")
Back to Journals » OncoTargets and Therapy » Volume 8
Polo-like kinase 2 acting as a promoter in human tumor cells with an abundance of TAp73
Authors Hu Z, Xu Z, Liao X, Yang X, Dong C, Luk K, Jin A, Lu H
Received 10 June 2015
Accepted for publication 8 October 2015
Published 20 November 2015 Volume 2015:8 Pages 3475—3488
DOI https://doi.org/10.2147/OTT.S90302
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jianmin Xu
ZhengBo Hu,1,* ZunYing Xu,1,* XiaoHong Liao,2,* Xiao Yang,1 Cao Dong,1 KuaDi Luk,3 AnMin Jin,1,* Hai Lu4,5,*
1Department of Orthopedics, Zhujiang Hospital of Southern Medical University, 2The State Key Laboratory of Respiratory Diseases, the First Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangdong, 3Department of Orthopedics and Traumatology, The University of Hong Kong, Hong Kong, SAR, 4Department of Orthopedics, the Third Affiliated Hospital of the Southern Medical University, Guangzhou, Guangdong, 5Academy of Orthopedics, Guangdong Province, People’s Republic of China
*These authors contributed equally to this work
Background: TAp73, a member of the p53 tumor suppressor family, is frequently overexpressed in malignant tumors in humans. TAp73 abundance and phosphorylation modification result in variations in transcriptional activity. In a previous study, we found that the antitumor function of TAp73 was reactivated by dephosphorylation in head and neck squamous cell carcinomas. Polo-like kinase 2 (PLK2) displayed a close relationship with the p53 family in affecting the fate of cells. Herein, we investigate the hypothesis that PLK2 phosphorylates TAp73 and inhibits TAp73 function.
Materials and methods: Head and neck squamous cell carcinoma cell lines and osteosarcoma cell lines were used as natural models of the different expression levels of TAp73. Phosphorylation predictor software Scansite 3.0 and the predictor GPS-polo 1.0 were used to analyze the phosphorylation sites. Coimmunoprecipitation, phosphor-tag Western blot, metabolic labeling, and indirect immunofluorescence assays were used to determine the interactions between PLK2 and TAp73. TAp73 activity was assessed by Western blot and reverse transcription polymerase chain reaction, which we used to detect P21 and PUMA, both downstream genes of TAp73. The physiological effects of PLK2 cross talk with TAp73 on cell cycle progress and apoptosis were observed by flow cytometry and terminal deoxynucleotidyl transferase dUTP nick end labeling assays.
Results: PLK2 binds to and phosphorylates TAp73. PLK2 phosphorylates TAp73 at residue Ser48 and prohibits TAp73 translocation to the nucleus. Additionally, PLK2 inhibition combined with a DNA-damaging drug upregulated p21 and PUMA mRNA expression to a greater extent than DNA-damaging drug treatment alone. Inhibiting PLK2 in TAp73-enriched cells strengthened the effects of the DNA-damaging drug on both G1 phase arrest and apoptosis. Pretreatment with TAp73-siRNA weakened these effects.
Conclusion: These findings reveal a novel PLK2 function (catalyzed phosphorylation of TAp73) which suppresses TAp73 functions. PLK2 promotes the survival of human tumor cells, a novel insight into the workings of malignant tumors characterized by TAp73 overexpression, and one that could speed the development of therapies.
Keywords: antitumor therapy, DNA damaging reagent, phosphorylation, PLK2, TAp73
Introduction
TAp73 is a member of the p53 family, the most important tumor-suppressing family, and has a structure similar to p53. It is able to activate some of the p53 target genes, such as p21 (a cell cycle inhibitor) and PUMA (a proapoptosis gene), which regulate cell survival.1 TAp73 is frequently overexpressed in carcinomas and sarcomas1 and is, under certain conditions, an accepted marker of malignant tumors. These mechanisms are not yet completely clear. Among the inconsistencies, we noted differences in expression levels of TAp73 between human tumor cell lines such as head and neck squamous cell carcinoma (HNSCC) cell lines originating from epithelial carcinoma and osteosarcoma cell lines originating from mesenchymal sarcoma.
Previous studies have demonstrated a close interaction between the p53 family and the Polo-like kinase (PLK) family in a number of human tumors, including osteosarcoma. However, the mechanisms, as well as the effects, remain to be elucidated. Since Polo-like kinase 2 (PLK2) was first reported in 1991,2 five members of the PLK family (PLKs 1–5) have been reported.3 PLKs play a key role in the regulation of cell cycle progression. Although many studies have determined that PLK1 plays a prosurvival role in human tumors,4,5 controversy still surrounds the functions of PLK2.6,7 We identified several phosphorylation sites on TAp73 using the PhosphoSite database. Interestingly, these TAp73 phosphorylation sites appear to have differing functions in certain cells. As is well known, the reversible protein phosphorylation mediated by phosphokinase helps harmonize a large number of principal cell processes.8–10 We have previously demonstrated that this kinase 2 (CK2) phosphorylates and inhibits the tumor-suppressing function of TAp73 in HNSCCs.11 Prior to our study, Songyang et al12 found that CK2 displays a consensus sequence identical to PLK2. More recently, studies have demonstrated that PLK2 can recognize substrates similar to CK2 even more efficiently.13,14 We speculated that PLK2, similar to CK2, may have a kinase-like function in its effect on TAp73.
We conducted this research so as to determine whether PLK2 activates or deactivates TAp73.
