")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Polyhydroxylated fullerene attenuates oxidative stress-induced apoptosis via a fortifying Nrf2-regulated cellular antioxidant defence system
Authors Ye S, Chen M, Jiang Y, Chen M, Zhou T, Wang Y , Hou Z, Ren L
Received 1 November 2013
Accepted for publication 24 January 2014
Published 29 April 2014 Volume 2014:9(1) Pages 2073—2087
DOI https://doi.org/10.2147/IJN.S56973
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Shefang Ye,1 Min Chen,1 Yuanqin Jiang,1,2 Mingliang Chen,3 Tong Zhou,1 Yange Wang,1 Zhenqing Hou,1 Lei Ren1
1Department of Biomaterials, Research Center of Biomedical Engineering, College of Materials, Xiamen University, Xiamen, People's Republic of China; 2First Affiliated Hospital of Xiamen University, Xiamen, People's Republic of China; 3Key Laboratory of Marine Biogenetic Resources, Third Institute of Oceanography, State Oceanic Administration, Xiamen, People's Republic of China
Abstract: Polyhydroxylated derivatives of fullerene C60, named fullerenols (C60[OH]n), have stimulated great interest because of their potent antioxidant properties in various chemical and biological systems, which enable them to be used as a new promising pharmaceutical for the future treatment of oxidative stress-related diseases, but the details remain unknown. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a principal transcription factor that regulates expression of several antioxidant genes via binding to the antioxidant response element and plays a crucial role in cellular defence against oxidative stress. In this study we investigated whether activation of the Nrf2/antioxidant response element pathway contributes to the cytoprotective effects of C60(OH)24. Our results showed that C60(OH)24 enhanced nuclear translocation of Nrf2 and upregulated expression of phase II antioxidant enzymes, including heme oxygenase-1 (HO-1), NAD(P)H: quinine oxidoreductase 1, and γ-glutamate cysteine ligase in A549 cells. Treatment with C60(OH)24 resulted in phosphorylation of p38 mitogen-activated protein kinases (p38 MAPK), extracellular signal-regulated kinases, and c-Jun-N-terminal kinases. By using inhibitors of cellular kinases, we showed that pretreatment of A549 cells with SB203580, a specific inhibitor of p38 MAPK, abolished nuclear translocation of Nrf2 and induction of HO-1 protein induced by C60(OH)24, indicating an involvement of p38 MAPK in Nrf2/HO-1 activation by C60(OH)24. Furthermore, pretreatment with C60(OH)24 attenuated hydrogen peroxide-induced apoptotic cell death in A549 cells, and knockdown of Nrf2 by small interfering ribonucleic acid diminished C60(OH)24-mediated cytoprotection. Taken together, these findings demonstrate that C60(OH)24 may attenuate oxidative stress-induced apoptosis via augmentation of Nrf2-regulated cellular antioxidant capacity, thus providing insights into the mechanisms of the antioxidant properties of C60(OH)24.
Keywords: fullerenol, Nrf2, oxidative stress, cytoprotection, A549 cells
Introduction
Accumulating evidence suggests that oxidative stress is implicated in several disease states (ie, cancer, aging, and neurological and vascular disorders).1 It occurs when the redox balance is disrupted by excessive production of reactive oxygen species (ROS) and/or by the presence of inadequate antioxidant defences.2 Excess amounts of ROS, such as superoxide anion (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (HO−), have been shown not only to damage cells by peroxidizing lipids and disrupting DNA and proteins but also to exert signaling functions and modulate gene transcription, thereby resulting in cellular dysfunction and apoptosis.3 Therefore, therapeutic strategies aimed at preventing ROS-induced oxidative damage might be promising for the treatment of oxidative stress-related diseases.4,5
One such therapeutic approach is represented by the induction of phase II antioxidant or detoxifying enzymes using nutritional or pharmacological intervention, which promotes activation of a nuclear factor erythroid 2-related factor 2 (Nrf2)/antioxidant response element (ARE) signaling pathway.6 Nrf2 is an essential transcription factor that regulates ARE-mediated expression of phase II antioxidant enzymes. Under unstressed physiological conditions, Nrf2 is bound to the repressor Kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm and easily degraded by ubiquitin–proteasome.7 Upon activation, the Nrf2/Keap1 complex is dissociated, and Nrf2 is released from Keap1 and translocates into the nucleus, where it forms a heterodimer with its obligatory partner Maf and binds to the ARE sequence to activate transcription of a battery of antioxidant and cytoprotective genes, including heme oxygenase-1 (HO-1), NAD(P)H: quinine oxidoreductase 1 (NQO1), and γ-glutamate cysteine ligase.8 Therefore, agents that modulate the Nrf2/ARE pathway would be expected to have beneficial effects in ameliorating oxidative stress-related diseases by upregulation of phase II antioxidant enzymes.7,8
The use of nanoparticles in biomedicine is currently an active area of research because nanoparticles hold great promise as novel and effective antioxidants for oxidative stress-related diseases.9,10 The advantages of using nanoparticles as antioxidants over currently available antioxidants are due to the possibility that they have a versatile surface that can be decorated with active agents,11 can penetrate the cell membrane,12 can quench radicals without the need for assistance from other detoxifying molecules,13 and possess higher physical stability in biological media,14 thus extending their applications to the treatment of oxidative damage. In this context, there is a considerable number of reports describing that various fullerene (C60) derivatives and surfactant-coated C60 derivatives modified through either covalent or noncovalent approaches had been reported to be biocompatible and to exhibit properties as powerful antioxidants to decompose ROS generated under oxidative stress in different model systems.14–17 Polyhydroxylated fullerenes, also known as fullerols [C60(OH)n], belong to a group of antioxidant nanoparticles that exhibit strong antioxidant activity in both in vitro and in vivo biological systems.13 It has been demonstrated that the anti-inflammatory,18 antiapoptotic,16,17 radioprotective,19,20 and neuroprotective effects21,22 and the enzyme-inhibitory activity of fullerols13 have been attributed to their ability to scavenge free radicals, such as superoxide anion radical, hydroxy radical, and nitrous oxide radical. It has since been well established that induction of Nrf2-regulated phase II antioxidant enzymes by nutritional or pharmacological intervention is an effective approach to combat the toxicities of ROS and to protect cells against oxidative stress.8 It is possible that the mechanisms of protective effects of fullerols may also involve upregulation of Nrf2-regulated phase II antioxidant enzymes, which putatively account in part for the antioxidant capacity of fullerols.
In the present study, we hypothesize that C60(OH)24 induces endogenous phase II antioxidant enzymes via Nrf2/ARE-dependent mechanisms and attenuates oxidative stress-mediated cell death, using the human alveolar epithelial A549 cells as a model. We found that C60(OH)24 activates Nrf2 via p38 mitogen-activated protein kinases (MAPKs) signal transduction pathways in A549 cells, which, in turn, upregulates phase II antioxidant enzymes, conferring protection against the cytotoxicity induced by H2O2.
Materials and methods
Materials
Dulbecco’s Modified Eagle’s Medium (DMEM), penicillin, streptomycin, fetal bovine serum (FBS), and the small interfering ribonucleic acid (siRNA) reagents for Nrf2 were obtained from Life Technologies (Carlsbad, CA, USA). Nrf2 consensus oligonucleotides were purchased from Promega Corporation (Fitchburg, WI, USA). γ-[32P] adenosine triphosphate was the product of NEN Life Science (Boston, MA, USA). Antibodies against Nrf2, HO-1, poly ADP-ribose polymerase (PARP), lamin A, β-actin, horseradish peroxidase- and fluorescein isothiocyanate-conjugated secondary antibodies, diamidino-2-phenylindole (DAPI), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and propidium iodide (PI) were purchased from Santa Cruz Biotechnology, Inc., (Santa Cruz, CA, USA). Antibodies against phospho-p38, phospho-Jun-N-terminal kinases (JNK), phospho-extracellular signal-related kinases (ERK1/2), p38, JNK, ERK1/2, cleaved caspase-3, and PARP were obtained from Cell Signaling Technology (Beverly, MA, USA). p38 inhibitor (SB203580), JNK inhibitor (SP600125), and ERK1/2 inhibitors (PD98059 and U0126) were purchased from Calbiochem (La Jolla, CA, USA). N-acetylcysteine (NAC) was from Sigma-Aldrich (St Louis, MO, USA). All other chemicals were of the highest commercial grade available.
Characterization of fullerol C60(OH)24
C60(OH)24, with a purity of more than 99.5%, was purchased from the MER Corporation (Tuscon, AZ, USA). A test solution of C60(OH)24 was prepared in the DMEM, and the highest solubility of C60(OH)24 in culture medium was up to 10 mM. A series of working solutions was freshly made by diluting the stock solution with cell culture medium before use. To investigate the size and shape of the C60(OH)24 nanoparticles, 100 μM of C60(OH)24 solution prepared either in phosphate buffered saline (PBS) (pH 7.0) or in culture medium containing 10% FBS was evaporated on a 400-mesh carbon-coated copper grid and characterized by transmission electron microscopy (TEM; Hitachi H-7500, Tokyo, Japan). The size distribution and zeta potentials of C60(OH)24 nanoparticles were measured by dynamic light scattering (DLS) using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK).
