")
Back to Journals » International Journal of Nanomedicine » Volume 12
Poly((D,L)lactic-glycolic)acid–star glucose nanoparticles for glucose transporter and hypoglycemia-mediated tumor targeting
Authors Park JH, Cho HJ, Kim DD
Received 29 July 2017
Accepted for publication 9 September 2017
Published 11 October 2017 Volume 2017:12 Pages 7453—7467
DOI https://doi.org/10.2147/IJN.S147668
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Ju-Hwan Park,1 Hyun-Jong Cho,2 Dae-Duk Kim1
1College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, Seoul, 2College of Pharmacy, Kangwon National University, Chuncheon, Gangwon, Republic of Korea
Abstract: Poly((D,L)lactic-glycolic)acid–star glucose (PLGA-Glc) polymer-based nanoparticles (NPs) were fabricated for tumor-targeted delivery of docetaxel (DCT). NPs with an approximate mean diameter of 241 nm, narrow size distribution, negative zeta potential, and spherical shape were prepared. A sustained drug release pattern from the developed NPs was observed for 13 days. Moreover, drug release from PLGA-Glc NPs at acidic pH (endocytic compartments and tumor regions) was significantly improved compared with that observed at physiological pH (normal tissues and organs). DCT-loaded PLGA-Glc NPs (DCT/PLGA-Glc NPs) exhibited an enhanced antiproliferation efficiency rather than DCT-loaded PLGA NPs (DCT/PLGA NPs) in Hep-2 cells, which can be regarded as glucose transporters (GLUTs)-positive cells, at ≥50 ng/mL DCT concentration range. Under glucose-deprived (hypoglycemic) conditions, the cellular uptake efficiency of the PLGA-Glc NPs was higher in Hep-2 cells compared to that observed in PLGA NPs. Cy5.5-loaded NPs were prepared and injected into a Hep-2 tumor-xenografted mouse model for in vivo near-infrared fluorescence imaging. The PLGA-Glc NPs group exhibited higher fluorescence intensity in the tumor region than the PLGA NPs group. These results imply that the PLGA-Glc NPs have active tumor targeting abilities based on interactions with GLUTs and the hypoglycemic conditions in the tumor region. Therefore, the developed PLGA-Glc NPs may represent a promising tumor-targeted delivery system for anticancer drugs.
Keywords: PLGA-Glc, nanoparticles, glucose transporter, hypoxia, tumor targeting
Introduction
Recently, nanosize particulate systems have been widely used in anticancer drug delivery and cancer imaging. Diverse organic (natural and synthetic) and inorganic materials have been introduced as the matrix of nanovehicles.1–7 Among various organic polymers, poly((D,L)lactic-glycolic)acid (PLGA) is the most popular material because of its biocompatibility and biodegradability and was approved by the US Food and Drug Administration. However, PLGA does not possess a tumor targeting moiety. Therefore, various approaches have been introduced to endow PLGA nanovehicles with tumor targeting abilities.8,9 Among these methods, functional groups or therapeutics (ie, small chemicals, peptides, and proteins) have been conjugated or adsorbed onto the surface of PLGA nanovehicles.10–13 Surface modification of PLGA nanoparticles (NPs) can modulate the in vivo fate of NPs and the antitumor efficacy of drugs in cancer therapy.14 Recently, the synthesis of a glucose-functionalized PLGA (PLGA-Glc) polymer via amide bond formation between the amino group of glucosamine and the carboxylic acid group of PLGA and its application in drug delivery have been reported.15,16 Glycosylation of PLGA NPs can enhance blood compatibility by reducing plasma protein adsorption and increase the cellular uptake of NPs.16
The rapid growth of a tumor results in increased vascular permeability, which is used as a passive targeting strategy by nanocarriers using the so-called “enhanced permeability and retention (EPR) effect”.17 Moreover, this rapid growth results in hypoxic conditions in the tumor region and triggers the activation of several genes. For example, hypoxia-inducible factor 1 (HIF-1) is upregulated in many malignant tumors. HIF-1 increases the expression of glycolytic enzymes and glucose transporters (GLUTs). It is important to note that the elevated expression of energy-independent GLUTs was observed in the cellular membrane of many types of cancers including breast, brain, cervical, colorectal, esophageal, hepatic, laryngeal, ovarian, and renal carcinomas.18–26 Along with the increased expression of GLUTs in cancer cells, neoplastic cells are known to accelerate metabolism and increase ATP requirements. Therefore, cancer cells have high rates of aerobic glycolysis, inducing the accumulation of lactate in cancer cells.27 Due to the increase in lactate secretion, tumor tissue generally exhibits more acidic extracellular pH rather than normal physiological organs and tissues. Acidification of tumor tissue has been widely used for the design of tumor microenvironment-responsive nanocarriers.28–31 Moreover, a low glucose concentration due to an increase in aerobic glycolysis in a hypoxic tumor microenvironment indicates the absence of the highly competitive inhibition of glucose transport via GLUTs. Therefore, NPs whose surface is modified with glucose would preferentially locate in the tumor region because of GLUTs targeting and EPR effect. A low glucose concentration in the tumor region is expected to contribute to improved cellular uptake of these NPs via GLUTs-mediated endocytosis, which would enhance the release of anticancer drugs from the NPs at low pH in cancer cells because of the accumulation of lactate.
PLGA-Glc is a star copolymer where the hydroxyl groups of the D-glucose core are extended by the PLGA domains (Figure S1).32 Although the sustained release and stability of encapsulated proteins in the millicylinder of PLGA-Glc have been reported and a microsphere depot has been marketed for the controlled release of a peptide drug,15 there has been no report on the tumor targeting effect of PLGA-Glc NPs. Herein, we report PLGA-Glc NPs as tumor-targeted drug delivery systems based on a combined strategy using the EPR effect, GLUTs targeting, and hypoglycemic conditions of the tumor microenvironment. The physicochemical properties of the NPs containing docetaxel (DCT), as well as the drug release from NPs, antiproliferation efficacy of DCT-loaded NPs, intracellular uptake of NPs, and in vivo near-infrared fluorescence (NIRF) imaging, were investigated.
Materials and methods
Materials
DCT was purchased from LC Laboratories (Woburn, MA, USA). PLGA (weight average molecular weight [average Mw]: 27 kDa) and PLGA-Glc (average Mw: 38 kDa) were obtained from PolySciTech (Akina Inc., West Lafayette, IN, USA). Poly(vinyl alcohol) (PVA) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) were purchased from Sigma Aldrich (St Louis, MO, USA). Tween 80 was purchased from Tokyo Chemical Industry, Co., Ltd. (Tokyo, Japan). Methanol and acetonitrile were obtained from Fisher Scientific Korea Ltd. (Seoul, Korea), and methylene chloride was acquired from Daejung Chemical (Shiheung, Korea). Dulbecco’s Modified Eagle’s Medium (DMEM) with high glucose (4.5 g/L) or no glucose, fetal bovine serum (FBS), and other cell culture reagents were obtained from Gibco BRL (Grand Island, NY, USA). All the other chemicals and reagents were of analytical grade.
Preparation of DCT-loaded NPs
PLGA NPs and PLGA-Glc NPs were prepared by oil-in-water (o/w) emulsification–solvent evaporation method. PLGA or PLGA-Glc (40 mg) was dissolved with DCT (4 mg) in methylene chloride (1 mL). This organic phase was added into 2% (w/v) PVA solution (20 mL), and homogenized in the ice using a probe-type sonicator (Sonics and Material Inc., Newtown, CT, USA) for 20 minutes. After the sonication, the prepared o/w emulsion was stirred for 1 hour to remove the remaining organic solvent and to make NPs. The stirred NP suspension was centrifuged at 13,200 rpm for 10 minutes. After that, supernatant PVA solution was eliminated and the NPs pellet was redispersed with distilled water (DW) by vortex-mixing for 5 minutes. The prepared NPs suspension was then lyophilized.
Characterization of NPs
The hydrodynamic diameter and zeta potential values of both PLGA NPs and PLGA-Glc NPs were measured using the electrophoretic light scattering method (ELS-Z; Otsuka Electronics, Hirakata, Japan). The morphology of the NPs was observed using a scanning electron microscope (SEM; SUPRA 55VP; Carl Zeiss, Oberkochen, Germany). Prior to observation, the NPs were coated with platinum using a sputter coater (Bal-Tec/SCD005; BalTec Maschinenbau AG, Pfäffikon, Switzerland). To measure the encapsulation efficiency of DCT, an aliquot of the prepared NP suspension was mixed with dimethyl sulfoxide (DMSO) to disrupt the matrix of the NP. After diluting the mixed solution with acetonitrile, the drug concentration was determined by injecting the mixture (20 μL) into a reversed-phase C-18 column (Gemini, 250×4.6 mm, 5 μm; Phenomenex, Torrance, CA, USA) on a high-performance liquid chromatography (HPLC) system, as reported.33 The HPLC system was equipped with a 717 autosampler, a 1525 binary pump, and a 2487 dual absorbance detector (Waters, Milford, MA, USA). A mixture of acetonitrile and DW (65:35, v/v) was used as the mobile phase at a flow rate of 1 mL per minute. The absorbance wavelength was set at 230 nm.
