | ||
Hou Z, Zhan C, Jiang Q, et al. Both FA- and mPEG-conjugated chitosan nanoparticles for targeted cellular uptake and enhanced tumor tissue distribution. Nanoscale Res Lett. 2011;6(1):563. | ||
Zhang HZ, Li XM, Gao FP, Liu LR, Zhou ZM, Zhang QQ. Preparation of folate-modified pullulan acetate nanoparticles for tumor-targeted drug delivery. Drug Deliv. 2010;17(1):48–57. | ||
Sutton D, Nasongkla N, Blanco E, Gao J. Functionalized micellar systems for cancer targeted drug delivery. Pharm Res. 2007;24(6):1029–1046. | ||
Oerlemans C, Bult W, Bos M, Storm G, Nijsen JF, Hennink WE. Polymeric micelles in anticancer therapy: targeting, imaging and triggered release. Pharm Res. 2010;27(12):2569–2589. | ||
Bisht S, Feldmann G, Soni S, et al. Polymeric nanoparticle-encapsulated curcumin (“nanocurcumin”): a novel strategy for human cancer therapy. J Nanobiotechnology. 2007;5:3. | ||
Jin X, Mo R, Ding Y, Zheng W, Zhang C. Paclitaxel-loaded N-octyl-O-sulfate chitosan micelles for superior cancer therapeutic efficacy and overcoming drug resistance. Mol Pharm. 2014;11(1):145–157. | ||
Jeong YI, Kim do H, Chung CW, et al. Doxorubicin-incorporated polymeric micelles composed of dextran-b-poly(DL-lactide-co-glycolide) copolymer. Int J Nanomedicine. 2011;6:1415–1427. | ||
Nishiyama N, Okazaki S, Cabral H, et al. Novel cisplatin-incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res. 2003;63(24):8977–8983. | ||
Vega J, Ke S, Fan Z, Wallace S, Charsangavej C, Li C. Targeting doxorubicin to epidermal growth factor receptors by site-specific conjugation of C225 to poly(l-glutamic acid) through a polyethylene glycol spacer. Pharm Res. 2003;20(5):826–832. | ||
Torchilin VP, Lukyanov AN, Gao Z, Papahadjopoulos-Sternberg B. Immunomicelles: targeted pharmaceutical carriers for poorly soluble drugs. Proc Natl Acad Sci U S A. 2003;100(10):6039–6044. | ||
Nasongkla N, Shuai X, Ai H, et al. cRGD-functionalized polymer micelles for targeted doxorubicin delivery. Angew Chem Int Ed Engl. 2004;43(46):6323–6327. | ||
Yoo HS, Park TG. Folate receptor targeted biodegradable polymeric doxorubicin micelles. J Control Release. 2004;96(2):273–283. | ||
Park EK, Lee SB, Lee YM. Preparation and characterization of methoxy poly(ethylene glycol)/poly(epsilon-caprolactone) amphiphilic block copolymeric nanospheres for tumor-specific folate-mediated targeting of anticancer drugs. Biomaterials. 2005;26(9):1053–1061. | ||
Han X, Liu J, Liu M, et al. 9-NC-loaded folate-conjugated polymer micelles as tumor targeted drug delivery system: preparation and evaluation in vitro. Int J Pharm. 2009;372(1–2):125–131. | ||
Wang Y, Yu L, Han L, Sha X, Fang X. Difunctional pluronic copolymer micelles for paclitaxel delivery: synergistic effect of folate-mediated targeting and pluronic-mediated overcoming multidrug resistance in tumor cell lines. Int J Pharm. 2007;337(1–2):63–73. | ||
Jeong YI, Seo SJ, Park IK, et al. Cellular recognition of paclitaxel-loaded polymeric nanoparticles composed of poly(gamma-benzyl L-glutamate) and poly(ethylene glycol) diblock copolymer endcapped with galactose moiety. Int J Pharm. 2005;296(1–2):151–161. | ||
Farokhzad OC, Jon S, Khademhosseini A, Tran TN, Lavan DA, Langer R. Nanoparticle-aptamer bioconjugates: a new approach for targeting prostate cancer cells. Cancer Res. 2004;64(21):7668–7672. | ||
Bharali DJ, Khalil M, Gurbuz M, Simone TM, Mousa SA. Nanoparticles and cancer therapy: a concise review with emphasis on dendrimers. Int J Nanomedicine. 2009;4:1–7. | ||
Morgan MT, Carnahan MA, Immoos CE, et al. Dendritic molecular capsules for hydrophobic compounds. J Am Chem Soc. 2003;125(50):15485–15489. | ||