Cell culture
The human type II alveolar epithelial A549 cell line was purchased from the cell bank of the Chinese Academy of Sciences (Shanghai, People’s Republic of China). Cells were maintained routinely in DMEM supplemented with 10% heat-inactivated FBS, L-glutamine (2 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. The medium was changed every other day, and the cells were plated at an appropriate density according to the scale of each experiment. For all experiments, the cells were grown to approximately 70%–80% confluence.
Cell viability assay
Cell viability was determined using MTT assay. Briefly, A549 cells were grown on 96-well plates at a density of 5 × 104 for 24 hours. After treatment with increasing doses of C60(OH)24 (10–200 μM) for 48 hours and 72 hours, cells were incubated with MTT solution (0.5 mg/mL, 1 × PBS) for 2 hours at 37°C. The reaction was then terminated by the addition of 100 μL dimethyl sulfoxide. The formazan crystals resulting from mitochondrial enzymatic activity on MTT substrate were solubilized with 200 μL of dimethyl sulfoxide, and absorbance at 570 nm was measured using a microplate reader (Model 680, Bio-Rad Laboratories, Hercules, CA, USA). For assessment of cell membrane integrity, lactate dehydrogenase (LDH) release in exposure medium was measured with LDH assay following the manufacturer’s instructions (Sigma-Aldrich). The results are given relative to the untreated control.
Apoptosis assay and cell cycle assay
The terminal deoxynucleotidyl transferase-mediated deoxyuridine 5-triphosphate nick end labeling (TUNEL) assay (Life Technologies) was conducted to identify apoptotic cell death. A549 cells were seeded in six-well plates (2 mL, 5 × 105 cells/well) and incubated overnight at 37°C to allow the cells to adhere. After treatment with 100 μM C60(OH)24 for 24 hours, 48 hours, and 72 hours, cells were fixed in 4% paraformaldehyde for 30 minutes at room temperature. After rinsing with PBS, the cells were permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 5 minutes on ice and incubated with the TUNEL reagent for 1 hour at 37°C in the dark. The cells were then rinsed twice with PBS and stained with 1 μg/mL DAPI for 15 minutes at 37°C. Following staining, the apoptotic features of cell death were examined under a fluorescence microscope (Nikon Eclipse 80i, Tokyo, Japan). For cell cycle analysis, cells were harvested and fixed in 70% ethanol and stored at 4°C overnight. The fixed cells were centrifuged at 1,000 × g for 10 minutes and washed with cold PBS three times and then incubated with 50 μg/mL PI containing 10 μg/mL ribonuclease A in the dark at 37°C for 30 minutes. The DNA content of cells was quantified by flow cytometry (Beckman-Coulter Epics XL, Miami, FL, USA).
Measurement of intracellular ROS
The intracellular ROS level was determined using a peroxide-sensitive fluorescent probe, 2,7-dichlorodihydro fluorescent diacetate (DCFH-DA; Molecular Probes, Eugene, OR, USA). DCFH-DA enters the cells via passive diffusion, where it reacts with ROS, resulting in the formation of the highly fluorescent compound dichlorofluorescein (DCF).23 Briefly, cells attached to plastic dishes were harvested by 0.25% trypsin, washed twice with cold PBS, and incubated with 10 μM DCFH-DA for 30 minutes at 37°C, and then analyzed using a flow cytometer (Beckman-Coulter Epics XL, Miami, FL, USA).
Western blot analysis
After treatment with C60(OH)24 nanoparticles, cells were harvested, washed twice with ice-cold PBS, and lysed in modified radioimmunoprecipitation buffer (10 mM Tris–HCl [pH 7.5], 1 mM ethylenediaminetetraacetic acid, 150 mM NaCl, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate, 0.1% sodium deoxycholate) containing protease inhibitors. Cytoplasmic and nuclear cell lysates were separated by using the Active Motif nuclear extract kit (Active Motif, Carlsbad, CA, USA) following the manufacturer’s protocol. The protein concentration was determined using the protein assay reagent (Bio-Rad Laboratories). For Western blot analysis, an equal amount of protein (30 μg) were loaded on to 12% sodium dodecyl sulfate–polyacrylamide gels and electrophoretically transferred on to a polyvinylidene difluoride membrane (Millipore Corporation, Bedford, MA, USA). After blocking, the membranes were then incubated overnight at 4°C with specific primary antibodies. After washing three times with Tris Buffered Saline with Tween 20 (TBST), the membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 1 hour. The blots were developed using enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ, USA) according to the manufacturer’s protocol. Densitometry data analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Reverse transcription-polymerase chain reaction
Total RNA was isolated from A549 cells using the Trizol reagent (Life Technologies). For cDNA preparation, DNase I-treated (25 Kunitz units) total RNA (2 μg) was incubated at 37°C for 90 minutes by the First-Strand cDNA Synthesis Kit according to the manufacturer’s instructions (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Oligonucleotide primer sets used were as follows: HO-1, 5′-GGAACTTTCAGAAGGGCCAG-3′ (sense), and 5′-GTCCTTGGTGTCATGGGTCA-3′ (antisense); NQO1, 5′-GTATCCTGCCGAGTCTGTT-3′ (sense), and 5′-GATCCCTTGCAGAGAGTACA-3′ (antisense); γ-glutamate cysteine ligase catalytic subunit (GCSc), 5′-TCACCTCCTATTGAAGATGG-3′ (sense); 5′-GGTTACTATTTGGTTTTACCTGT-3′ (antisense); glyceraldehyde-3-phosphate dehydrogenase (GAPDH): 5′-GCCAAGGTCATCCATGACAAC-3′ (sense) and 5′-AGTGTAGCCCAGGATGCCCTT-3′ (antisense). The amplified samples were run in a 1% agarose gel with ethidium bromide and the bands were visualized under ultraviolet illumination.
Immunofluorescence staining
A549 cells were fixed with paraformaldehyde, permeabilized with 0.5% Triton X-100 in PBS, and then incubated with blocking buffer (PBS, 5% goat serum, and 0.3% Triton X-100) for 30 minutes. The cells were then labeled with primary antibodies against Nrf2 in blocking buffer at 4°C overnight, followed by incubation with a fluorescein isothiocyanate-conjugated secondary antibody. Thereafter, cells were nuclear-stained via 15-minute incubation in a blocking solution containing 0.25 mg/mL DAPI. Fluorescent-labeled cells were imaged with a fluorescent microscope (Leica DMR, Solms, Germany).
Electrophoretic mobility shift assay
Nuclear extracts from cells incubated with C60(OH)24 nanoparticles were prepared as described previously.24 Electrophoretic mobility shift assay was performed using a synthetic double-stranded oligonucleotide containing the Nrf2-binding domain (ARE), which was labeled with [γ-32P] adenosine triphosphate using the T4 polynucleotide kinase and purified using a ProbeQuant™ G-50 Micro Columns following the manufacturer’s protocol (Amersham Pharmacia Biotech Inc., Piscataway, NJ). The double-stranded sequence was 5′-TTTTCTGCTGAGTCAAGGGTCCG-3′ and 3′-AAAAGACGACTCAGTTCCAGGC-5′. Prior to the addition of 32P-labeled oligonucleotide (50,000 cpm), 10 μg of the nuclear extract was incubated for 30 minutes on ice in the gel-shift assay binding buffer (20% glycerol, 5 mM MgCl2, 2.5 mM ethylenediaminetetraacetic acid, 250 mM NaCl, 2.5 mM dithiothreitol, and 50 mM Tris–HCl, pH 7.5 with 0.25 μg/ml poly[dI-dC]). DNA–protein complexes were resolved in 5% polyacrylamide gel electrophoresis and the bands were visualized by autoradiography.
siRNA transfection
A549 cells were grown to 50%–70% confluence in DMEM supplemented with 10% FBS. Cells were transfected with the Nrf2–siRNA or siRNA control with Lipofectamine RNAi Max (Life Technologies) according to the manufacturer’s protocol. The final concentration of the siRNA was 20 nmol/L. The knockdown efficiency was validated by Western blot analysis. The Nrf2–siRNA duplex with the following sense and antisense sequences was used: 5′-GUAAGAAGCCAGAUGUUAAdUdU-3′ (sense) and 3′-dUdUCAUUCUUCGGUCUACAATT-5′ (antisense). To confirm the specificity of the inhibition, the nontargeting siRNA (siRNA control; 5′-UAGCGACUAAACACAUCAAUU-3′) was used as a negative control. After 48 hours of transfection, the transfection solution was removed and the cells were rinsed with PBS and treated with C60(OH)24 nanoparticles in the presence or absence of H2O2. Cell samples were collected for cell viability.