Materials and methods
Bioinformatics analysis
We used the online bioinformatics phosphorylation predictor Scansite 3.0 (http://scansite3.mit.edu) to predict the phosphorylation sites of TAp73.15 The predictor GPS-polo 1.0 was used to identify PLK binding and phosphorylation sites in TAp73.16 The known phosphorylation sites in TAp73 were collected from www.PhosphoSitePlus.org.17
Cell lines
We used four human tumor cell lines such as osteosarcoma cell lines Saos2 and MG63 (ATCC), UM-SCC1, and UM-SCC22A (squamous cell carcinoma cell lines were previously described in our research)18 from the University of Michigan. The Ethical Commission of South Medical University stated that these cell lines are commercialized and can be purchased from many cell banks; thus, ethical permission was not required for the use of these cell lines in this study. Cells were cultured in McCoy’s 5A medium (Thermo Fisher Scientific, Waltham, MA, USA) or Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum. The medium was replaced with fresh medium as necessary, and the cultures were maintained at 37°C in the presence of 5% CO2.
Transfection
Forty-eight hours before the experiments, the small interfering RNA (200 pmol of siTAp73 or siPLK2; Thermo Fisher Scientific Dharmacon Products, Lafayette, CO, USA) and appropriate expression vectors (20 μg of Flag-pcDNA3-PLK2, Flag-pcDNA3-TAp73, or Flag-pcDNA3-TAp73S48A, obtained from Dr Zhi-Min Yuan, Harvard University, USA) were transfected into cells as described previously19 using Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific) following the manufacturer’s protocols.11 The TAp73 S48A mutant, in which Ser-48 was substituted with Ala (GENEWIZ, Inc., USA), was confirmed by DNA sequencing. Empty pcDNA3 (Thermo Fisher Scientific) and control siRNA (contr-si) were used as control.
Drug treatment and antibodies
Cells were seeded at a density of 60%–70% and cultured for 24 hours before 12-hour drug treatments. DNA-damaging reagents, cisplatin (CDDP, Qiru Corp., People’s Republic of China), adriamycin (ADM, Sigma-Aldrich, St Louis, MO, USA), and ifosfamide (Sigma-Aldrich), were diluted to the indicated concentrations in cell culture medium. The PLK2 inhibitor ELN58264620,21 (Elan Pharmaceuticals, San Francisco, CA, USA) was diluted to 5 μg/mL in cell culture medium. Dimethyl sulfoxide was used as a control. The following antibodies were used for Western blot (WB) and coimmunoprecipitation (co-IP): anti-PLK2 (EPR10070, ab154794, Abcam, USA), anti-TAp73 (38C674.2, ab79078, Abcam, USA), anti-PUMA (4976S, CST, MA, USA), anti-p21 (SC-397, Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-Flag (F3165, Sigma-Aldrich), and anti-β-actin (A5441, Sigma-Aldrich). The following antibodies were used for immunofluorescence assay: primary antibodies against PLK2 (EPR10070, ab154794, Abcam, MA, USA) and TAp73 (1E8, ab118985, Abcam, USA) and secondary antibodies against PLK2 (anti-rabbit 594, ZF-0416, ZSGB-BIO, People’s Republic of China) and TAp73 (anti-mouse-488, ZF-0412, ZSGB-BIO, People’s Republic of China).
Real-time reverse transcription polymerase chain reaction
Reverse transcription polymerase chain reaction (RT-PCR) was performed as described in our previous research.11,18 Briefly, the isolated RNA was subjected to reverse transcription and PCR, and β-actin mRNA was used as an internal control. The relative gene expression levels (2−ΔΔCt) were normalized. Table 1 shows the primer and siRNA sequences.
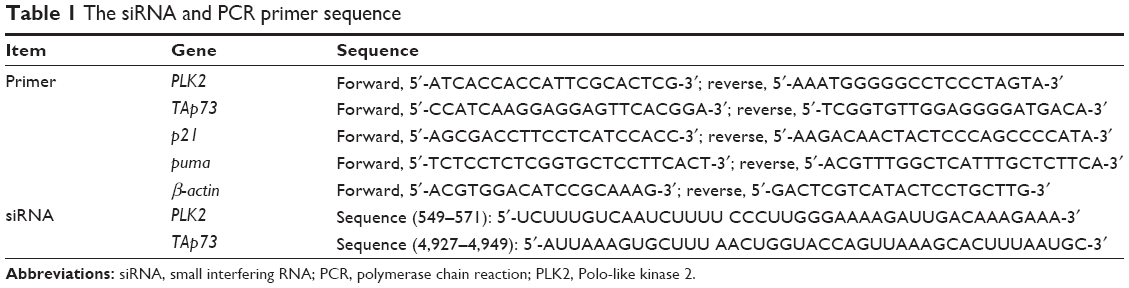 | Table 1 The siRNA and PCR primer sequence |
Coimmunoprecipitation, Western blot, and Phos-tag Western blot
Co-IP and WB were performed as described in our previous research.11,18 Phosphorylated TAp73 was tested by Phos-tag WB, as previously described.22 Briefly, cell lysates were harvested using 1× RIPA (radioimmunoprecipitation assay) lysis buffer (0.5% Nonidet P-40, 50 mM NaCl containing protease inhibitors). The protein concentration was determined by protein assay reagents (Bio-Rad Laboratories Inc., Hercules, CA, USA) and spectrophotometer quantification by Beckman DU-640 (Beckman Coulter, Pasadena, CA, USA). Total protein (25 μg) was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a pure nitrocellulose membrane. Primary antibodies (1:500) were incubated in Tris-buffered saline (pH 7.4) with 0.1% Tween-20 at 4°C for 12 hours. Signals were generated by horseradish peroxidase-conjugated secondary antibodies (1:2,000; Bio-Rad Laboratories Inc.) in Tris-buffered saline (pH 7.4) with 5% nonfat milk and 0.1% Tween-20 at room temperature for 1 hour. Immunoblots were detected. For immunoprecipitation, cell lysate (500 μg) was incubated at 4°C for 12 hours with TAp73 (1:100), PLK2 (1:200), flag (1:200) antibody, 20 μL protein G+/protein A agarose (Oncogene Research Products), or anti-flag affinity agarose gel (Sigma-Aldrich). The immunocomplex subsets were subjected to SDS-PAGE and immunoblotting. For TAp73 Phos-tag WB, the immunocomplex subsets were subjected to SDS-PAGE in which gels were made using a Phos-tag™ Acrylamide (50 μmol/L; Wako Pure Chemical Industries, Ltd., Osaka, Japan), containing MnCl2 (30 μmol/L), and then transferred to membranes and immunoblotted with TAp73 antibody. For testing exogenous TAp73 phosphorylate, both TAp73 and PLK2 immunocomplex subsets (ratio 1:5) from an anti-flag affinity agarose gel were incubated together for indicated times at 37°C in kinase buffer. The reactions were stopped using 2× loading buffer and the mixtures were used for phos-tag WB, as described earlier.