Morgan MT, Carnahan MA, Finkelstein S, et al. Dendritic supramolecular assemblies for drug delivery. Chem Commun (Camb). 2005;34:4309–4311. | ||
Morgan MT, Nakanishi Y, Kroll DJ, et al. Dendrimer-encapsulated camptothecins: increased solubility, cellular uptake, and cellular retention affords enhanced anticancer activity in vitro. Cancer Res. 2006;66(24):11913–11921. | ||
Wang F, Bronich TK, Kabanov AV, Rauh RD, Roovers J. Synthesis and evaluation of a star amphiphilic block copolymer from poly(epsilon-caprolactone) and poly(ethylene glycol) as a potential drug delivery carrier. Bioconjug Chem. 2005;16(2):397–405. | ||
Neerman MF, Chen HT, Parrish AR, Simanek EE. Reduction of drug toxicity using dendrimers based on melamine. Mol Pharm. 2004;1(5):390–393. | ||
Padilla De Jesús OL, Ihre HR, Gagne L, Fréchet JM, Szoka FC Jr. Polyester dendritic systems for drug delivery applications: in vitro and in vivo evaluation. Bioconjug Chem. 2002;13(3):453–461. | ||
Malik N, Evagorou EG, Duncan R. Dendrimer-platinate: a novel approach to cancer chemotherapy. Anticancer Drugs. 1999;10(8):767–776. | ||
Patri AK, Kukowska-Latallo JF, Baker JR Jr. Targeted drug delivery with dendrimers: comparison of the release kinetics of covalently conjugated drug and non-covalent drug inclusion complex. Adv Drug Deliv Rev. 2005;57(15):2203–2214. | ||
Majoros IJ, Myc A, Thomas T, Mehta CB, Baker JR Jr. PAMAM dendrimer-based multifunctional conjugate for cancer therapy: synthesis, characterization, and functionality. Biomacromolecules. 2006;7(2):572–579. | ||
Levine DH, Ghoroghchian PP, Freudenberg J, et al. Polymersomes: a new multi-functional tool for cancer diagnosis and therapy. Methods. 2008;46(1):25–32. | ||
Xu J, Zhao Q, Jin Y, Qiu L. High loading of hydrophilic/hydrophobic doxorubicin into polyphosphazene polymersome for breast cancer therapy. Nanomedicine. 2014;10(2):349–358. | ||
Li S, Byrne B, Welsh J, Palmer AF. Self-assembled poly(butadiene)-b-poly(ethyleneoxide) polymersomes as paclitaxel carriers. Biotechnol Prog. 2007;23(1):278–285. | ||
Ahmed F, Pakunlu RI, Brannan A, Bates F, Minko T, Discher DE. Biodegradable polymersomes loaded with both paclitaxel and doxorubicin permeate and shrink tumors, inducing apoptosis in proportion to accumulated drug. J Control Release. 2006;116(2):150–158. | ||
Ahmed F, Pakunlu RI, Srinivas G, et al. Shrinkage of a rapidly growing tumor by drug-loaded polymersomes: pH-triggered release through copolymer degradation. Mol Pharm. 2006;3(3):340–350. | ||
Petersen MA, Hillmyer MA, Kokkoli E. Bioresorbable polymersomes for targeted delivery of cisplatin. Bioconjug Chem. 2013;24(4):533–543. | ||
Upadhyay KK, Mishra AK, Chuttani K, et al. The in vivo behavior and antitumor activity of doxorubicin-loaded poly(γ-benzyl l-glutamate)-block-hyaluronan polymersomes in Ehrlich ascites tumor-bearing BalB/c mice. Nanomedicine. 2012;8(1):71–80. | ||
Demirgöz D, Pangburn TO, Davis KP, Lee S, Bates FS, Kokkoli E. PR_b-targeted delivery of tumor necrosis factor-α by polymersomes for the treatment of prostate cancer. Soft Matter. 2009;5:2011–2019. | ||
Christie RJ, Nishiyama N, Kataoka K. Delivering the code: polyplex carriers for deoxyribonucleic acid and ribonucleic acid interference therapies. Endocrinology. 2010;151(2):466–473. | ||
Zhao GX, Tanaka H, Kim CW, et al. Histidinylated poly-l-lysine-based vectors for cancer-specific gene expression via enhancing the endosomal escape. J Biomater Sci Polym Ed. 2014;25(5):519–534. | ||
Chen L, Wang H, Zhang Y, Wang Y, Hu Q, Ji J. Bioinspired phosphorylcholine-modified polyplexes as an effective strategy for selective uptake and transfection of cancer cells. Colloids Surf B Biointerfaces. 2013;111:297–305. | ||