Statistical analysis
Results are presented as the means ± standard deviation of the triplicate experiments. Comparisons between groups were evaluated by two-sided Student’s t-test or one-way analysis of variance. A difference was considered significant at P<0.05.
Results
Characterization of C60(OH)24 nanoparticles
Prior to the in vitro study of the molecular mechanism of antioxidant affects, characterization of the C60(OH)24 nanoparticles was performed using TEM and DLS methods. The C60(OH)24 nanoparticles were found to be easily dissolved and aggregated either in PBS buffer (pH 7.0) or in culture medium. The images obtained with TEM revealed that the diameter of C60(OH)24 nanoparticles aggregated in PBS buffer (Figure 1A) was smaller than that in culture medium supplemented with 10% FBS (Figure 1B). The size distribution was further investigated using a DLS method (Figure 1C and D), showing that the average diameter distributed was about 96 nm in PBS buffer and 142 nm in culture medium, respectively, which approximately matched the average size obtained by TEM. The precipitation of C60(OH)24 nanoparticles was not observed in the culture medium during the testing periods, and the clear aqueous solution remained stable for at least 1 week at room temperature (Figure 1E). The C60(OH)24 nanoparticles exhibited a lower negative zeta potential in PBS buffer (−35 mV) than in cell culture medium (−27 mV), suggesting that serum protein adsorption may affect the surface charge of nanoparticles.
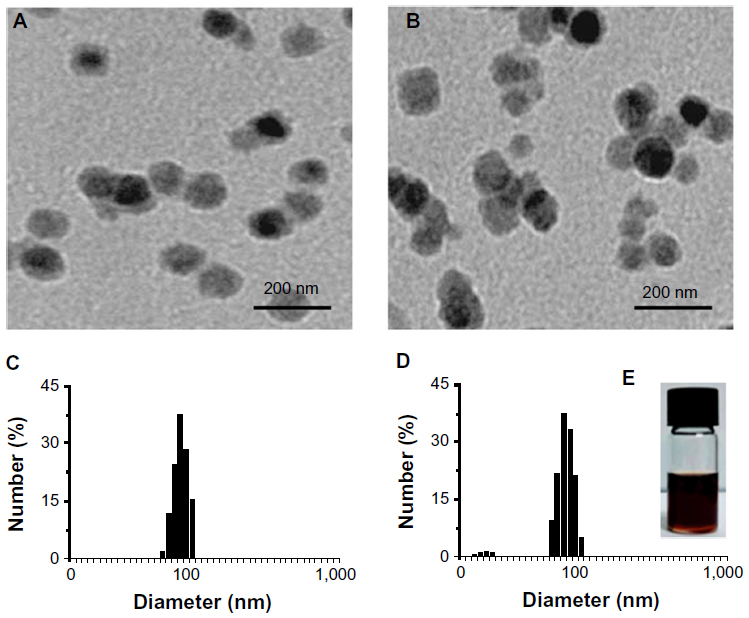 | Figure 1 Characterization of C60(OH)24 nanoparticles. Representative transmission electron microscopy images of C60(OH)24 aggregation in (A) phosphate buffered saline (pH 7.0) and (B) culture medium supplemented with 10% fetal bovine serum. The concentration of C60(OH)24 was 100 μM. (C) The size distribution of C60(OH)24 nanoparticles in (C) phosphate buffered saline and (D) culture medium by dynamic light scattering. (E) Photograph of C60(OH)24 nanoparticles (100 μM) dispersed in culture medium for 1 week at room temperature. |
C60(OH)24 induced HO-1 expression in A549 cells
To initially screen the sensitivity of A549 cells to C60(OH)24, cultures were incubated with increasing doses of C60(OH)24 (10 μM, 25 μM, 50 μM, 100 μM, and 200 μM) for 48 hours and 72 hours, and the cell viability was determined by MTT and LDH assays. As shown in Figure 2A and B, treatment of cells with C60(OH)24 did not affect the survival of A549 cells within the tested concentration range, even as high as 200 μM, for up to 72 hours. Therefore, a concentration of less than 200 μM was considered cell-friendly and biocompatible under normal physiological conditions in principle. To compare the effects of C60(OH)24 in previous in vitro studies,14,25,26 the cytotoxic effects toward A549 cells were further evaluated using 100 μM of C60(OH)24 by TUNEL and DCFH-DA assays, and results clearly showed that C60(OH)24 did not cause significant apoptotic cell death and ROS production for up to 72 hours (Figure 2C). To explore the potential ability of C60(OH)24 to induce phase II antioxidant enzymes, we investigated the possibility that C60(OH)24 nanoparticles might alter the expression of the antioxidant enzyme HO-1, an important component of the cellular defence against oxidative stress. A549 cells were treated with C60(OH)24 nanoparticles at 10 μM, 50 μM, and 100 μM for up to 24 hours, and the results obtained from Western blot analysis demonstrated that treatment with C60(OH)24 nanoparticles induced the protein expression of HO-1 in a concentration- and time-dependent manner (Figure 3A–D). The protein expression of HO-1 was elevated from 6 hours after treatment with 100 μM C60(OH)24 nanoparticles and kept to be upregulated until 24 hours. In accordance with Western blot analysis of HO-1 expression, the mRNA expression of HO-1, γ-GCS, and NQO-1 after treatment of C60(OH)24 exhibited a similar time-dependent manner (Figures 3E and 4F).
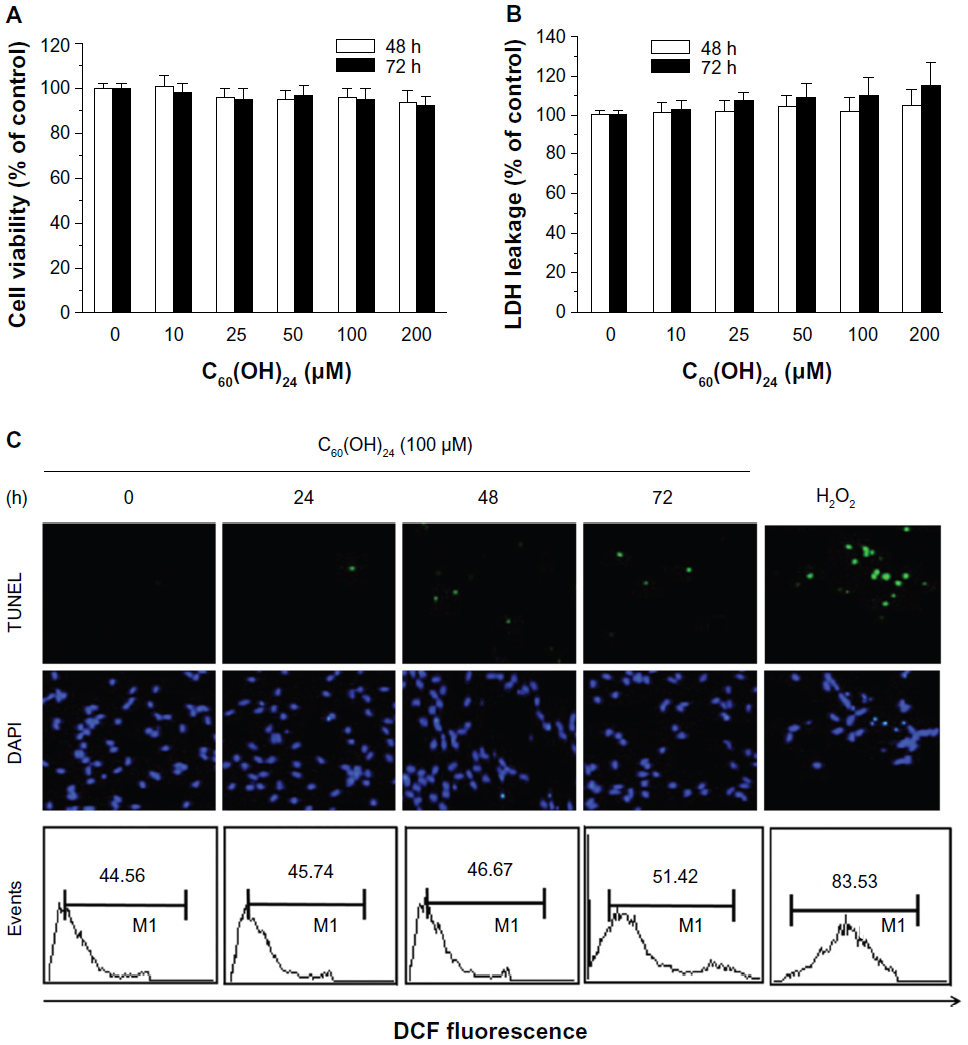 | Figure 2 A549 cells were incubated with increasing doses of C60(OH)24 (10–200 μM) for 48 hours and 72 hours, respectively, and the cell viability was determined by (A) 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide and (B) lactate dehydrogenase assays. Data are presented as the mean ± standard deviation of triplicate independent experiments. (C) A549 cells were treated with 100 μM of C60(OH)24 for indicated times, then apoptotic cell death and intracellular reactive oxygen species production were evaluated by TUNEL and DCFH-DA assays. Representative images from three independent experiments are shown. |