Indirect immunofluorescence microscopy
Indirect immunofluorescence experiments were performed according to the experimental protocols in the previous research.23 Briefly, UM-SCC1 cells with or without transfection cultivated on coverslips in six-well plates were treated with ADM for 2 hours and then rinsed with phosphate-buffered saline (PBS) and fixed with 3% paraformaldehyde at room temperature for 20 minutes. They were washed again with PBS and blocked with 3% bovine serum albumin (Sigma-Aldrich) for 10 minutes. Cells were then incubated with primary antibody (1:100) at room temperature for 60 minutes, washed three times using PBS, and then incubated with secondary antibody (1:200) at room temperature for 50 minutes. After a final PBS wash, cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Peterborough, UK) for 10 minutes, and the coverslips were mounted using Dako Fluorescence Mounting Medium (Dako Denmark A/S, Glostrup, Denmark). At least 200 cells were observed overall within discrete areas of each sample, and the samples were prepared from three independent wells of examined cell lines. Samples were observed using an Olympus BH2-RFCA microscope and an Olympus LSM-GB200 confocal microscope.
Metabolic labeling of PLK2 assay
To determine the half-life of endogenous PLK2, pulse-chase experiments were performed in Saos2 cells following previously reported protocols.24 Briefly, 2.5 mL of Saos2, transfected Saos2, or TAp73-Knockdown Saos2 cells (5×105 mL) per well was planted in six-well plates, which were then cultivated for 24 hours. Total protein was collected after cultivation in methionine-free medium containing 100 μCi/mL trans 35S-labeled methionine for 30 minutes. The newborn PLK2 from Saos2 and TAp73-knockdown Saos2 cells, stimulated by ADM (25 μg/mL) at indicated time points, was harvested to SDS-PAGE and radioautography, respectively.
Cell cycle analysis
Flow cytometric analysis of the cell cycle phase was conducted as described previously.25 Briefly, cell samples were washed in PBS, trypsinized, harvested by centrifugation, washed in PBS, and fixed in ethanol (50%) overnight at 4°C. Cells were harvested and resuspended in PBS. Fifty microliters of RNAase (100 μg/mL) was added to each sample and they were then incubated for 30 minutes at 37°C. Thereafter, propidium iodide (2 mg/mL) was added to each sample, and they were analyzed by flow cytometry (Becton Dickinson Coulter Epic XL flow cytometer), where over 10,000 events were recorded.
Apoptosis assay
Cell apoptosis was assessed according to the manufacturer’s instructions provided in the TUNEL kit (a terminal deoxynucleotidyl transferase dUTP nick end labeling kit; Hoffman-La Roche Ltd., Basel, Switzerland).
Statistical analysis
Data are expressed as mean ± standard deviation (SD). Differences between two groups were compared using either a chi-squared test or a two-tailed Student’s t-test. Differences between more than two groups were analyzed using one-way analysis of variance in conjunction with Dunnett’s post hoc test or a chi-squared test, and for repeated measurements, analysis of variance was used. Each sample was assayed in triplicate. Differences were considered significant when P<0.05.
Results
Theoretically, Ser48 within TA1 domain of TAp73 is the most feasible target after integrating predictor software and online data
The Scansite 3.0 program provides predictions for all of the known kinases that can phosphorylate a given site. TAp73 was analyzed as a substrate. A total of 21 sites were identified under matching with high thresholds (Figure S1A). TAp73 was also considered as a substrate using the software GPS-polo 1.0 based on the high threshold match. The results predicted PLKs that could phosphorylate a given site. A total of two binding and four phosphorylation sites were identified (Figure S1B). Table 2 summarizes the predicted sites distributed in TAp73 domains. Integrating the aforementioned predictions with the PLK2 functions and characteristics, the residue Ser48 within the TA1 domain of TAp73 was deemed to have the most potential.
 | Table 2 Distribution of predicted sites from Scansite 3.0 and GPS 1.0 |
There are 14 additional known phosphorylation sites in TAp73, according to the PhosphoSitePlus database (Figure 1). Our analysis of these known sites indicates that a phosphorylation site within the TA domain would negatively regulate TAp73 function (Table 3). Accordingly, we hypothesized that PLK2 phosphorylates the Ser48 residue of TAp73 and inhibits TAp73 transcriptional functionality.