Nie Y, Schaffert D, Rödl W, Ogris M, Wagner E, Günther M. Dual-targeted polyplexes: one step towards a synthetic virus for cancer gene therapy. J Control Release. 2011;152(1):127–134. | ||
van Steenis JH, van Maarseveen EM, Verbaan FJ, et al. Preparation and characterization of folate-targeted PEG-coated pDMAEMA-based polyplexes. J Control Release. 2003;87(1–3):167–176. | ||
Wang JL, Tang GP, Shen J, et al. A gene nanocomplex conjugated with monoclonal antibodies for targeted therapy of hepatocellular carcinoma. Biomaterials. 2012;33(18):4597–4607. | ||
Han L, Tang C, Yin C. Effect of binding affinity for siRNA on the in vivo antitumor efficacy of polyplexes. Biomaterials. 2013;34(21):5317–5327. | ||
Dohmen C, Edinger D, Fröhlich T, et al. Nanosized multifunctional polyplexes for receptor-mediated siRNA delivery. ACS Nano. 2012;6(6):5198–5208. | ||
Kim SH, Lee SH, Tian H, Chen X, Park TG. Prostate cancer cell-specific VEGF siRNA delivery system using cell targeting peptide conjugated polyplexes. J Drug Target. 2009;17(4):311–317. | ||
Zhang L, Chan JM, Gu FX, et al. Self-assembled lipid-polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano. 2008;2(8):1696–1702. | ||
Wong HL, Rauth AM, Bendayan R, et al. A new polymer-lipid hybrid nanoparticle system increases cytotoxicity of doxorubicin against multidrug-resistant human breast cancer cells. Pharm Res. 2006;23(7):1574–1585. | ||
Wong HL, Bendayan R, Rauth AM, Xue HY, Babakhanian K, Wu XY. A mechanistic study of enhanced doxorubicin uptake and retention in multidrug resistant breast cancer cells using a polymer-lipid hybrid nanoparticle system. J Pharmacol Exp Ther. 2006;317(3):1372–1381. | ||
Wong HL, Rauth AM, Bendayan R, Wu XY. In vivo evaluation of a new polymer-lipid hybrid nanoparticle (PLN) formulation of doxorubicin in a murine solid tumor model. Eur J Pharm Biopharm. 2007;65(3):300–308. | ||
Hu CM, Kaushal S, Tran Cao HS, et al. Half-antibody functionalized lipid-polymer hybrid nanoparticles for targeted drug delivery to carcinoembryonic antigen presenting pancreatic cancer cells. Mol Pharm. 2010;7(3):914–920. | ||
Liu Y, Li K, Pan J, Liu B, Feng SS. Folic acid conjugated nanoparticles of mixed lipid monolayer shell and biodegradable polymer core for targeted delivery of docetaxel. Biomaterials. 2010;31(2):330–338. | ||
Chavanpatil MD, Khdair A, Gerard B, et al. Surfactant-polymer nanoparticles overcome P-glycoprotein-mediated drug efflux. Mol Pharm. 2007;4(5):730–738. | ||
Bellocq NC, Pun SH, Jensen GS, Davis ME. Transferrin-containing, cyclodextrin polymer-based particles for tumor-targeted gene delivery. Bioconjug Chem. 2003;14(6):1122–1132. | ||
Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6(9):688–701. | ||
Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in medicine: therapeutic applications and developments. Clin Pharmacol Ther. 2008;83(5):761–769. | ||
Thakor AS, Gambhir SS. Nanooncology: the future of cancer diagnosis and therapy. CA Cancer J Clin. 2013;63(6):395–418. | ||
Heidel JD, Davis ME. Clinical developments in nanotechnology for cancer therapy. Pharm Res. 2011;28(2):187–199. | ||
Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med. 2012;63:185–198. | ||
Sanna V, Pala N, Sechi M. Targeted therapy using nanotechnology: focus on cancer. Int J Nanomedicine. 2014;9:467–483. | ||
Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 2009;6(3):659–668. | ||
Hrkach J, Von Hoff D, Mukkaram Ali M, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4(128):128ra39. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.