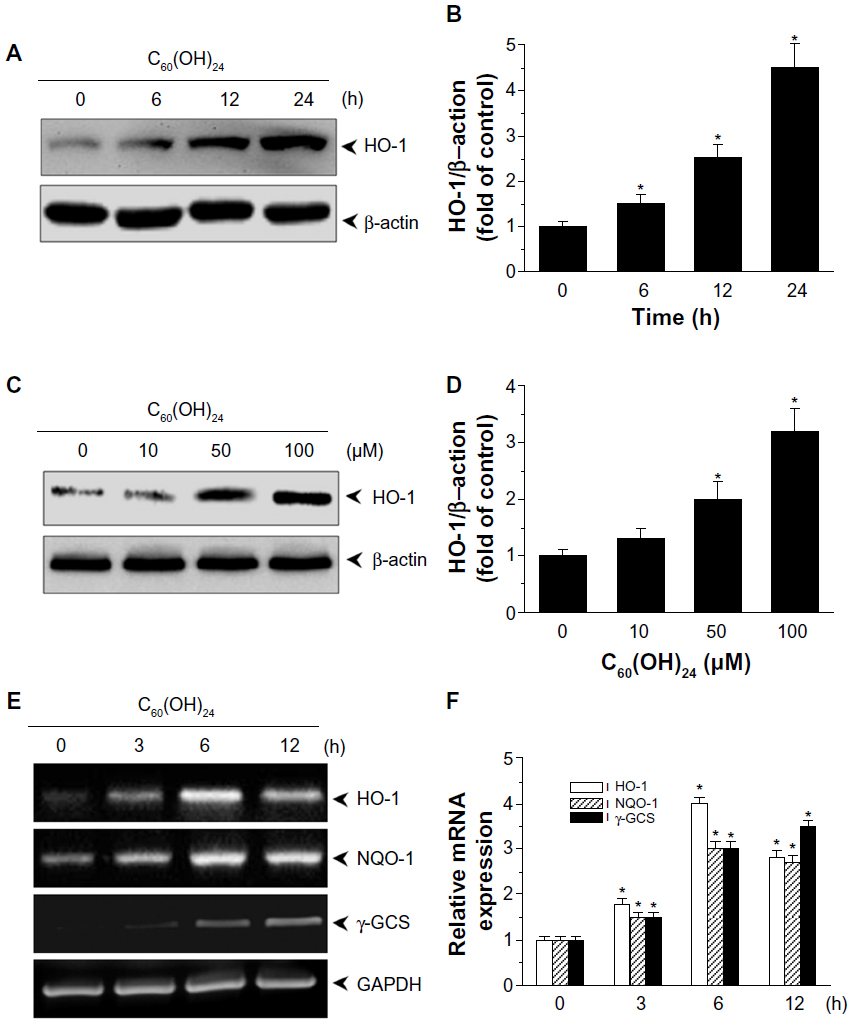 | Figure 3 C60(OH)24 upregulated phase II antioxidant enzymes in A549 cells. A549 cells were treated either with 100 μM C60(OH)24 for (A) indicated time periods or (C) increasing doses of C60(OH)24 (10 μM, 50 μM, and 100 μM) for 24 hours, and protein expression of HO-1 was examined by Western blot analysis. The relative protein expression of HO-1 was performed by densitometric analysis (B and D). Representative data from three independent experiments are shown. *P<0.05 versus control. (E) A549 cells were treated with 100 μM C60(OH)24 for 6 hours, and mRNA levels of HO-1, NQO1, and γ-GCSc were analyzed by reverse transcription-polymerase chain reaction. The relative mRNA expression of HO-1, NQO1, and γ-GCSc was performed by densitometric analysis (F). Representative data from three independent experiments are shown. *P<0.05 versus control. |
C60(OH)24 upregulated cellular antioxidant defence capacity via activation of Nrf2
To gain further insights into the molecular mechanisms underlying the induction of phase II enzymes by C60(OH)24 in A549 cells, the possible involvement of transcription factor Nrf2 was examined as an upstream regulator of the cellular antioxidant enzymes. We first attempted to examine the nuclear accumulation of Nrf2 protein in the C60(OH)24-stimulated A549 cells. The results obtained from Western blot analysis showed that treatment with 100 μM C60(OH)24 for 3 hours resulted in significant nuclear Nrf2 accumulation, accompanied with a decrease of cytosolic Nrf2, in a time-dependent manner (Figure 4A–C). The nuclear translocation of Nrf2 from cytosol was confirmed by immunolocalization of anti-Nrf2 antibody using confocal microscopy (Figure 4D). To elucidate the role of Nrf2–ARE binding in the transcriptional activation of the HO-1 gene, electrophoretic mobility shift assay was further performed using the oligonucleotides that harbor the Nrf2-specific ARE sequence. Treatment of A549 cells with C60(OH)24 resulted in an increased Nrf2 DNA-binding activity, with significant effect occurring at 2 hours post-treatment with C60(OH)24 (Figure 4E). Since Keap1 modification by ROS and electrophiles could account for Nrf2 activation,27 we determined whether C60(OH)24 could activate Nrf2 through the generation of ROS. The intracellular ROS level was increased 1 hour after treatment with C60(OH)24, which remained elevated at 3 hours and then gradually decreased to the basal level at 6 hours. Pretreatment was for 1 hour with NAC at 2.5 mM, which eliminated Nrf2 induction by C60(OH)24 at 6 hours after treatment (Figure S2), suggesting that transient ROS production is involved in Nrf2 activation by C60(OH)24.
Activation of signal transduction by C60(OH)24
To elucidate the plausible signal transduction pathways involved in the C60(OH)24 nanoparticles-induced Nrf2 activation and HO-1 expression, we examined the phosphorylation of several upstream kinases. Treatment with C60(OH)24 resulted in an increased level of p38 MAPK and ERK1/2 phosphorylation at 15 minutes after treatment, which gradually increased to 2 hours (Figure 5A and B). The level of phosphorylated JNK was also seen at 60 minutes after C60(OH)24 treatment (Figure 5A and B). In order to determine which signal transduction pathways could contribute to the C60(OH)24-mediated Nrf2 nuclear translocation and HO-1 expression, we pretreated cells with their specific inhibitors of cellular kinases prior to C60(OH)24 treatment. As a result, pretreatment for 1 hour with a p38 inhibitor, SB203580 (10 μM), attenuated C60(OH)24-mediated Nrf2 nuclear translocation and HO-1 induction, whereas a JNK-specific inhibitor, SP600125, and ERK1/2 inhibitors, PD98059 and U0126, showed little or no effect on Nrf2 nuclear translocation and HO-1 induction (Figure 5C and D). It is worth noting that both Nrf2 nuclear translocation and HO-1 induction were not affected by pretreatment with the inhibitors of cellular kinases alone (Figure S1). These results indicated that the p38 MAPK signaling pathway is involved in C60(OH)24-stimulated Nrf2/HO-1 upregulation.