In vitro stability of developed NPs was assessed by measuring their hydrodynamic size in the mixture of FBS and phosphate-buffered saline (PBS) (pH 7.4) (1:1, v/v). The mean diameters of NPs were measured at 0, 3, 6, and 24 hours postincubation.
In vitro release test
In vitro release of DCT from NPs was evaluated using a tube-type dialysis bag (molecular weight cutoff: 12–14 kDa). An aliquot (1 mL) of the DCT-loaded NP suspension containing DCT (0.1 mg) was placed into the dialysis bag, and both ends of the membrane were sealed. The NP-loaded dialysis bag was immersed in 20 mL of PBS (pH 7.4 for DCT/PLGA NPs and pH 5.5, 6.8, and 7.4 for DCT/PLGA-Glc NPs, with 0.5% Tween 80) and shaken in a water bath for 13 days. The temperature of the water bath was set to 37°C, and the shaking speed was set to 50 rpm. At each sampling point, an aliquot (0.2 mL) of the release medium was collected, and an equal volume of fresh medium was added. The DCT concentration in each sample was quantitatively analyzed using an HPLC system.
In vitro cytotoxicity test of blank NPs
The in vitro cytotoxicity of blank NPs and PLGA-Glc NPs without DCT was determined from the cell viability after incubation with various concentrations of NPs. Hep-2 cells (Korea Cell Line Bank, Seoul, Korea) were cultured with DMEM containing 10% FBS and 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C in a 5% CO2 atmosphere. The cells (1×104 cells in 0.1 mL culture media) were seeded in a 96-well plate and incubated for 1 day, and then, the culture medium was removed. Next, blank NPs suspended in a serum-free culture medium (containing glucose) at various concentrations (10–1,000 μg/mL) were applied to the cells. After 24, 48, and 72 hours of incubation, the cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethonyphenol)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)-based CellTiter 96 Aqueous One Solution Cell Proliferation Assay Reagent (Promega Corp., Madison, WI, USA). The NP suspension in each well was removed, and the MTS solution was added. After incubation for 4 hours at 37°C, the absorbance at 490 nm was measured to calculate the cell viability (%).
Antiproliferation assay
The antiproliferation efficacy of DCT-loaded NPs was tested in Hep-2 cells by the MTS assay. Hep-2 cells were seeded onto a 96-well plate at a density of 1×104 cells per well and incubated for 1 day. DCT/PLGA NPs and DCT/PLGA-Glc NPs, including 1, 10, 50, 100, and 500 ng/mL DCT, were applied to the cells and they were incubated for 48 hours in glucose-free cell culture media. Then, cell viability was measured by MTS-based CellTiter 96 Aqueous One Solution Cell Proliferation Assay Reagent. The absorbance at 490 nm of tested sample was compared with that of control (no treatment) group to obtain cell viability.
In vitro cellular uptake study
Flow cytometry analysis was employed to compare the cellular uptake efficiency of each NP formulation, where DiI was encapsulated as a fluorescent dye. Rather than DCT (4 mg), DiI (1 mg) was used to prepare the NPs with PLGA (40 mg). Hep-2 cells (6×105 cells per well) were seeded in a six-well plate with culture medium. After incubation of the cells for 1 day, PLGA NPs or PLGA-Glc NPs were loaded into the cells. The DiI concentration in the NP suspension was fixed at 1 μg/mL. Each NP formulation was suspended in culture media either with or without glucose. After incubation for 4 hours under normoxia conditions, the cells were washed with PBS and detached from the plate. Each cell suspension was centrifuged at 13,200 rpm for 5 minutes, and the cell pellet was resuspended with PBS containing 2% (v/v) FBS. The prepared cell suspensions were analyzed by FACSCalibur fluorescence-activated cell sorter (FACS™) equipped with CELLQuest software (Becton Dickinson Biosciences, San Jose, CA, USA).
Cellular distribution of the developed NPs was also observed by confocal laser scanning microscopy (CLSM). Hep-2 cells (at a density of 1×105 cells per well) were seeded in a four-well culture slide (1.7 cm2 of surface area; BD Falcon, Bedford, MA, USA) and incubated for 1 day. Similar to the flow cytometry test, culture media with or without glucose were used. The DiI concentration in the NP suspension was fixed at 1 μg/mL. The cells were incubated for 4 hours and washed with PBS by gently shaking. The cells were fixed with 4% formaldehyde for 10 minutes and completely dried under an air stream. Then, VECTASHIELD mounting medium, which contains 4′,6-diamidino-2-phenylindole (H-1200; Vector Laboratories Inc., Burlingame, CA, USA), was applied to the fixed cells to stain the nucleus of the cells. The treated cells were observed by CLSM (LSM 710; Carl-Zeiss, Thornwood, NY, USA).
NIRF imaging
Tumor targeting of the PLGA and PLGA-Glc NPs was observed by NIRF imaging after intravenous injection into Hep-2 tumor-xenografted mouse models. Cy5.5 (FKR-675; BioActs, Incheon, Korea) was loaded into the PLGA NPs and PLGA-Glc NPs according to the method described in the previous section except that 0.5 mg of Cy5.5 (dissolved in 0.1 mL of DMSO) was used instead of 4 mg of DCT. Prior to the intravenous injection, lyophilized NPs were dispersed in normal saline, and the fluorescence intensity of the dye in the NPs was normalized by Optix MX3 (ART Advanced Research Technologies Inc., Saint-Laurent, QC, Canada). Female BALB/c nude mice (5 weeks old; Charles River, Wilmington, MA, USA) were used to prepare the Hep-2 tumor-xenografted mouse model. Mice were maintained in a light-controlled room maintained at 22°C±2°C and 55%±5% relative humidity. The animal study was performed according to the “Guide for the Care and Use of Laboratory Animals” released by the National Research Council (USA). The experimental protocols were approved by the Animal Care and Use Committee of the College of Pharmacy, Seoul National University. The Hep-2 cell suspension (2×106 cells in 0.1 mL of cell culture medium) was injected subcutaneously into the back of the mice. After the tumor volume reached 150-200 mm3, Cy5.5-loaded NPs (dispersed in 0.1 mL of normal saline) were injected into the tail vein of the mouse. The tumor volume (V, mm3) was calculated using the following formula: V =0.5× longest diameter × (shortest diameter).2 A laser diode with a wavelength of 670 nm was used to excite the Cy5.5 dye. Fluorescence images were acquired at 2, 4, 6, and 24 hours after the intravenous injection. The ex vivo images were obtained by measuring the fluorescence intensity in dissected tumor tissue 24 hours after injection. The tumor targetability of the developed NPs was evaluated by determining the fluorescence intensity in the tumor region.
Statistical analysis
The experiments in this study were performed at least three times, and the data are reported as the mean ± SD. Statistical analysis was conducted using a Student’s t-test. A p-value <0.05 was considered statistically significant.
Results and discussion
Preparation and characterization of DCT-loaded NPs
Figure 1 describes the tumor-targeting strategy of the PLGA-Glc NPs via passive (ie, EPR effect) and active (ie, GLUTs) targeting followed by GLUTs-mediated endocytosis under hypoglycemic conditions. The chemical structure of PLGA-Glc is shown in Figure S1. As presented in its chemical structure, five PLGA units were conjugated to one glucose ring. Its chemical property was confirmed by proton nuclear magnetic resonance (1H-NMR) and Fourier-transform-infrared spectroscopy (FT-IR) analysis (Figure S1). In the 1H-NMR spectrum of PLGA-Glc, the chemical shifts for the PLGA segment remained with peak signals corresponding to −CHCH3 in lactic acid, −CHCH3 in lactic acid, and −CH2− in glycolic acid at 1.6, 5.2, and 4.8 ppm, respectively.13 The FT-IR spectrum also confirmed the successful synthesis of PLGA-Glc with a C=O stretching band at 1,760 cm−1, indicating the ester bond linkage between glucose and the PLGA unit.