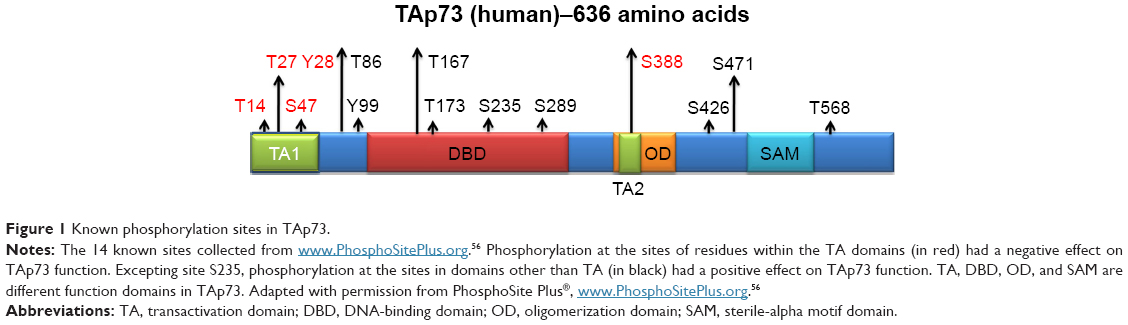 | Figure 1 Known phosphorylation sites in TAp73. |
 | Table 3 Distribution of total 14 known phosphorylation sites |
PLK2 binds to enriched TAp73
To test our hypothesis, co-IP experiments between endogenous PLK2 and TAp73 were conducted in human tumor cells to determine whether PLK2 and TAp73 are capable of physically binding to each other. UM-SCC cell lines (UM-SCC1 and UM-SCC22A) originate from epithelial tumor cells, whereas osteosarcoma cell lines (Saos2 and MG63) originate from mesenchymal tumor cells. Counter to our expectation, co-IP between PLK2 and TAp73 was negative in Saos2 and UM-SCC1 under normal culture conditions (Figure 2A). However, PLK2 does bind directly to TAp73 in MG63 and UM-SCC22A. In addition, the process was also apparent in Saos2 and UM-SCC1 when pretreated for 12 hours with the DNA-damaging drugs CDDP or ADM (Figure 2B). This was surprising because we previously found high levels of TAp73 in UM-SCC 22A cell lines and