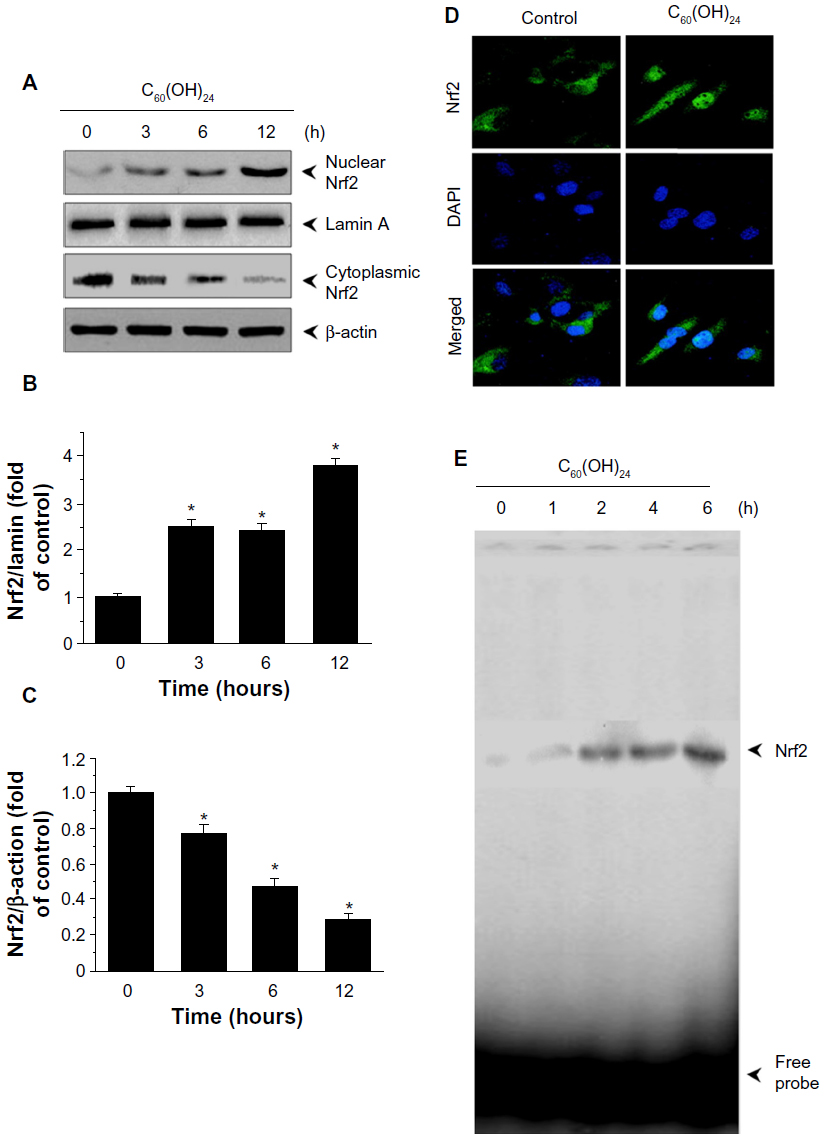 | Figure 4 Effect of C60(OH)24 on Nrf2 activation in A549 cells. Cells were treated with 100 μM C60(OH)24 for indicated time periods, and the nuclear and cytoplasmic levels of Nrf2 were examined by Western blot analysis (A). The relative protein expression of Nrf2 was performed by densitometric analysis (B and C). Representative data from three independent experiments are shown. *P<0.05 versus control. (D) Immunolocalization of Nrf2 using confocal microscopy after 6 hours of treatment of A549 cells with 100 μM C60(OH)24. (E) A549 cells were treated with 100 μM C60(OH)24 for indicated time periods, and electrophoretic mobility shift assay was performed to analyze DNA-binding activity of Nrf2. Representative images from three independent experiments are shown. |
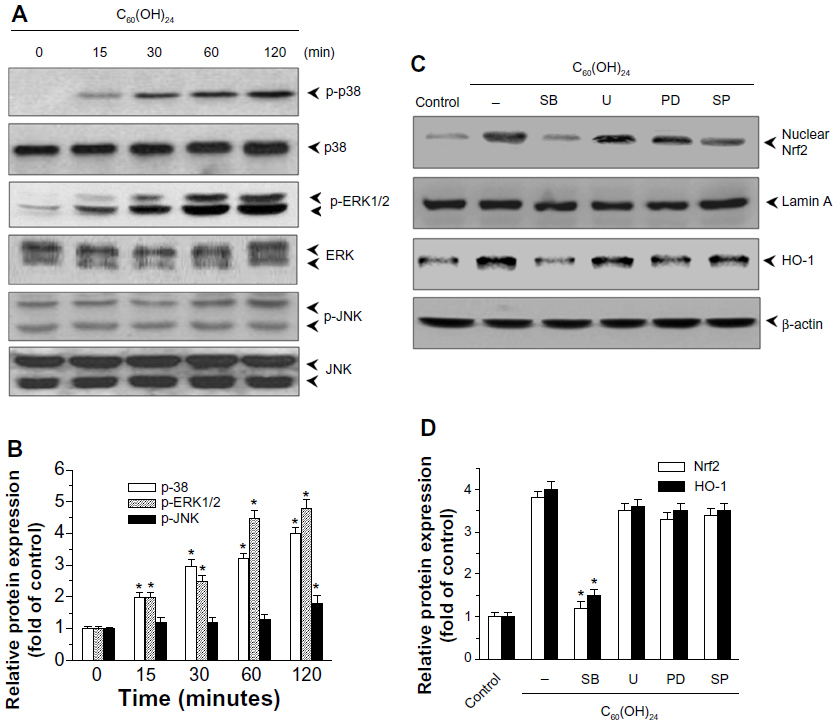 | Figure 5 Effects of C60(OH)24 on activation of MAPK pathway in A549 cells. (A) A549 cells were treated with 100 μM C60(OH)24 for indicated time periods, and cell lysates were prepared and subjected to Western blot analysis for phosphorylated and total p38, ERK1/2, and JNK protein expression, and further analyzed by densitometric analysis (B). A549 cells were pretreated for 1 hour with SB203580 (10 μM), SP600125 (10 μM), U0126 (5 μM), or PD98059 (10 μM), and then treated for 12 hours with 100 μM C60(OH)24. Cell lysates were prepared and subjected to Western blot analysis for nuclear Nrf2 and HO-1 protein expression. The relative protein expression of Nrf2 and HO-1 was performed by densitometric analysis. A representative blot from three independent experiments is shown. *P<0.05 versus control. |
C60(OH)24 protected A549 cells from H2O2-induced apoptosis
As shown in Figure 6A, pretreatment of A549 cells with C60(OH)24 for 24 hours, prior to addition of H2O2, protected against the H2O2-induced cell death in a concentration-dependent manner as determined by the MTT assay, which was confirmed by examination of cell morphology using inverted phase-contrast microscopy (Figure 6B). The C60(OH)24 nanoparticles were also evaluated for their ability to protect against H2O2-induced apoptosis by performing PI staining followed by flow cytometry; apoptotic cells were identified by their sub-G1 DNA content. As shown in Figure 6C, addition of H2O2 to cell medium caused a significant increase in the sub-G1 population; however, pretreatment of cells with 100 μM C60(OH)24 nanoparticles resulted in approximately 80% of restoration of the sub-G1 population. The antiapoptotic effects by C60(OH)24 were further confirmed by Western blot analysis of the cleaved caspase-3 and PARP (Figure 6D). Of note, C60(OH)24 alone at the tested doses (50 μM and 100 μM) was found to be nontoxic to A549 cells. These results indicated that C60(OH)24 exerted the protective effect on A549 cells mainly by inhibiting H2O2-mediated apoptotic cell death.