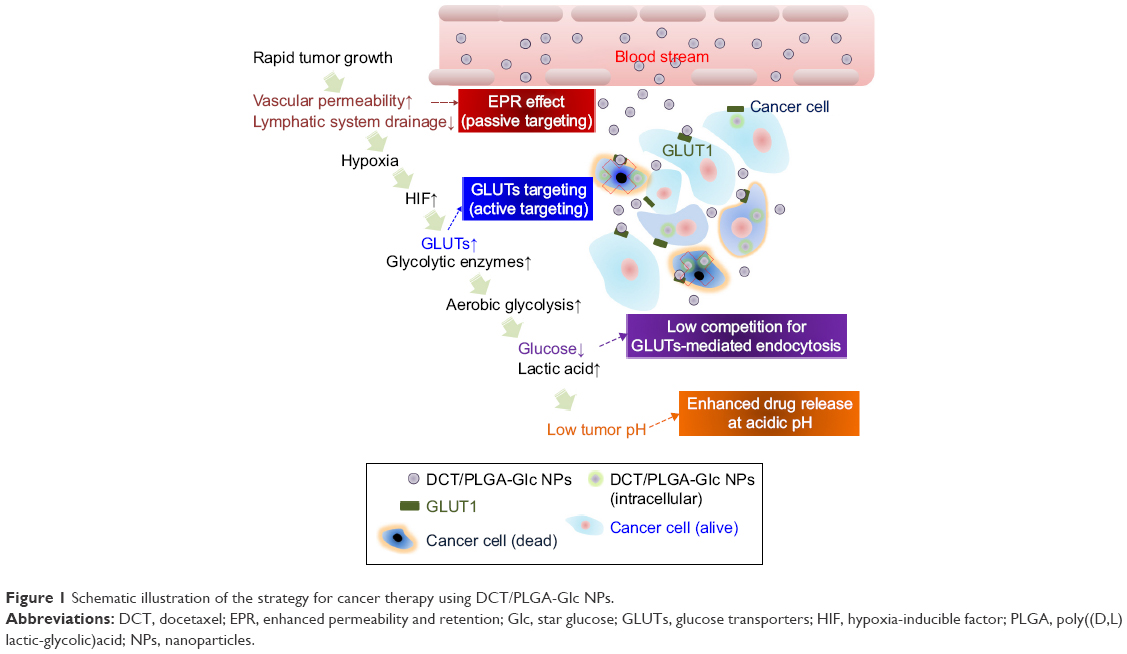 | Figure 1 Schematic illustration of the strategy for cancer therapy using DCT/PLGA-Glc NPs. |
For anticancer drug delivery, PLGA NPs (control) and PLGA-Glc NPs were successfully prepared using the emulsification-solvent evaporation method, and their physicochemical properties were investigated. Both NPs exhibit mean diameter of 241–256 nm, negative zeta potentials, and narrow size distributions (polydispersity index <0.1), as shown in Table 1 and Figure 2A. DCT was quantitatively determined by HPLC assay to measure its encapsulation efficiency in NPs. The encapsulation efficiency values of both NPs were over 50% in this study (Table 1). The spherical shape of both NPs was observed as shown in the field emission scanning electron microscope images (Figure 2B). The in vitro stability of DCT-loaded NPs in serum-included aqueous buffer was evaluated by measuring their hydrodynamic size. As shown in Figure 3, both DCT/PLGA NPs and DCT/PLGA-Glc NPs maintained their initial hydrodynamic size after incubating for 24 hours. It is expected that developed NPs hardly form aggregates in the blood stream after intravenous administration. The observed particle size range of the prepared NPs appears to be suitable for passive tumor targeting via the EPR effect.34
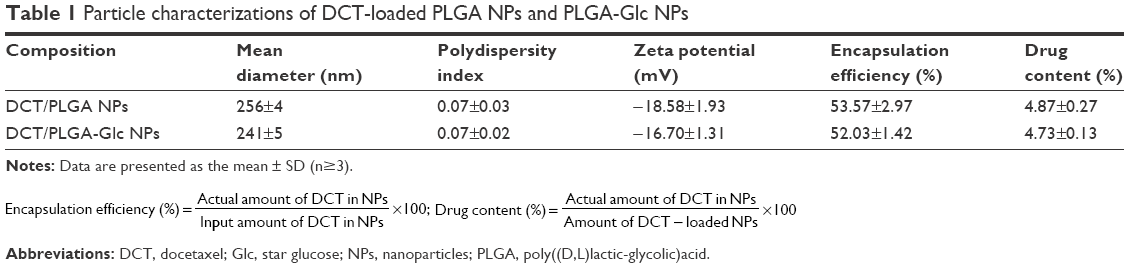 | Table 1 Particle characterizations of DCT-loaded PLGA NPs and PLGA-Glc NPs |
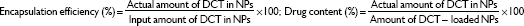
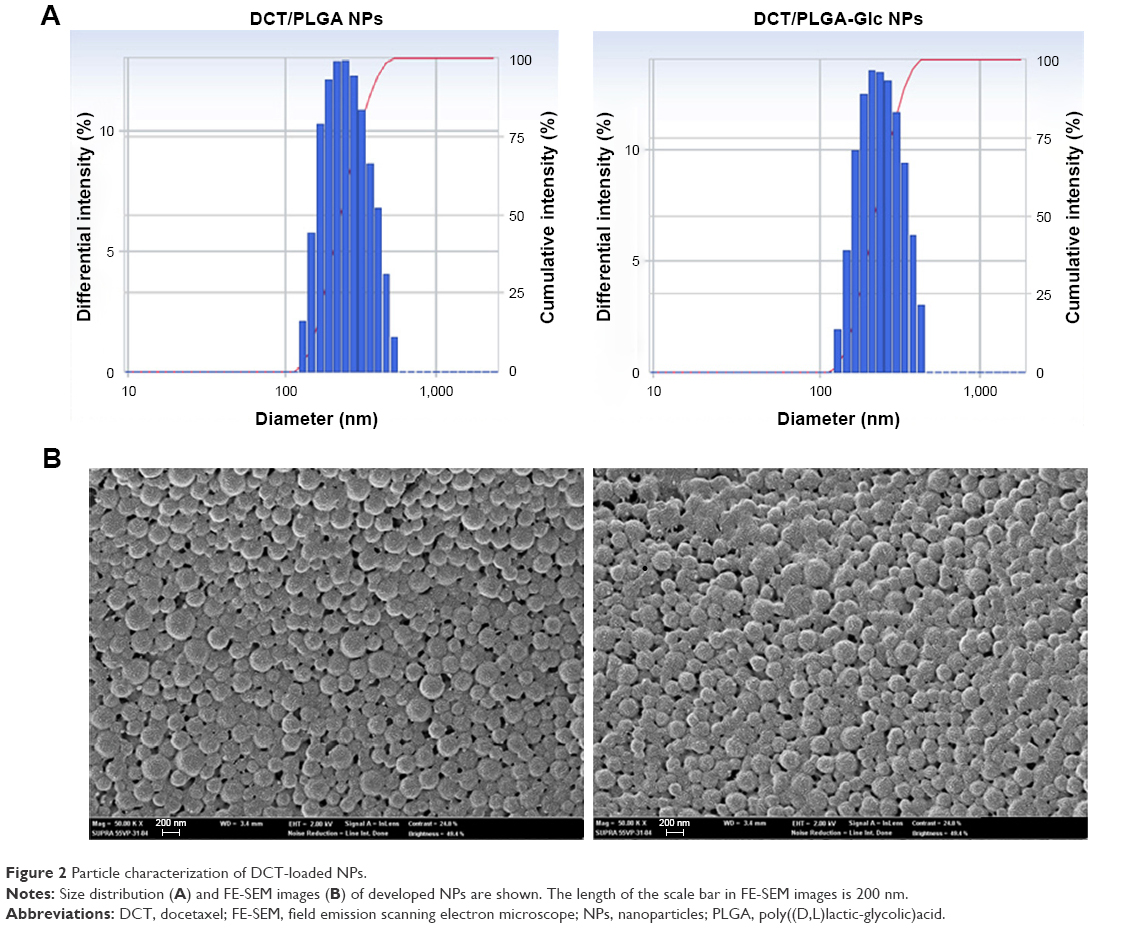 | Figure 2 Particle characterization of DCT-loaded NPs. |
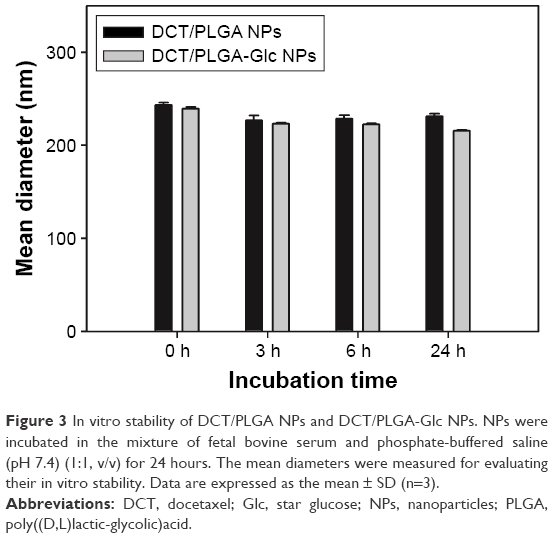 | Figure 3 In vitro stability of DCT/PLGA NPs and DCT/PLGA-Glc NPs. NPs were incubated in the mixture of fetal bovine serum and phosphate-buffered saline (pH 7.4) (1:1, v/v) for 24 hours. The mean diameters were measured for evaluating their in vitro stability. Data are expressed as the mean ± SD (n=3). |
Moreover, the presence of a glucose ring on the surface of the PLGA-Glc NPs was confirmed by electron spectroscopy for chemical analysis, as shown in Figure S2 and Table S1. The total chemical composition of carbon as C−C and C−H from the glucose ring was higher in the PLGA-Glc NPs than in the PLGA NPs (Table S1), indicating the existence of a glucose ring on the surface of the PLGA-Glc NPs. Therefore, the glucose ring of the copolymer on the surface of the PLGA-Glc NPs is expected to exhibit an active tumor-targeting effect via its interaction with the GLUTs that are expressed in cancer cells.