low levels of TAp73 in UM-SCC1 cell lines.18 We measured the expression level of TAp73 in these cells under normal culture conditions and found that MG63 was similar to UM-SCC22A, expressing a high level of TAp73 compared with Saos2, a result that was similar to the levels found in UM-SCC1 (Figure 3A and B).
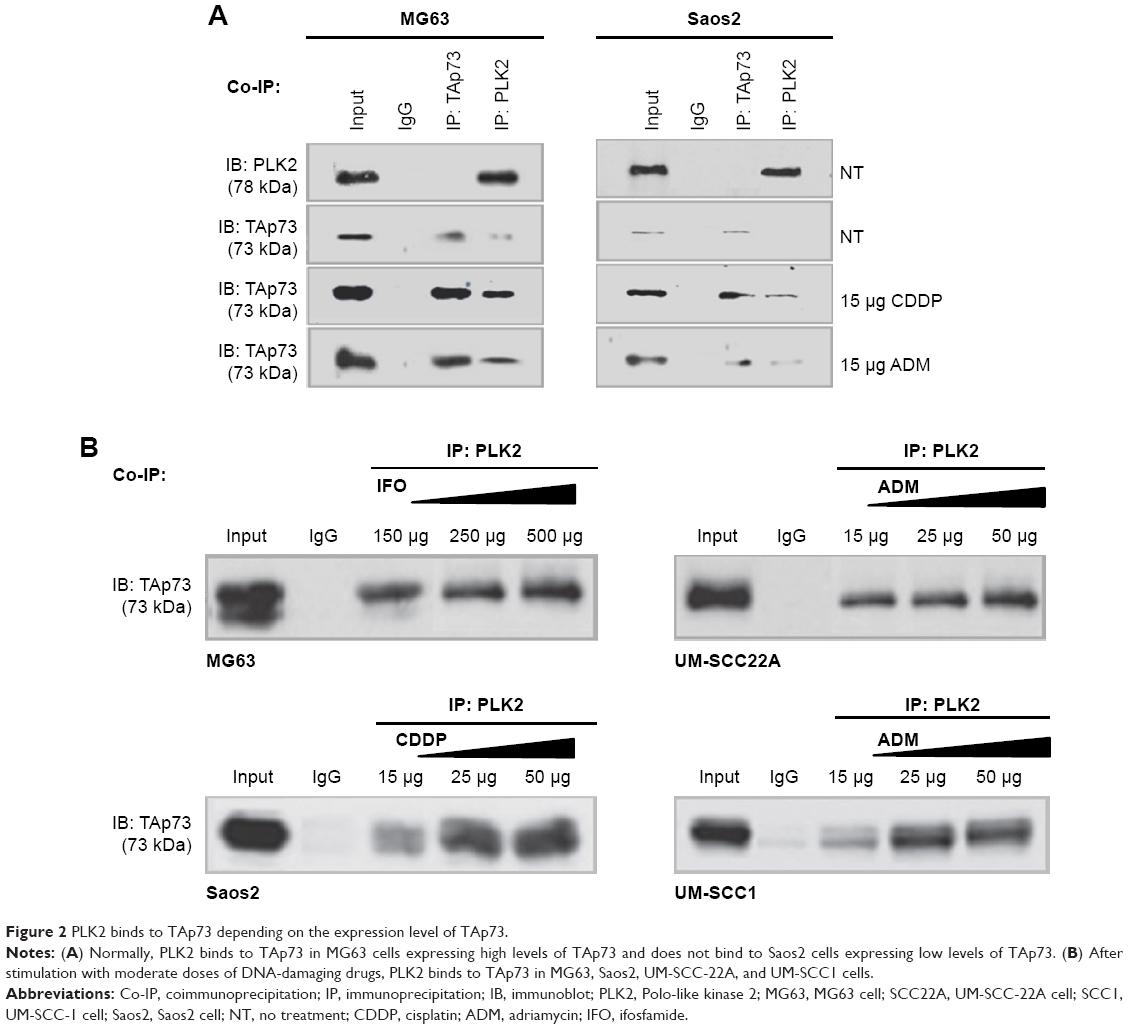 | Figure 2 PLK2 binds to TAp73 depending on the expression level of TAp73. |
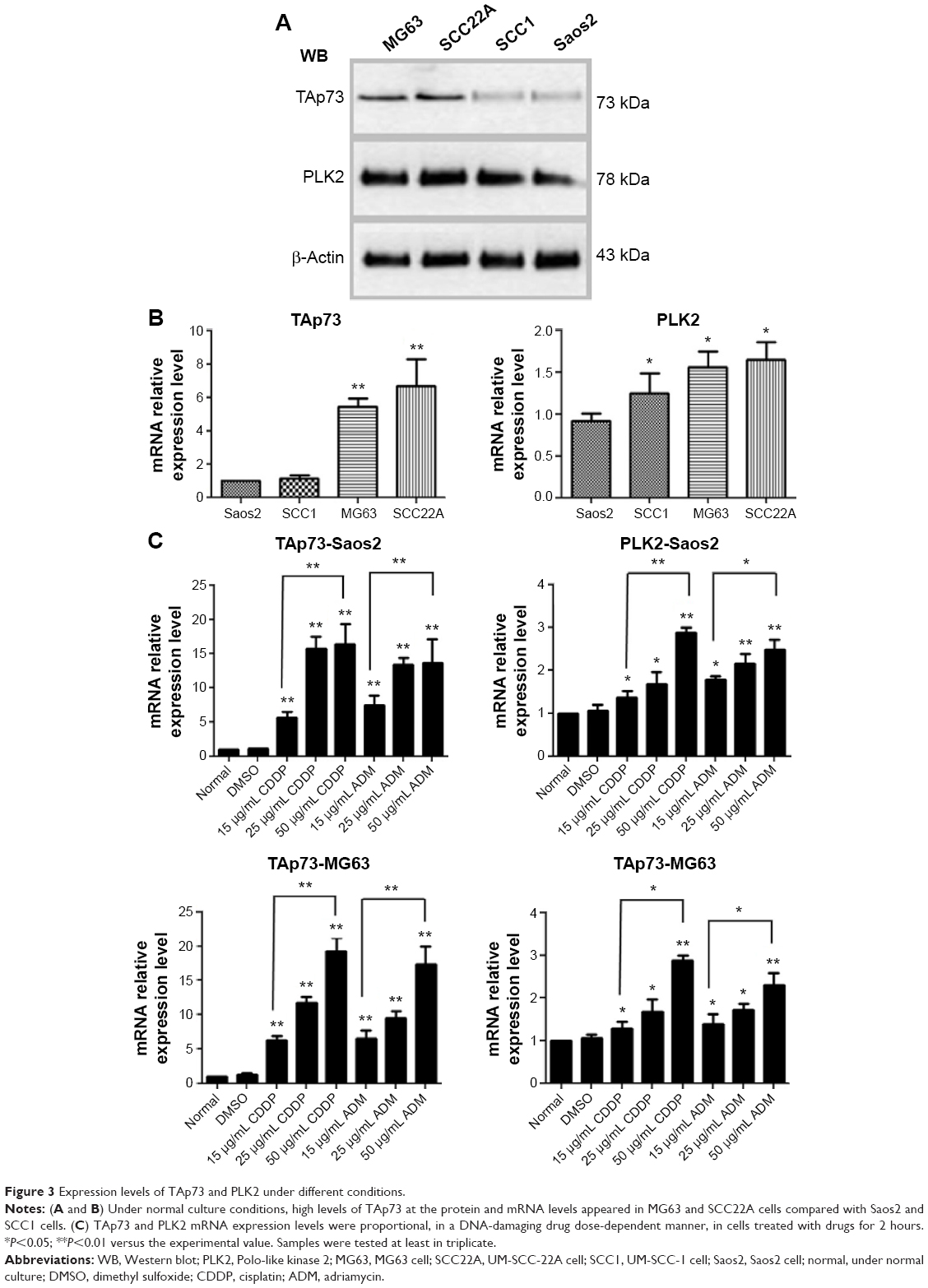 | Figure 3 Expression levels of TAp73 and PLK2 under different conditions. |
Previous research has demonstrated that DNA damage upregulates endogenous TAp7326 and PLK227 expression. We determined that DNA-damaging drugs significantly upregulated both TAp73 and PLK2 in a dose-dependent manner (Figure 3C). We then applied, at random, the DNA-damaging drugs CDDP, ADM, and ifosfamide to stimulate upregulation of endogenous TAp73 and PLK2. Our co-IP results indicated that PLK2 binds to TAp73 in cells with high levels of TAp73.