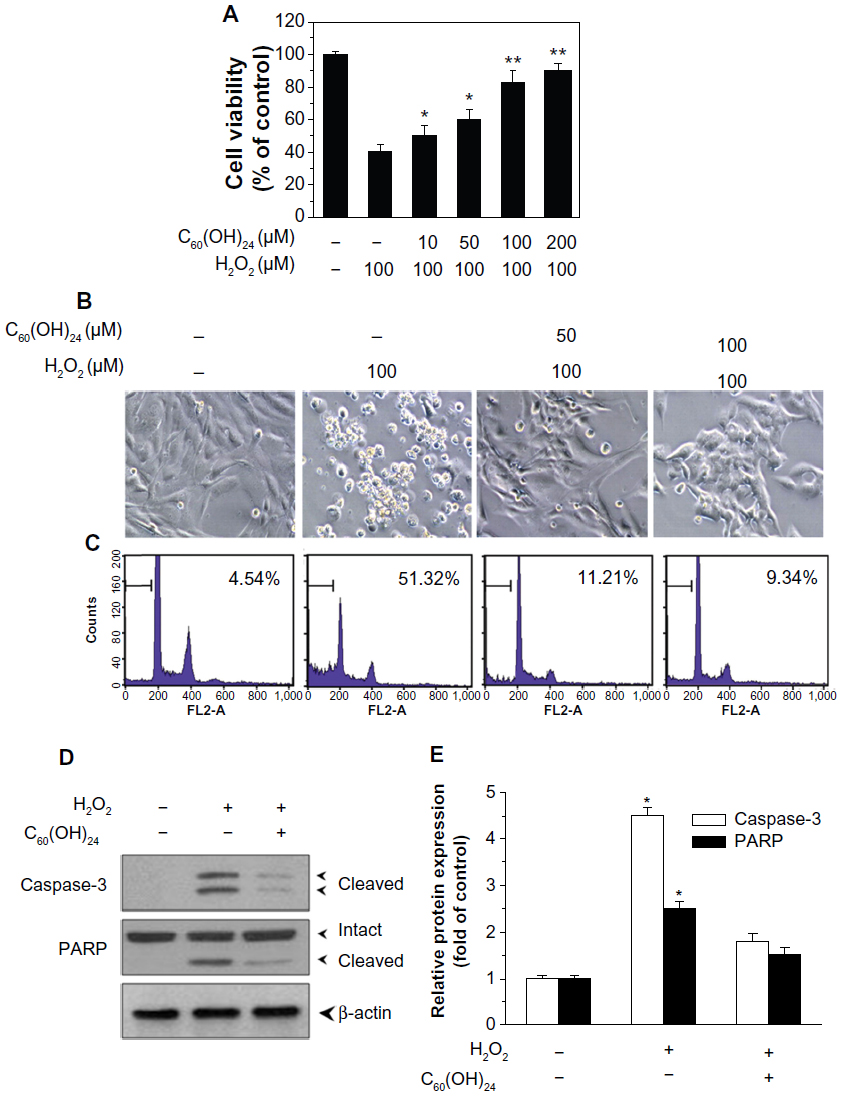 | Figure 6 Cytoprotective effects of C60(OH)24 in A549 cells. (A) A549 cells were pretreated for 24 hours with or without increasing doses of C60(OH)24, and then treated for 20 hours with 100 μM H2O2. The cell viability was measured by the MTT assay. Data represent the mean ± standard deviation of results in three independent experiments. *P<0.05 compared with untreated control **P<0.01 compared with untreated control. (B) Cell morphologic phenotypes of A549 cells were examined using a phase-contrast microscope. (C) The apoptotic cell death was analyzed by PI staining with flow cytometry (D and E) and Western blot analysis of the expression of cleaved caspase-3 and PARP. Representative images from three independent experiments are shown. |
The protective effect of C60(OH)24 involves the Nrf2 antioxidant pathway
In order to provide direct evidence for the involvement of Nrf2 activation and HO-1 induction in C60(OH)24-mediated cytoprotection, we transfected A549 cells with either Nrf2-siRNA or siRNA control for 48 hours, followed by treatment with 100 μM C60(OH)24 for an additional 24 hours. As shown in Figure 7A, cells did not show any remarkable morphological change at 48 hours postinfection. The efficiency of the Nrf2 siRNA in knocking down Nrf2 was verified by Western blot analysis. As shown in Figure 7B and C, the Nrf2–siRNA treatment significantly decreased the levels of Nrf2 in nuclear extracts from cells treated with C60(OH)24. After knockdown of Nrf2 expression, the induction of HO-1 protein expression by C60(OH)24 was also apparently abolished (Figure 7B and C). We subsequently exposed Nrf2–siRNA-transfected A549 cells to C60(OH)24 and then challenged them with 100 μM H2O2. As expected, C60(OH)24 protected siRNA control-treated A549 cells against H2O2-induced cell death, but was significantly less effective in protecting Nrf2–siRNA-transfected A549 cells from cell death induced by H2O2 (Figure 7D). Taken together, these results suggested that C60(OH)24 protects against H2O2-induced cell death partly through Nrf2 activation.
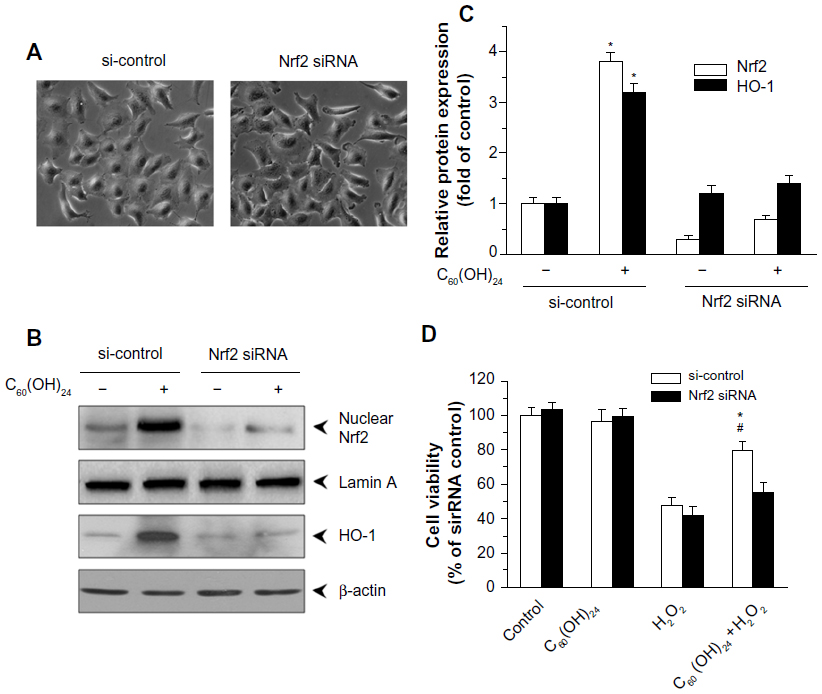 | Figure 7 Nrf2 activation contributed to C60(OH)24-mediated cytoprotective effects. A549 cells were transiently transfected with control or Nrf2 siRNA for 48 hours, and then cell morphology was examined using phase-contrast microscopy (A). After transfection, the cells were treated with 100 μM C60(OH)24 for an additional 6 hours. Nuclear extracts were analyzed for Nrf2 levels by Western blot and densitometric analysis (B and C). (B and C) Transient transfection of A549 cells with Nrf2 siRNA inhibited the HO-1 protein expression. The levels of HO-1 protein were determined by Western blot analysis and densitometric analysis in the control or Nrf2 siRNA cells following C60(OH)24 treatment for 12 hours. (D) A549 cells were transfected with control or Nrf2 siRNA for 48 hours, then subjected to 100 μM C60(OH)24 for 24 hours, and subsequently exposed to 100 μM H2O2 for an additional 20 hours. Cell survival was assessed by the MTT assay, and calculated as a ratio to siRNA control without treatments. Data represent the mean ± standard deviation of results in three independent experiments. *P<0.05 compared with si-control + H2O2 group; #P<0.05 compared with Nrf2 siRNA + H2O2 + C60(OH)24 group. |
Discussion
Recently, a variety of functionalized carbon-based nanomaterials such as carbon nanotubes, fullerenes, graphene, and/or their derivatives have been studied for the treatment of many diseases, including inflammation, cancer, arthritis, neurodegenerative diseases, and traumatic brain injury.28–34 Researchers therefore envisage a major breakthrough in disease treatment by using an emerging class of particle-based pharmaceutical, such as nanoparticles.33,34 C60(OH)24 belongs to a group of antioxidant nanoparticles and exhibits strong antioxidative activity in various chemical and biological systems. Although several previous studies have shown that activation of the Nrf2/ARE pathway can protect cells against oxidative stress-induced cell death in vitro and in vivo, it remains unclear whether C60(OH)24 could protect cells against oxidative stress-induced cell death by activating the Nrf2/ARE pathway. In the present study, we demonstrated that C60(OH)24 attenuates oxidative stress-induced cell death at least partially by activating Nrf2-mediated cellular antioxidant defence capacity through the p38 MAPK signaling pathway.
Induction of endogenous antioxidant enzymes provides a major mechanism by which cells combat the toxicities of ROS. The transcription factor Nrf2 is an essential mediator on the expression of antioxidant enzymes and stress-inducible proteins, and activation of Nrf2 in cells provides an indirect way to enhance antioxidant capacity,35 thereby preventing cell dysfunction from free radical production. Among the target genes whose expression is upregulated by Nrf2, HO-1 is the key enzyme that exerts a protective or adaptive effect under conditions of oxidative stress by converting the pro-oxidant heme to biologically active antioxidant by-products such as carbon monoxide and bilirubin.36 C60(OH)24 has been well known for its possible antioxidant role and protective effects against oxidant damage. In the present study, we tested the ability of C60(OH)24 to activate Nrf2 signaling pathways in lung epithelial A549 cells. Our data showed that C60(OH)24 treatment resulted in increased expression of HO-1 mRNA and protein, which was preceded by nuclear translocation of Nrf2. Furthermore, C60(OH)24 induced expression of other phase II enzymes, including γ-GCS and NQO-1, whose genes are known to be upregulated by Nrf2 activation.37 γ-GCSc associates with γ-GCS modifier subunit (γ-GCSm) to form γ-GCS, and NQO-1 catalyzes the two-electron reduction of quinone compounds.38 These results collectively demonstrated that C60(OH)24 upregulates expression of phase II enzymes as a result of Nrf2/ARE activation. However, the molecular mechanisms of Nrf2 activation by C60(OH)24 still need to be elucidated. Previous studies suggested that C60 derivatives could penetrate cell membrane and gain access to various intracellular compartments, including organelles, proteins, and DNA, which might result in regulation of a series of intracellular stress-sensitive kinases.14 MAPK is one of the most common signaling pathways that serve to coordinate the cellular response to a variety of extracellular stimuli. The three major MAP kinase cascades are represented by p38 MAPK, ERK1/2, and JNK. All these kinases are likely to be involved in the upstream pathways leading to Nrf2 activation.39 Therefore, we investigated the upstream signaling mechanisms responsible for C60(OH)24-mediated Nrf2/HO-1 induction. Our results demonstrated that C60(OH)24 increased phosphorylation of p38 MAP kinase, and pretreatment with a p38 inhibitor, SB203580, diminished nuclear Nrf2 translocation and HO-1 induction caused by C60(OH)24. Since it has been reported that Nrf2 phosphorylation by protein kinases facilitates its nuclear translocation,40,41 these results suggest that Nrf2 phosphorylation by p38 MAPK may participate in Nrf2/ARE activation by C60(OH)24. Oxidative modification of cysteine sulfhydryl groups of Keap1 by ROS has been shown to change its conformation, resulting in Nrf2 release.27 Our results showed that C60(OH)24 treatment transiently increased the intracellular ROS level, whereas pretreatment with NAC markedly abolished C60(OH)24-induced Nrf2 activation and HO-1 expression (Figure S2). It is therefore likely that ROS transiently generated by C60(OH)24 modify sulfhydryl groups of Keap1, thereby activating Nrf2/ARE signaling.