In vitro DCT release
When DCT release from PLGA and PLGA-Glc NPs was monitored using a dialysis bag, a sustained-release profile was observed for 13 days (Figure 4). Sustained release of an anticancer drug can extend systemic exposure, which can be advantageous for cancer therapy. Notably, no significant difference in the release amount of DCT was observed between PLGA NPs and PLGA-Glc NPs at a pH of 7.4. Therefore, the effect of the star-shaped PLGA-Glc polymer compared to the PLGA polymer on the drug release from NPs at a pH of 7.4 appeared to be negligible. Moreover, for drug release from PLGA-Glc NPs at different pH values (5.5, 6.8, and 7.4), the released amounts of DCT increased at acidic pH. The released amounts of DCT for 13 days from PLGA-Glc NPs at a pH of 7.4 and 5.5 were 35.38%±3.60% and 59.94%±1.64%, respectively (p<0.05). The accelerated degradation of PLGA-Glc at acidic pH may explain the improved drug release of DCT from NPs at acidic pH compared to that at neutral pH. It is reported that degradation of PLGA is faster at acidic pH than that at neutral pH.35,36 As PLGA-Glc contains PLGA units, it is expected that it would also show an accelerated degradation at acidic pH. Due to the faster degradation property of PLGA derivatives, drug release seemed to be improved at acidic pH, compared to the neutral pH, in our previous study.37 This result implies that more drug can be released in the tumor region (pH 6.8) as well as endosomal and lysosomal environments (pH 5.5) than under normal physiological conditions (pH 7.4).38 Therefore, the antitumor efficacy of the developed DCT-loaded PLGA-Glc NPs should be improved. The drug release profiles for the PLGA-Glc NPs suggest that it would be useful to apply as an injectable formulation for anticancer drug delivery.
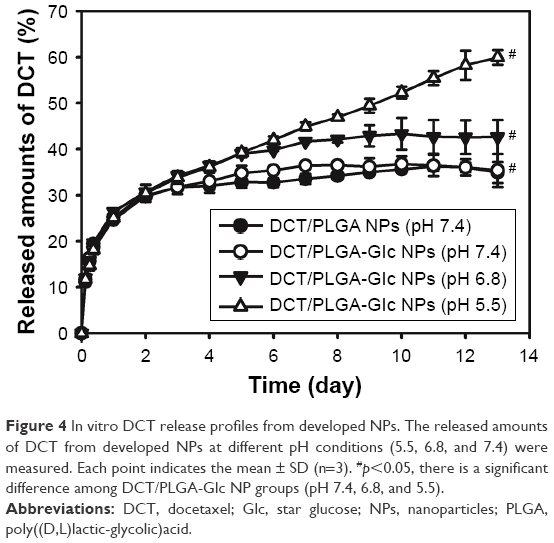 | Figure 4 In vitro DCT release profiles from developed NPs. The released amounts of DCT from developed NPs at different pH conditions (5.5, 6.8, and 7.4) were measured. Each point indicates the mean ± SD (n=3). #p<0.05, there is a significant difference among DCT/PLGA-Glc NP groups (pH 7.4, 6.8, and 5.5). |
In vitro cytotoxicity of blank NPs
The cytotoxicity of blank (without loading drug) PLGA NPs and PLGA-Glc NPs was assessed using Hep-2 (human laryngeal carcinoma) cells, which are human squamous cell carcinomas of the head and neck (SCCHN). DCT in combination with other anticancer drugs (ie, cisplatin and fluorouracil) has been approved for the treatment of locally advanced SCCHN. In particular, several isotypes of GLUTs were expressed in Hep-2 cells including GLUT-1, which is the most ubiquitously distributed isotype and the main transporter of glucose.22 Therefore, to test the targeting strategy based on the interaction between glucose and GLUTs, Hep-2 cells have been selected to study the cytotoxicity of NPs.26 Both groups of blank NPs (up to 1 mg/mL concentration) were incubated with Hep-2 cells for 24, 48, and 72 hours. Then, the cell viability was measured using the MTS assay. As shown in Figure 5, negligible cytotoxicity was observed in the tested concentration range and incubation period. The hemocompatibility of glucose-functionalized PLGA has been previously reported,16,26 and the result of the cytotoxicity test for PLGA-Glc NPs revealed that they can be used without severe cytotoxicity for cancer therapy.
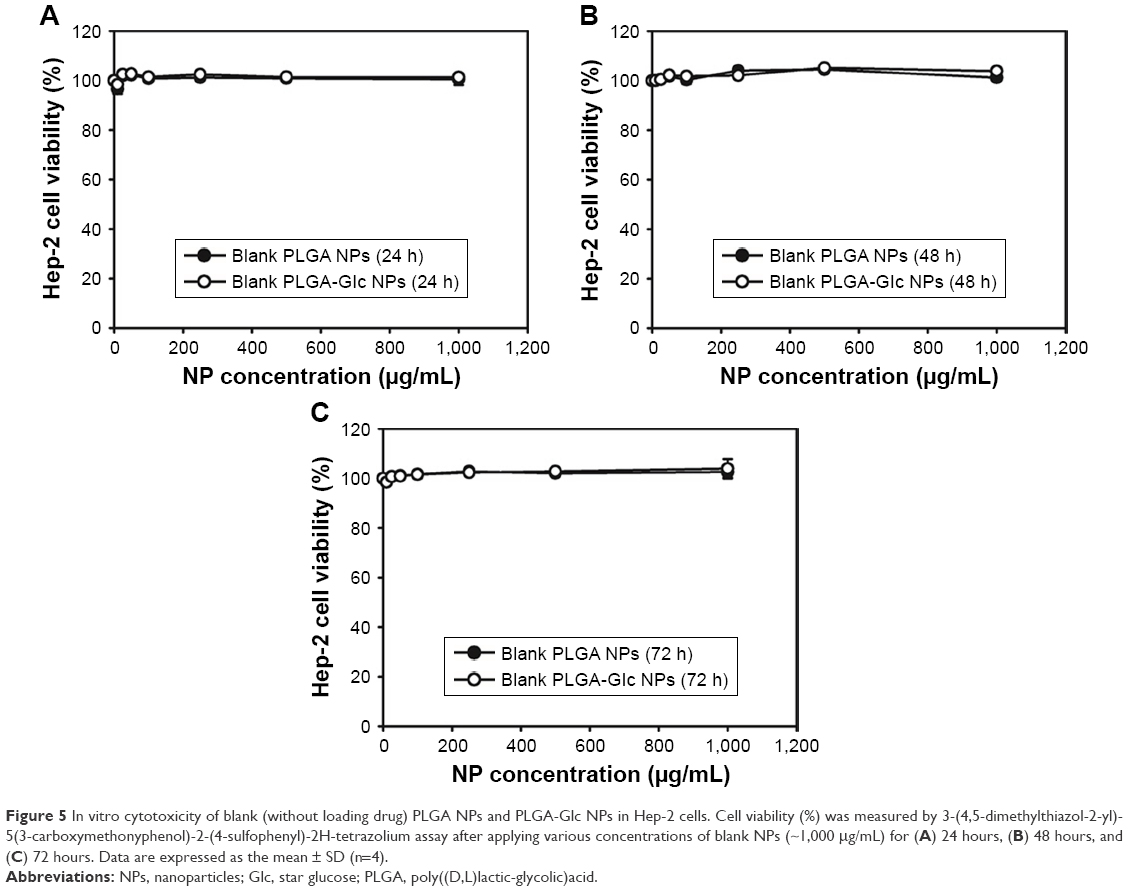 | Figure 5 In vitro cytotoxicity of blank (without loading drug) PLGA NPs and PLGA-Glc NPs in Hep-2 cells. Cell viability (%) was measured by 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethonyphenol)-2-(4-sulfophenyl)-2H-tetrazolium assay after applying various concentrations of blank NPs (~1,000 μg/mL) for (A) 24 hours, (B) 48 hours, and (C) 72 hours. Data are expressed as the mean ± SD (n=4). |
Antiproliferation assay
Antiproliferation efficiency of DCT-loaded NPs was tested in Hep-2 cells by MTS-based assay (Figure 6). The antitumor efficacies of DCT in head and neck cancer cells (ie, Hep-2 cells) have been already reported.39,40 At 50, 100, and 500 ng/mL DCT concentrations, the cell viability of DCT/PLGA-Glc NPs was significantly lower than that of DCT/PLGA NPs (p<0.05). Also, the half-maximum inhibitory concentration value of DCT/PLGA-Glc NPs group (356.5±6.2 ng/mL) was significantly lower than that of DCT/PLGA NPs group (457.3±43.9 ng/mL) (p<0.05). In glucose-free cell culture media, DCT/PLGA-Glc NPs seemed to be efficiently internalized into the cells, compared to DCT/PLGA NPs. Higher accumulation efficiency of DCT/PLGA-Glc NPs rather than that of DCT/PLGA NPs may lead to the improved antiproliferation efficacy. Specific cellular uptake mechanisms of PLGA-Glc NPs were elucidated by the following cellular uptake experiments.