PLK2 does catalyze TAp73 phosphorylation at Ser48 residue
We investigated whether PLK2 was able to phosphorylate TAp73 protein via Phos-tag™ WB assay. We tested this in UM-SCC1 cells with low expression levels of TAp73. Endogenous phosphorylation of TAp73 in UM-SCC1 clearly occurred when cells whose Tap73 was lowered by PLK2 inhibition were stimulated using CDDP or ADM (50 μg/mL). We confirmed these results in MG63 cells with high expression levels of TAp73. In fact, in MG63 cells treated with or without DNA-damaging drugs, PLK2 inhibition using siRNA or the selective inhibitor ELN582646 reduced endogenous phosphorylation of TAp73 (Figure 4A). Because many kinases promote TAp73 phosphorylation, an in vitro phosphorylation assay was performed. Flag-PLK2 and Flag-TAp73 were detected (Figure 4B), purified, and then incubated together in kinase buffer for the indicated amount of time. Identical amounts of these samples were subjected to Phos-tag WB assay, which demonstrated that ectopic Flag-TAp73 phosphorylation occurred and also increased in a time-dependent way (Figure 4C). To the contrary, we also found that phosphorylation levels were close to zero in purified mutant TAp73 (S48A) incubated with Flag-PLK2 when it was subjected to Phos-tag™ WB assay (Figure 4D).
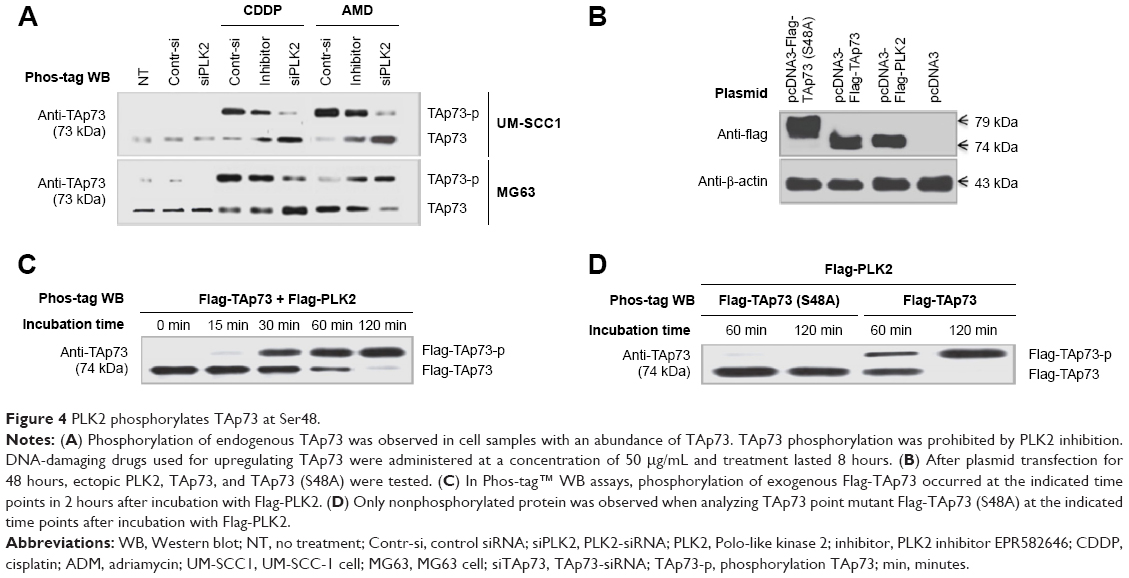 | Figure 4 PLK2 phosphorylates TAp73 at Ser48. |
PLK2 suppresses the transcriptional activity of enriched TAp73
Although previous experiments have shown that PLK2 phosphorylates higher levels of TAp73, it remains to be determined whether or not the transcriptional activity of TAp73 is suppressed. As mentioned earlier, two of the main target genes of TAp73 are p21 and PUMA, which are related to the cell cycle and cell apoptosis. We used RT-PCR to assess the transcriptional activity of TAp73 in MG63 and Saos2. The mRNA expression levels of p21 and PUMA increased in response to PLK2 inhibition in all cells treated with CDDP or ADM (Figure 5A–D). In MG63 cells, PLK2 inhibition alone was able to increase the mRNA expression levels of p21 and PUMA (Figure 5C and D). Moreover, the increases in p21 and PUMA expression were greater in the copresence of PLK2 inhibition and DNA-damaging drugs when compared with the increase in the presence of DNA-damaging drugs alone, and this increase was markedly attenuated by pretreatment with siTAp73. However, the PLK2-dependent increase in p21 and PUMA expression did not occur in Saos2 cells that are left untreated with DNA-damaging drugs (Figure 5A and B).