Since the transcription factor Nrf2 is a master regulator for the expression of several antioxidant genes, there can be a hypothesis that the mechanism of the protective effect that is observed with C60(OH)24 treatment is associated with a consequence of the activation of Nrf2. In the present study, pretreatment with C60(OH)24 attenuated H2O2-induced apoptotic cell death in A549 cells in a dose-dependent manner. Furthermore, siRNA knockdown of Nrf2 diminished C60(OH)24-mediated cytoprotective effects, providing direct evidence for the involvement of HO-1 induction and Nrf2/ARE activation in C60(OH)24-mediated cytoprotection. Our results are similar to previous studies demonstrating that Nrf2 plays a critical role in protecting cells against oxidative stress.42,43 Previously, accumulating evidence has suggested that C60(OH)24 is efficient in protecting various cell types from ROS-mediated damage in vitro and in vivo. In an animal irradiated model that is associated with oxidative stress, pretreatment with C60(OH)24 showed radioprotective effects by scavenging ROS and increasing the antioxidant enzyme activities.19,20 It has also been reported that C60(OH)24 shows hepatoprotective effects in doxorubicin-treated rats by acting as an antioxidant.44,45 Hence, it is possible to propose that C60(OH)24 pretreatment prevented deleterious effects of ROS by direct ROS scavenging or increasing the antioxidant enzyme activities. Previous studies have shown that the radical-scavenging abilities of C60(OH)24 have been attributed to the molecular properties of fullerenols, including large electron affinity and formation of electron-deficient areas on the C60,13 and these properties of C60(OH)24 may lead to direct ROS scavenging similar to that catalyzed by superoxide dismutase.15,46 However, in the present study, our results have clearly indicated that C60(OH)24 protects against H2O2-induced cell death through activation of Nrf2/ARE signaling and induction of phase II antioxidant enzymes such as HO-1, γ-GCS, and NQO-1, thus providing an insight into mechanisms by which C60 derivatives exert protective effects against cell death induced by oxidative stress.
Conclusion
Taken together, the results obtained from this study imply that enhancement of the cellular defence activities by induction of phase II detoxifying enzymes represents one of the important antioxidant mechanisms of C60(OH)24. C60(OH)24 potentiates cellular defence capacity against oxidative stress via Nrf2-regulated antioxidant or phase II detoxifying enzymes, and it can also block oxidative stress-mediated cell damage and dysfunctions, as schematically represented in Figure 8. In addition to induction of phase II antioxidant enzymes, Nrf2 has been shown to influence directly or indirectly expression of genes that are implicated in cell growth, apoptosis, inflammation, and cell adhesion. Therefore, further studies using in vivo models are warranted to identify other molecules in relation to activation of the Nrf2 pathway and to clarify potential crosstalk with upstream and downstream signaling molecules.
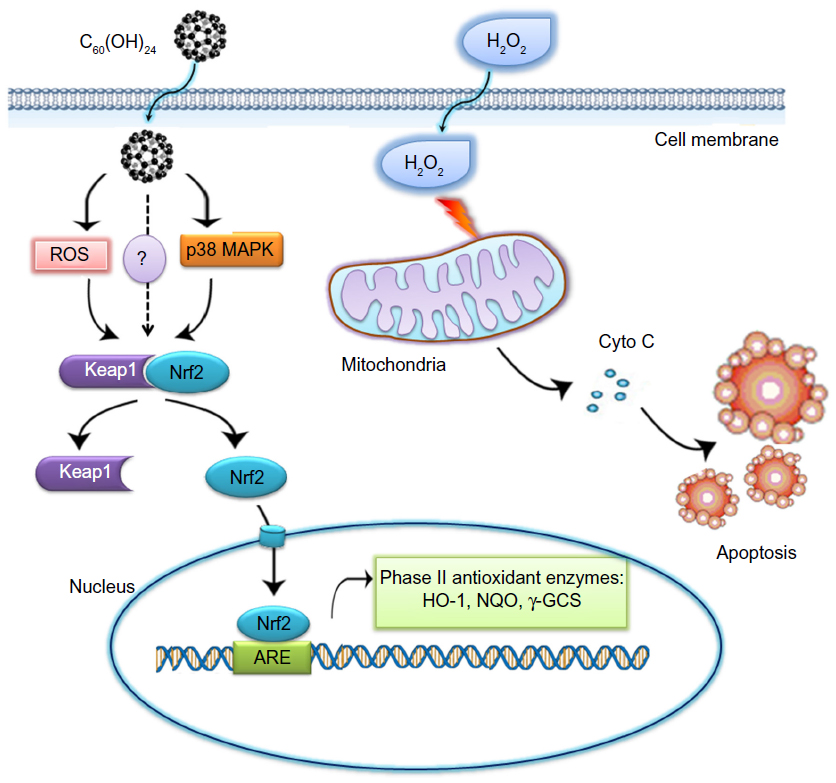 | Figure 8 A hypothetical mechanism of C60(OH)24-mediated cell protection from H2O2. Nrf2 is a transcription factor that regulates expression of many detoxification or antioxidant enzymes. It is plausible that C60(OH)24 transiently increases the intracellular level of ROS and/or activates p38 MAPK signaling pathway, which may possibly lead to facilitating the dissociation of Nrf2 from Keap. The resultant Nrf2/ARE activation induced phase II detoxification or antioxidant enzyme, thereby potentiating cellular defence capacity against H2O2-induced cell death. |
Acknowledgments
This study was financially supported by grants from the National Natural Science Foundation of China (31271071, 31371012, 30901175, and 81171448) and the National Basic Research Program of China (2013CB933703).
Disclosure
The authors report no conflicts of interest in this work.