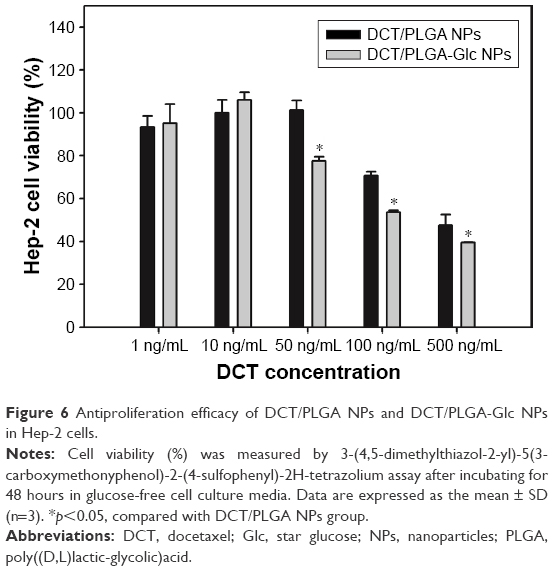 | Figure 6 Antiproliferation efficacy of DCT/PLGA NPs and DCT/PLGA-Glc NPs in Hep-2 cells. |
In vitro cellular uptake
Cellular uptake efficiency and intracellular distribution of PLGA and PLGA-Glc NPs were evaluated in Hep-2 cells (Figure 7). DiI, which is a fluorescent dye, was incorporated into the NPs for FACS and CLSM studies. To confirm the GLUTs-mediated internalization of the PLGA-Glc NPs, the cellular uptake studies were conducted in high-glucose or no-glucose cell culture media. As GLUTs were known to be expressed in Hep-2 cells,26 we did not confirm it again in this study. The glucose that is contained in the cell culture medium can be a competitive inhibitor for GLUT-mediated cellular uptake of PLGA-Glc NPs. As shown in Figure 7, the intracellular fluorescence intensity of PLGA-Glc NPs, which was observed by CLSM, in medium without glucose was higher than that in high-glucose-containing medium but no immense difference in the PLGA NPs was observed. The quantitative analysis of the uptake amounts of DiI-loaded NPs by FACS can further support this phenomenon. In the glucose-free medium (hypoglycemic condition) (Figures 7A and S3), the fluorescence intensity of the PLGA-Glc NPs group (53.27±0.81) was higher than that of the PLGA NPs group (16.20±0.06) (p<0.05). However, in a high-glucose-containing medium (Figures 7B and S3), the fluorescence intensity values of the PLGA-Glc NPs and PLGA NPs groups were 15.95±0.46 and 10.67±0.15, respectively. Moreover, the fluorescence intensity of the PLGA-Glc NPs group in the glucose-free medium was 3.34-fold higher than that in the high-glucose-containing medium (p<0.05). These data confirmed that the cellular uptake of the PLGA-Glc NPs was based on the interaction between the glucose moiety of the NPs and the GLUTs expressed in the cells, matching with a previous study that reported the specific interaction between a glucose ligand-carrying polymer and GLUT in cell lines.41 GLUTs-mediated internalization of PLGA-Glc NPs implies the feasibility of its application in tumor-targeted drug delivery. The tumor microenvironment can be characterized by hypoxia, a low glucose concentration, and a high lactate concentration.42,43 Deprivation of glucose in the tumor region in vivo can be relevant to the result for the glucose-free medium (Figure 7A) in this study. Therefore, the PLGA-Glc NPs are expected to be taken into the cell more efficiently compared to the PLGA NPs in the hypoxic tumor microenvironment. High expression level of GLUTs in cancer cells and a low glucose concentration in the hypoxic tumor region may contribute to the selective and improved cellular uptake of PLGA-Glc NPs. Enhanced internalization of PLGA-Glc NPs would lead to the improved antiproliferation efficacy (Figures 6 and 7).
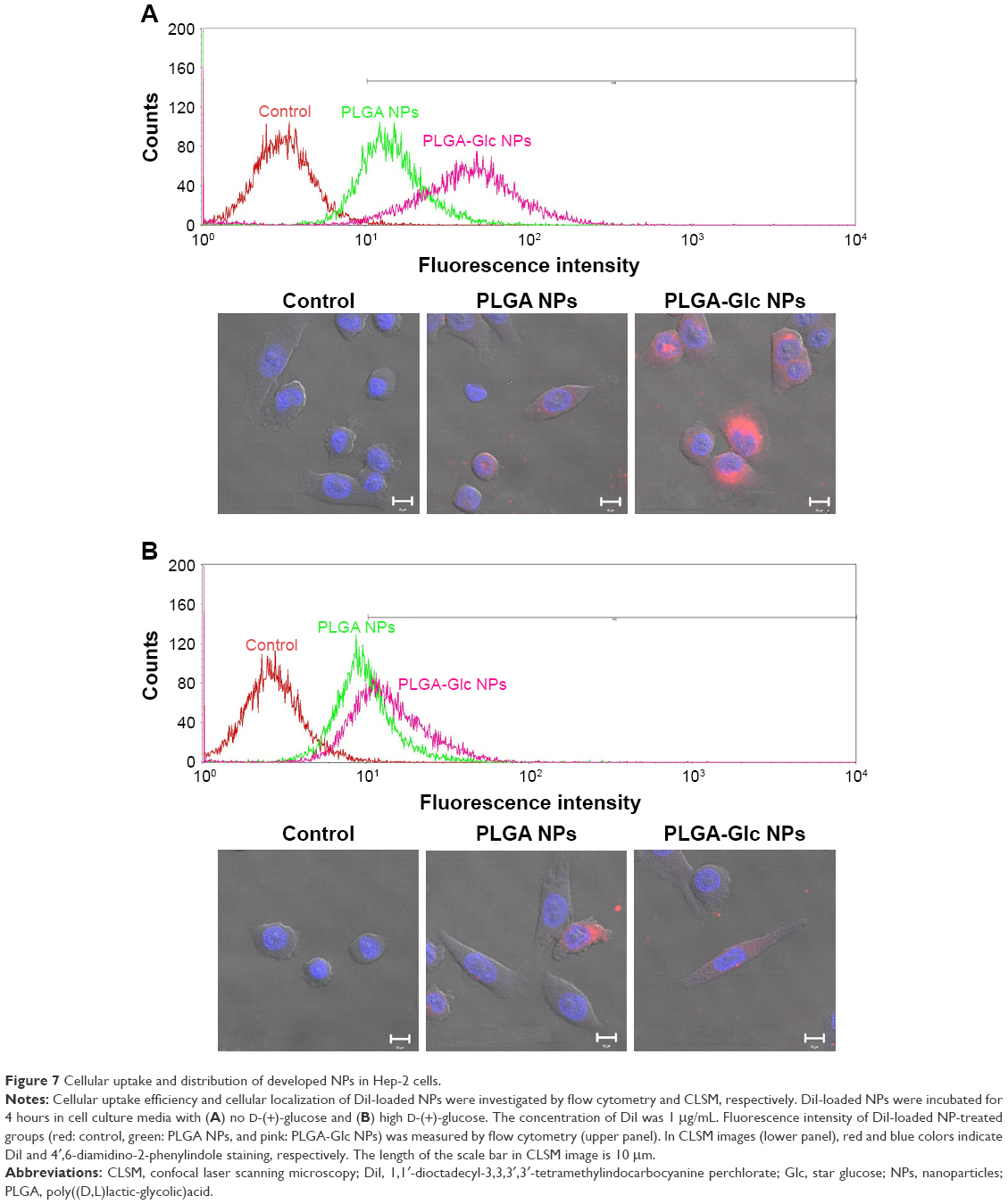 | Figure 7 Cellular uptake and distribution of developed NPs in Hep-2 cells. |
In vivo NIRF imaging
The tumor targetability of the PLGA NPs and PLGA-Glc NPs after intravenous injection into tumor-xenografted mouse was assessed via an in vivo NIRF imaging study. The Hep-2 tumor-xenografted mouse model was prepared, and Cy5.5-loaded NPs were injected intravenously into the mouse for NIRF imaging. The fluorescence intensity in the tumor region was scanned at 2, 4, 6, and 24 hours after injection (Figure 8A) and quantitatively analyzed (Figure 8B). The fluorescence intensity of the PLGA-Glc NPs group was 1.35-fold higher than that of the PLGA NPs group at 24 hours (p<0.05). In addition, in the ex vivo image (Figure 8C), a significant increase in the fluorescence intensity of the tumor tissue in the PLGA-Glc NPs group was observed compared to that of the PLGA NPs group. Passive tumor targeting, which is primarily based on the EPR effect, of the PLGA NPs has been widely used for anticancer drug delivery via the intravenous route.8,17,44 The results of the in vivo imaging study confirm that glucose modification of PLGA NPs (PLGA-Glc NPs) can be successfully applied for active targeting of PLGA NPs. The animal species-dependent difference in the expression levels of GLUTs in the plasma membrane of red blood cells (RBCs) might influence on the clinical application of the observed tumor targetability in animal models.45 However, it is reported that a large portion of GLUT1 expressed in the plasma membrane of RBCs is inactive (masked) form.46 Therefore, it is expected that injected PLGA-Glc NPs may hardly react with GLUTs in RBCs without their stimulation. Tumor targeting via the EPR effect and the interaction between the glucose moiety of PLGA-Glc NPs and GLUTs in cancer cells can produce a higher accumulation of NPs in the tumor region. Hypoxic and hypoglycemic conditions in the tumor microenvironment can result in lower competitive inhibition for GLUT-mediated uptake and subsequent enhanced cellular accumulation. These effects appear to contribute synergistically to the enhanced in vivo tumor targetability of PLGA-Glc NPs.