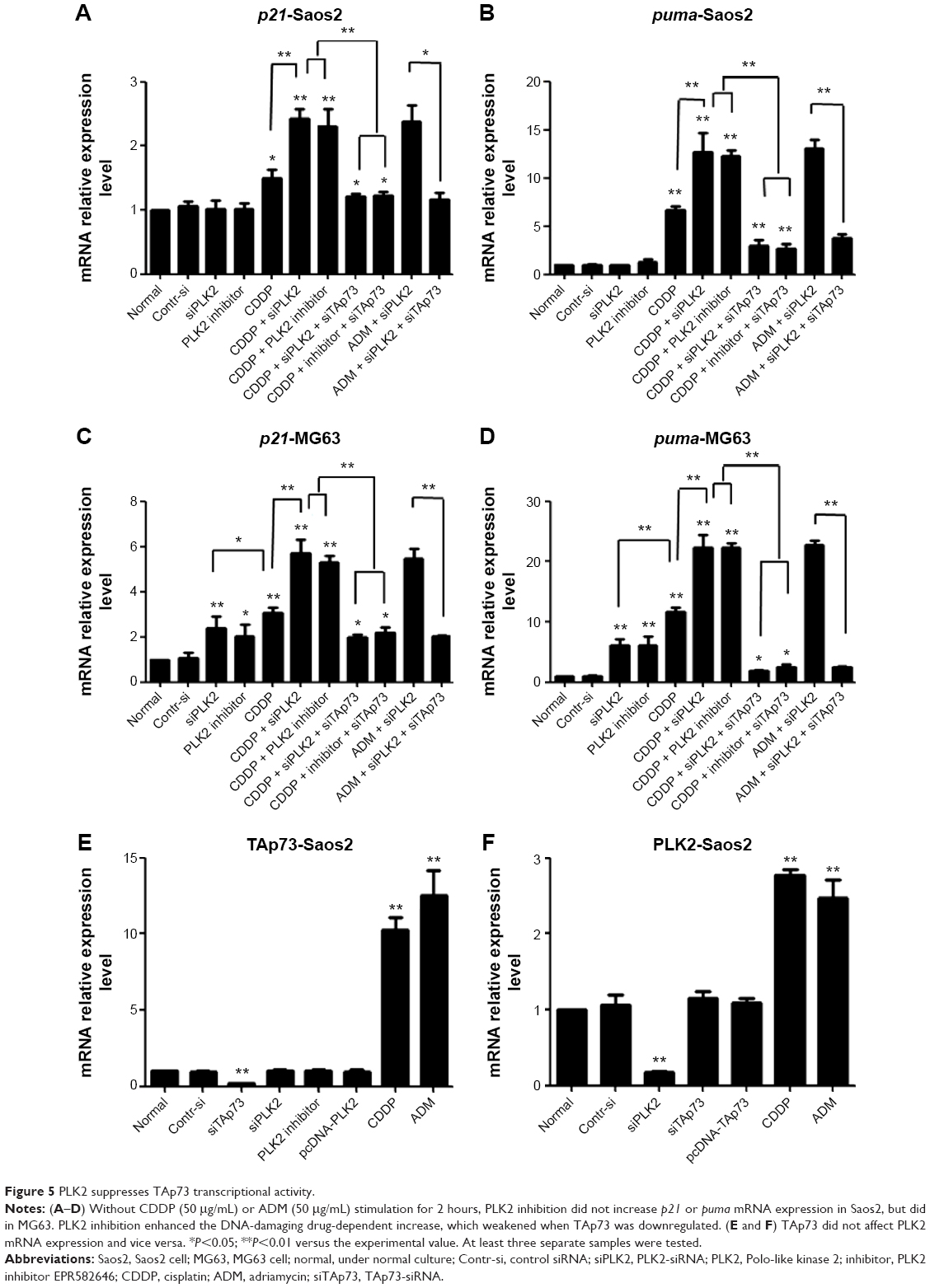 | Figure 5 PLK2 suppresses TAp73 transcriptional activity. |
The levels of protein expression of p21 and PUMA in MG63 were also tested by WB (Figure 6A). The resultant levels were similar to that of mRNA. A further immunofluorescence confocal microscopy experiment on UM-SCC1 treated with 50 μg/mL ADM showed that PLK2 interferes with TAp73 translocation to the nucleus (Figure 7). These results suggest that PLK2 suppresses TAp73 transcriptional activity even when there is a high level of TAp73.
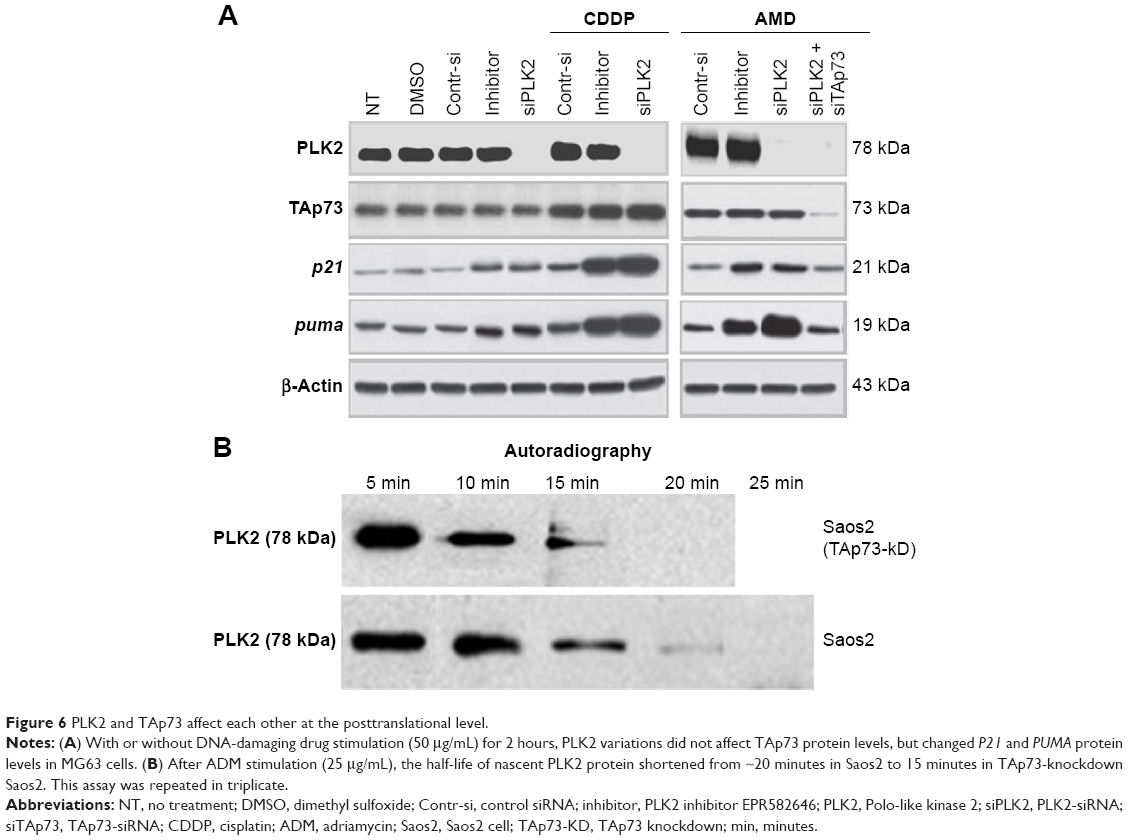 | Figure 6 PLK2 and TAp73 affect each other at the posttranslational level. |
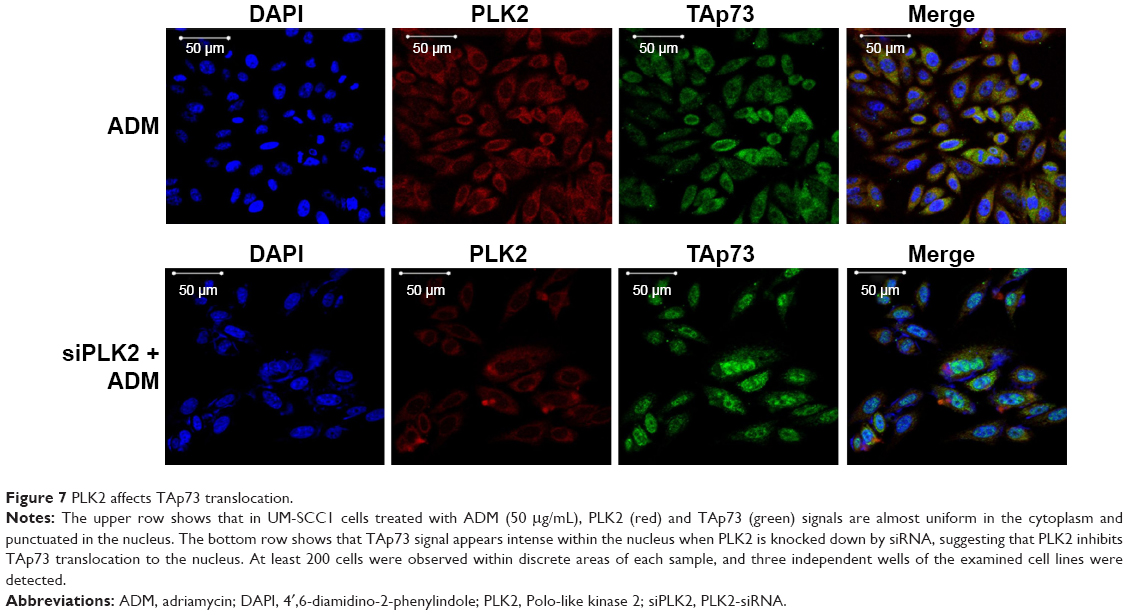 | Figure 7 PLK2 affects TAp73 translocation. |
Cross talk between PLK2 and TAp73 proteins occurs at a posttranslational level
A previous study reported that the TAp73 family protein p53 can directly transcribe PLK2 expression under certain conditions. However, whether or not TAp73 directly transcribes PLK2 expression remains unclear. When compared with cells under normal conditions, PLK2 mRNA expression did not vary when TAp73 was either overexpressed or silenced by siRNA in MG63 and Saos2 (Figure 5E) and vice versa (Figure 5F), indicating that TAp73 does not directly transcribe PLK2 mRNA expression. Likewise, PLK2 expression did not regulate TAp73 mRNA expression. We recorded similar results in HNSCC cell lines (data not shown). Thus, PLK2 and TAp73 do not directly cross talk at gene