References
Seo AY, Joseph AM, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci. 2010;123(Pt 15):2533–2542. | |
Yasuda K, Hartman PS, Ishii T, et al. Interrelationships between mitochondrial fusion, energy metabolism and oxidative stress during development in caenorhabditis elegans. Biochem Biophys Res Commun. 2011;404(3):751–755. | |
McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal. 2006;8(9–10):1775–1789. | |
Victor VM, Apostolova N, Herance R, Hernandez-Mijares A, Rocha M. Oxidative stress and mitochondrial dysfunction in atherosclerosis: mitochondria-targeted antioxidants as potential therapy. Curr Med Chem. 2009;16(35):4654–4667. | |
Pérez-Matute P, Zulet MA, Martínez JA. Reactive species and diabetes: counteracting oxidative stress to improve health. Curr Opin Pharmacol. 2009;9(6):771–779. | |
Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol. 2010;80(12):1895–1903. | |
Wu KC, McDonald PR, Liu JJ, Chaguturu R, Klaassen CD. Implementation of a high-throughput screen for identifying small molecules to activate the Keap1-Nrf2-ARE pathway. PLoS One. 2012;7(10):e44686. | |
Hur W, Gray NS. Small molecule modulators of antioxidant response pathway. Curr Opin Chem Biol. 2011;15(1):162–173. | |
Kim CK, Kim T, Choi IY, et al. Ceria nanoparticles that can protect against ischemic stroke. Angew Chem Int Ed. 2012;51(44):11039–11043. | |
Bakry R, Vallant RM, Najam-ul-Haq M, et al. Medicinal applications of fullerenes. Int J Nanomedicine. 2007;2(4):639–649. | |
Cheng LC, Jiang X, Wang J, Chen C, Liu RS. Nano-bio effects: interaction of nanomaterials with cells. Nanoscale. 2013;5(9):3547–3569. | |
Qiao R, Roberts AP, Mount AS, Klaine SJ, Ke PC. Translocation of C60 and its derivatives across a lipid bilayer. Nano Lett. 2007;7(3):614–619. | |
Markovic Z, Trajkovic V. Biomedical potential of the reactive oxygen species generation and quenching by fullerenes (C60). Biomaterials. 2008;29(26):3561–3573. | |
Lao F, Chen L, Li W, et al. Fullerene nanoparticles selectively enter oxidation-damaged cerebral microvessel endothelial cells and inhibit JNK-related apoptosis. ACS Nano. 2009;3(11):3358–3368. | |
Ali SS, Hardt JI, Dugan LL. SOD activity of carboxyfullerenes predicts their neuroprotective efficacy: a structure-activity study. Nanomedicine. 2008;4(4):283–294. | |
Lao F, Li W, Han D, et al. Fullerene derivatives protect endothelial cells against NO-induced damage. Nanotechnology. 2009;20(22):225103. | |
Misirkic MS, Todorovic-Markovic BM, Vucicevic LM, et al. The protection of cells from nitric oxide-mediated apoptotic death by mechanochemically synthesized fullerene (C(60)) nanoparticles. Biomaterials. 2009;30(12):2319–2328. | |
Roursgaard M, Poulsen SS, Kepley CL, Hammer M, Nielsen GD, Larsen ST. Polyhydroxylated C60 fullerene (fullerenol) attenuates neutrophilic lung inflammation in mice. Basic Clin Pharmacol Toxicol. 2008;103(4):386–388. | |
Stankov K, Borisev I, Kojic V, Rutonjski L, Bogdanovic G, Djordjevic A. Modification of antioxidative and antiapoptotic genes expression in irradiated K562 cells upon fullerenol C60(OH)24 nanoparticle treatment. J Nanosci Nanotechnol. 2013;13(1):105–113. | |
Bogdanović V, Stankov K, Icević I, et al. Fullerenol C60(OH)24 effects on antioxidative enzymes activity in irradiated human erythroleukemia cell line. J Radiat Res. 2008;49(3):321–327. | |
Zha YY, Yang B, Tang ML, et al. Concentration-dependent effects of fullerenol on cultured hippocampal neuron viability. Int J Nanomedicine. 2012;7:3099–3109. | |
Hu Z, Huang Y, Guan W, Zhang J, Wang F, Zhao L. The protective activities of water-soluble C(60) derivatives against nitric oxide-induced cytotoxicity in rat pheochromocytoma cells. Biomaterials. 2010;31(34):8872–8881. | |
Jiang Y, Zhang H, Wang Y, et al. Modulation of apoptotic pathways of macrophages by surface-functionalized multi-walled carbon nanotubes. PLoS One. 2013;8(6):e65756. | |
Yeligar SM, Machida K, Kalra VK. Ethanol-induced HO-1 and NQO1 are differentially regulated by HIF-1alpha and Nrf2 to attenuate inflammatory cytokine expression. J Biol Chem. 2010;285(46):35359–35373. | |
Cai X, Jia H, Liu Z, et al. Polyhydroxylated fullerene derivative C(60)(OH)(24) prevents mitochondrial dysfunction and oxidative damage in an MPP(+)-induced cellular model of Parkinson’s disease. J Neurosci Res. 2008;86(16):3622–3634. | |
Saitoh Y, Miyanishi A, Mizuno H, et al. Super-highly hydroxylated fullerene derivative protects human keratinocytes from UV-induced cell injuries together with the decreases in intracellular ROS generation and DNA damages. J Photochem Photobiol B. 2011;102(1):69–76. | |
Hamada N, Tanaka A, Fujita Y, et al. Involvement of heme oxygenase-1 induction via Nrf2/ARE activation in protection against H2O2–induced PC12 cell death by a metabolite of sesamin contained in sesame seeds. Bioorg Med Chem. 2011;19(6):1959–1965. | |
Hampel S, Kunze D, Haase D, et al. Carbon nanotubes filled with a chemotherapeutic agent: a nanocarrier mediates inhibition of tumor cell growth. Nanomedicine (Lond). 2008;3(2):175–182. | |
Lv M, Zhang Y, Liang L, et al. Effect of graphene oxide on undifferentiated and retinoic acid-differentiated SH-SY5Y cells line. Nanoscale. 2012;4(13):3861–3866. | |
Kang SG, Zhou G, Yang P, et al. Molecular mechanism of pancreatic tumor metastasis inhibition by Gd@C82(OH)22 and its implication for de novo design of nanomedicine. Proc Nat Acad Sci U S A. 2012;109(38):15431–15436. | |
Yudoh K, Karasawa R, Masuko K, Kato T. Water-soluble fullerene (C60) inhibits the development of arthritis in the rat model of arthritis. Int J Nanomedicine. 2009;4:217–225. | |
Yin JJ, Lao F, Fu PP, et al. The scavenging of reactive oxygen species and the potential for cell protection by functionalized fullerene materials. Biomaterials. 2009;30(4):611–621. | |
Amiri A, Memarpoor-Yazdi M, Shanbedi M, Eshghi H. Influence of different amino acid groups on the free radical scavenging capability of multi walled carbon nanotubes. J Biomed Mater Res A. 2012;101(8):2219–2228. | |
Bitner BR, Marcano DC, Berlin JM, et al. Antioxidant carbon particles improve cerebrovascular dysfunction following traumatic brain injury. ACS Nano. 2012;6(9):8007–8014. | |
Zhao CR, Gao ZH, Qu XJ. Nrf2–ARE signaling pathway and natural products for cancer chemoprevention. Cancer Epidemiol. 2010;34(5):523–533. | |
Dolinay T, Choi AM, Ryter SW. Heme oxygenase-1/CO as protective mediators in cigarette smoke-induced lung cell injury and chronic obstructive pulmonary disease. Curr Pharm Biotechnol. 2012;13(6):769–776. | |
Lin X, Yang H, Zhou L, Guo Z. Nrf2-dependent induction of NQO1 in mouse aortic endothelial cells overexpressing catalase. Free Radic Biol Med. 2011;51(1):97–106. | |
Sumi D, Manji A, Shinkai Y, Toyama T, Kumagai Y. Activation of the Nrf2 pathway, but decreased gamma-glutamylcysteine synthetase heavy subunit chain levels and caspase-3-dependent apoptosis during exposure of primary mouse hepatocytes to diphenylarsinic acid. Toxicol Appl Pharmacol. 2007;223(3):218–224. | |
Zhang DD. Mechanistic studies of the Nrf2–Keap1 signaling pathway. Drug Metab Rev. 2006;38(4):769–789. | |
Granado-Serrano AB, Martín MA, Bravo L, Goya L, Ramos S. Quercetin modulates Nrf2 and glutathione-related defenses in HepG2 cells: Involvement of p38. Chem Biol Interact. 2012;195(2):154–164. | |
Rodríguez-Ramiro I, Ramos S, Bravo L, Goya L, Martín Má. Procyanidin B2 induces Nrf2 translocation and glutathione S-transferase P1 expression via ERKs and p38–MAPK pathways and protect human colonic cells against oxidative stress. Eur J Nutr. 2012;51(7):881–892. | |
Huang TC, Chung YL, Wu ML, Chuang SM. Cinnamaldehyde enhances Nrf2 nuclear translocation to upregulate phase II detoxifying enzyme expression in HepG2 cells. J Agric Food Chem. 2011;59(9):5164–5171. | |
Minelli A, Conte C, Grottelli S, Bellezza I, Emiliani C, Bolaños JP. Cyclo(His-Pro) up-regulates heme oxygenase 1 via activation of Nrf2–ARE signalling. J Neurochem. 2009;111(4):956–966. | |
Injac R, Perse M, Cerne M, et al. Protective effects of fullerenol C60(OH)24 against doxorubicin-induced cardiotoxicity and hepatotoxicity in rats with colorectal cancer. Biomaterials. 2009;30(6):1184–1196. | |
Injac R, Perse M, Obermajer N, et al. Potential hepatoprotective effects of fullerenol C60(OH)24 in doxorubicin-induced hepatotoxicity in rats with mammary carcinomas. Biomaterials. 2008;29(24–25):3451–3460. | |
Osuna S, Swart M, Solà M. On the mechanism of action of fullerene derivatives in superoxide dismutation. Chemistry. 2010;16(10):3207–3214. |
Supplementary materials
| Figure S1 A549 cells were treated for 1 hour with SB203580 (10 μM), SP600125 (10 μM), U0126 (5 μM), or PD98059 (10 μM) alone. Cell lysates were prepared and subjected to Western blot analysis for phosphorylated and total p38, ERK1/2, and JNK proteins. |
| Figure S2
Effects of C60(OH)24 on the intracellular ROS level in A549 cells. (A) At indicated times after treatment of A549 cells with C60(OH)24 (100 μM), intracellular ROS levels were detected by flow cytometry using a fluorescent probe DCFH-DA. (B) A549 cells were pretreated for 1 hour with or without NAC (2.5 mM) and then treated for 6 hours with C60(OH)24 (100 μM). Cell lysates were prepared and subjected to Western blot analysis for Nrf2. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.