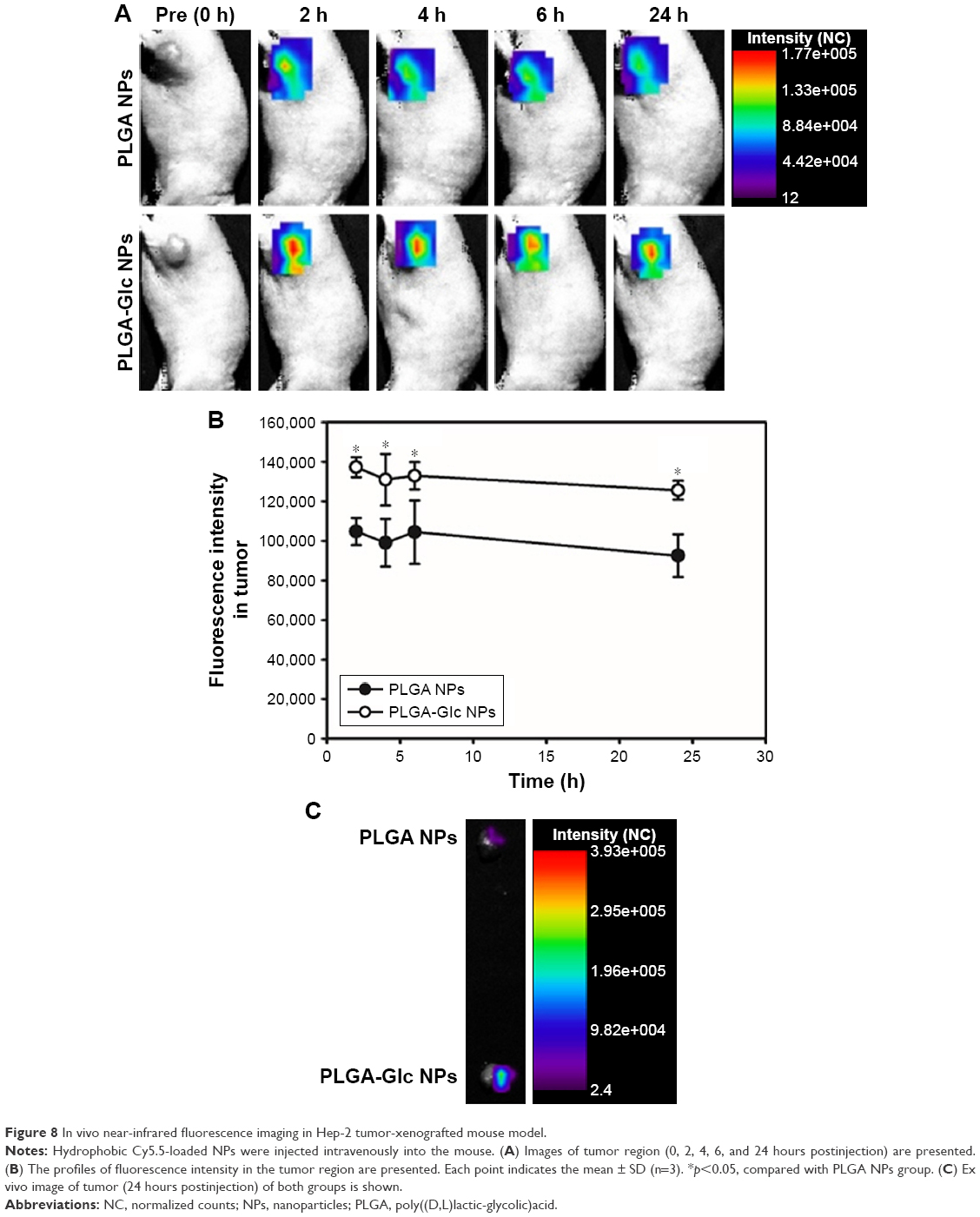 | Figure 8 In vivo near-infrared fluorescence imaging in Hep-2 tumor-xenografted mouse model. |
Conclusion
The PLGA-Glc polymer was used to fabricate tumor-targeting NPs via hypoxia-responsive mechanism. The DCT-loaded NPs with approximate mean diameters of 241–256 nm, narrow distributions, and spherical shapes were prepared. Sustained and pH-dependent drug release patterns from the NPs were observed in the drug release test. Cellular uptake studies of PLGA NPs and PLGA-Glc NPs in Hep-2 cells expressing GLUTs revealed that the cellular uptake of PLGA-Glc NPs was improved in the glucose-free cell culture medium (hypoglycemic condition). DCT/PLGA-Glc NPs exhibited an improved antiproliferation efficiency, compared with DCT/PLGA NPs, in Hep-2 cells, over 50 ng/mL DCT concentration. It is expected that GLUTs-mediated endocytosis of PLGA-Glc NPs with an EPR effect may contribute to the improved tumor targeting efficiency in the hypoxic/hypoglycemic tumor microenvironment. Therefore, the developed PLGA-Glc NP formulation can be a useful strategy for efficient tumor-targeted drug delivery.
Acknowledgments
This research was supported by the National Research Foundation of Korea (NRF), funded by the Korean government (MSIP) (No 2009–0083533 and NRF-2015R1A1A1A05027671).
Disclosure
The authors report no conflicts of interest in this work.
References
Cho HJ, Yoon HY, Koo H, et al. Self-assembled nanoparticles based on hyaluronic acid-ceramide (HA-CE) and Pluronic® for tumor-targeted delivery of docetaxel. Biomaterials. 2011;32(29):7181–7190. | ||
Jin YJ, Termsarasab U, Ko SH, et al. Hyaluronic acid derivative-based self-assembled nanoparticles for the treatment of melanoma. Pharm Res. 2012;29(12):3443–3454. | ||
Kim CH, Lee SG, Kang MJ, Lee S, Choi YW. Surface modification of lipid-based nanocarriers for cancer cell-specific drug targeting. J Pharm Invest. 2017;47(3):203–227. | ||
Kim HS, Lee DY. Photothermal therapy with gold nanoparticles as an anticancer medication. J Pharm Invest. 2017;47(1):19–26. | ||
Townsend SA, Evrony GD, Gu FX, Schulz MP, Brown RH Jr, Langer R. Tetanus toxin C fragment-conjugated nanoparticles for targeted drug delivery to neurons. Biomaterials. 2007;28(34):5176–5184. | ||
Termsarasab U, Cho HJ, Kim DH, et al. Chitosan oligosaccharide-arachidic acid-based nanoparticles for anti-cancer drug delivery. Int J Pharm. 2013;441(1–2):373–380. | ||
Youn YS, Kwag DS, Lee ES. Multifunctional nano-sized fullerenes for advanced tumor therapy. J Pharm Invest. 2017;47(1):1–10. | ||
Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumor delivery by EPR effect. Adv Drug Deliv Rev. 2011;63(3):170–183. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. Adv Drug Deliv Rev. 2012;161(2):505–522. | ||
Gullotti E, Park J, Yeo Y. Polydopamine-based surface modification for the development of peritumorally activatable nanoparticles. Pharm Res. 2013;30(8):1956–1967. | ||
Karra N, Nassar T, Ripin AN, Schwob O, Borlak J, Benita S. Antibody conjugated PLGA nanoparticles for targeted delivery of paclitaxel palmitate: efficacy and biofate in a lung cancer mouse model. Small. 2013;9(24):4221–4236. | ||
Bala I, Hariharan S, Kumar MN. PLGA nanoparticles in drug delivery: the state of the art. Crit Rev Ther Drug Carrier Syst. 2004;21(5):387–422. | ||
Zeng X, Tao W, Mei L, Huang L, Tan C, Feng SS. Cholic acid-functionalized nanoparticles of star-shaped PLGA-vitamin E TPGS copolymer for docetaxel delivery to cervical cancer. Biomaterials. 2013;34(25):6058–6067. | ||
Chang J, Paillard A, Passirani C, et al. Transferrin adsorption onto PLGA nanoparticles governs their interaction with biological systems from blood circulation to brain cancer cells. Pharm Res. 2012;29(6):1495–1505. | ||
Kang J, Lambert O, Ausborn M, Schwendeman SP. Stability of proteins encapsulated in injectable and biodegradable poly(lactide-co-glycolide)-glucose millicylinders. Int J Pharm. 2008;357(1–2):235–243. | ||
Thasneem YM, Sajeesh S, Sharma CP. Glucosylated polymeric nanoparticles: a sweetened approach against blood compatibility paradox. Colloids Surf B Biointerfaces. 2013;108:337–344. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | ||
Chan DA, Sutphin PD, Nguyen P, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3(94):94ra70. | ||
He C, Sun XP, Qiao H, et al. Downregulating hypoxia-inducible factor-2α improves the efficacy of doxorubicin in the treatment of hepatocellular carcinoma. Cancer Sci. 2012;103(3):528–534. | ||
Huang CY, Kuo WT, Huang YC, Lee TC, Yu LC. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis. 2013;4:e622. | ||
Jiang X, Xin H, Ren Q, et al. Nanoparticles of 2-deoxy-d-glucose functionalized poly(ethylene glycol)-co-poly(trimethylene carbonate) for dual-targeted drug delivery in glioma treatment. Biomaterials. 2014;35(1):518–529. | ||
Luo XM, Zhou SH, Fan J. Glucose transporter-1 as a new therapeutic target in laryngeal carcinoma. J Int Med Res. 2010;38(6):1885–1892. | ||
Semaan A, Munkarah AR, Arabi H, et al. Expression of GLUT-1 in epithelial ovarian carcinoma: correlation with tumor cell proliferation, angiogenesis, survival and ability to predict optimal cytoreduction. Gynecol Oncol. 2011;121(1):181–186. | ||
Shan XH, Hu H, Xiong F, et al. Targeting Glut1-overexpressing MDA-MB-231 cells with 2-deoxy-D-g1ucose modified SPIOs. Eur J Radiol. 2012;81(1):95–99. | ||
Xiong F, Zhu ZY, Xiong C, et al. Preparation, characterization of 2-deoxy-D-glucose functionalized dimercaptosuccinic acid-coated maghemite nanoparticles for targeting tumor cells. Pharm Res. 2012;29(4):1087–1097. | ||
Zhou SH, Fan J, Chen XM, Cheng KJ, Wang SQ. Inhibition of cell proliferation and glucose uptake in human laryngeal carcinoma cells by antisense oligonucleotides against glucose transporter-1. Head Neck. 2009;31(12):1624–1633. | ||
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. | ||
Chen J, Ding J, Xu W, et al. Receptor and microenvironment dual-recognizable nanogel for targeted chemotherapy of highly metastatic malignancy. Nano Lett. 2017;17(7):4526–4533. | ||
Zhao K, Li D, Xu W, et al. Targeted hydroxyethyl starch prodrug for inhibiting the growth and metastasis of prostate cancer. Biomaterials. 2017;116:82–94. | ||
Li D, Han J, Ding J, Chen L, Chen X. Acid-sensitive dextran prodrug: A higher molecular weight makes a better efficacy. Carbohydr Polym. 2017;161:33–41. | ||
Li D, Xu W, Li P, et al. Self-targeted polysaccharide prodrug suppresses orthotopic hepatoma. Mol Pharm. 2016;13(12):4231–4235. | ||
Kissel T, Brich S, Bantle I, Lancranjan F, Nimmerfall F, Vit P. Paraenteral depot-systems on the basis of biodegradable polyesters. J Control Release. 1991;16(1–2):27–42. | ||
Lee JY, Kim JS, Cho HJ, Kim DD. Poly(styrene)-b-poly(DL-lactide) copolymer-based nanoparticles for anticancer drug delivery. Int J Nanomedicine. 2014;9:2803–2813. | ||
Bae YH, Park K. Targeted drug delivery to tumors: Myths, reality and possibility. J Control Release. 2011;153(3):198–205. | ||
Yoo JY, Kim JM, Seo KS, Jeong YK, Lee HB, Khang G. Characterization of degradation behavior for PLGA in various pH condition by simple liquid chromatography method. Biomed Mater Eng. 2005;15(4):279–288. | ||
Zolnik BS, Burgess DJ. Effect of acidic pH on PLGA microsphere degradation and release. J Control Release. 2007;122(3):338–344. | ||
Lee SY, Cho HJ. Amine-functionalized poly(lactic-co-glycolic acid) nanoparticles for improved cellular uptake and tumor penetration. Colloids Surf B Biointerfaces. 2016;148:85–94. | ||
Ge Z, Liu S. Functional block copolymer assemblies responsive to tumor and intracellular microenvironments for site-specific drug delivery and enhanced imaging performance. Chem Soc Rev. 2013;42(17):7289–7325. | ||
Kogashiwa Y, Sakurai H, Kimura T, Kohno N. Docetaxel suppresses invasiveness of head and neck cancer cells in vitro. Cancer Sci. 2010;101(6):1382–1386. | ||
Liu T, Zhang M, Zhang H, et al. Combined antitumor activity of cucurbitacin B and docetaxel in laryngeal cancer. Eur J Pharmacol. 2008;587(1–3):78–84. | ||
Chung JH, Park KH, Seo BM, et al. Determination of specific interactions between glucose ligand carrying polymer and glucose transporter type-1 (GLUT-1) using different cell types. J Biomed Mater Res A. 2003;67(3):1055–1059. | ||
Höckel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93(4):266–276. | ||
Lee YJ, Moon MS, Kwon SJ, Rhee JG. Hypoxia and low glucose differentially augments TRAIL-induced apoptotic death. Mol Cell Biochem. 2005;270(1–2):89–97. | ||
Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148(2):135–146. | ||
Montel-Hagen A, Blanc L, Boyer-Clavel M, et al. The Glut1 and Glut4 glucose transporters are differentially expressed during perinatal and postnatal erythropoiesis. Blood. 2008;112(12):4729–4738. | ||
Zhang JZ, Ismail-Beigi F. Activation of Glut1 glucose transporter in human erythrocytes. Arch Biochem Biophys. 1998;356(1):86–92. |
Supplementary materials
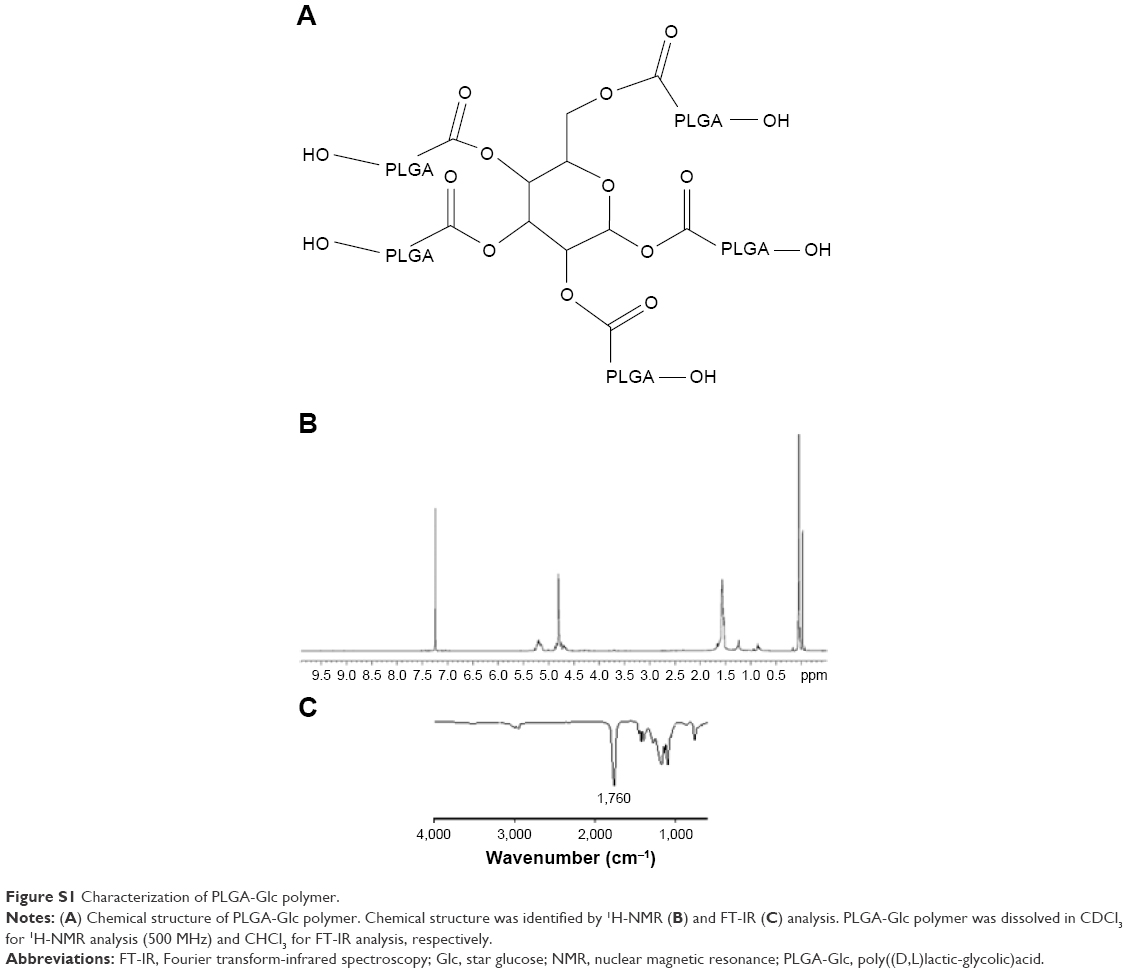 | Figure S1 Characterization of PLGA-Glc polymer. |
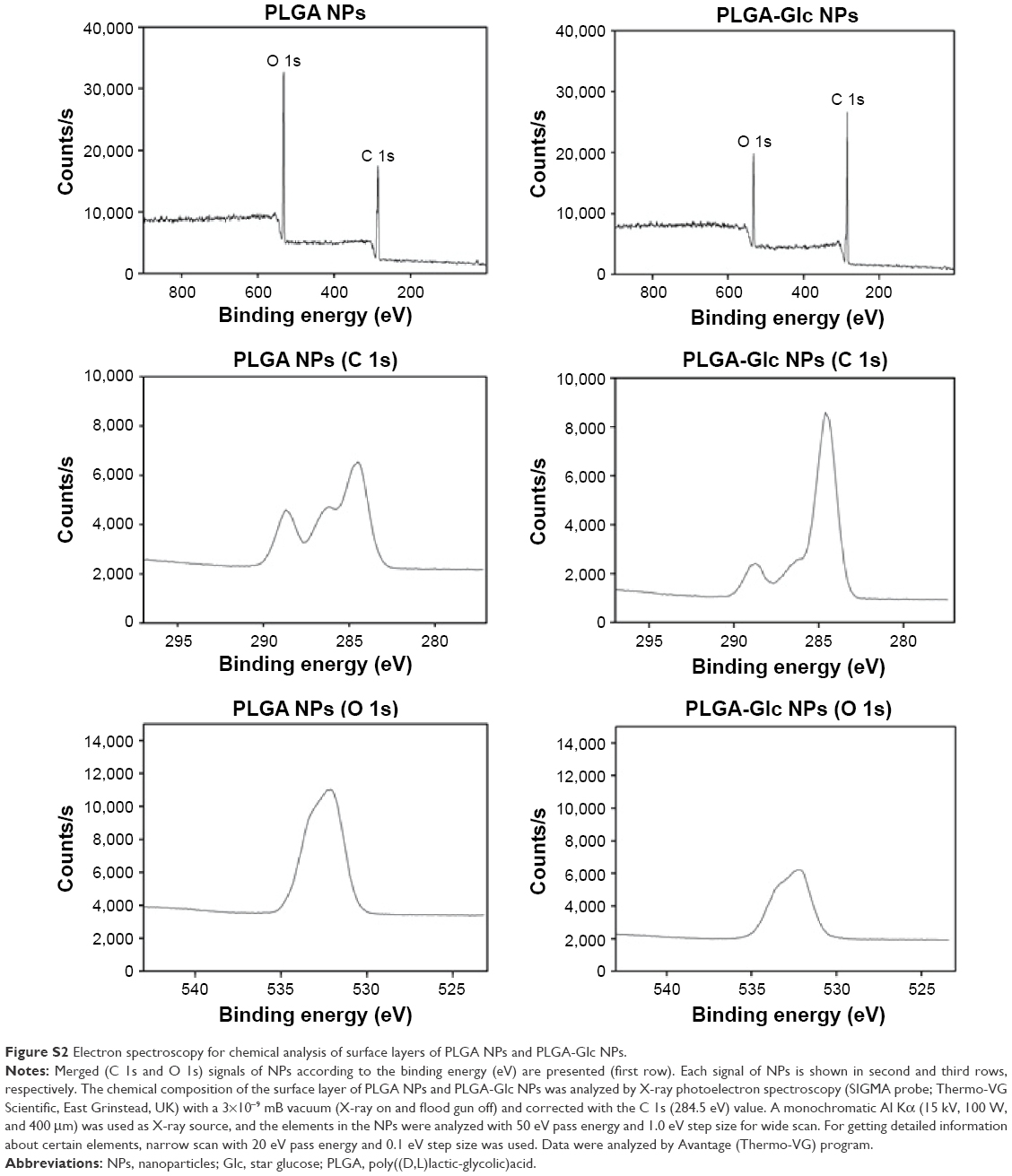 | Figure S2 Electron spectroscopy for chemical analysis of surface layers of PLGA NPs and PLGA-Glc NPs. |
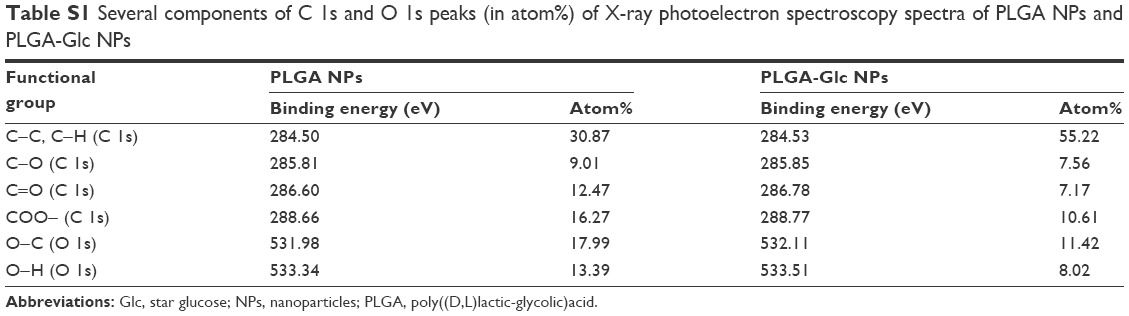 | Table S1 Several components of C 1s and O 1s peaks (in atom%) of X-ray photoelectron spectroscopy spectra of PLGA NPs and PLGA-Glc NPs |
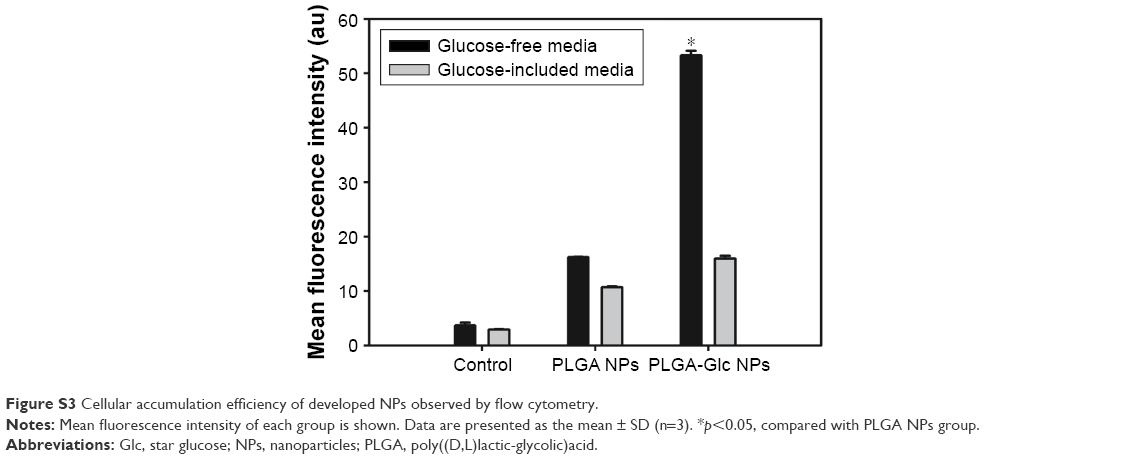 | Figure S3 Cellular accumulation efficiency of developed NPs observed by flow cytometry. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.