transcriptional and translational levels (Figures 5 and 6). Since PLK2 is able to phosphorylate TAp73 at the posttranslational level, we considered whether TAp73 had the capability directly to affect PLK2 at a posttranslational level. We measured nascent PLK2 protein levels by autoradiography assay of Saos2 in the presence of CDDP stimuli and found that the half-life of nascent PLK2 from Saos2 was prolonged when compared with that of nascent PLK2 from Saos2 cells where PAP73 had been silenced (Figure 6B). This suggested that the cross talk between PLK2 and TAp73 may occur at a posttranslational level.
PLK2 inhibition arrests cell cycle in the G1 phase and increases apoptosis through rescuing enriched TAp73 activity
Because PLK2 regulates the cell cycle G1/S transition and PLK2 prohibition in the presence of rich abundance of TAp73 increased p21 and PUMA expression, both of which play a key role in cell cycle arrest and cell apoptosis, we determined the physiological effects of PLK2 on TAp73 in MG63 with high levels of TAp73. The results of this analysis showed a significant increase in the proportion of G1 phase cells in the copresence of CDDP and PLK2 inhibition when compared with that resulting from CDDP treatment or PLK2 inhibition alone (Figure 8A and B). Consistent with FCM results, PLK2 inhibition increased MG63 cell apoptosis (Figure 8C). Additionally, we found that PLK2 inhibition enhances the effect of DNA-damaging drug-induced cell cycle arrest and apoptosis (Figure 8B and C). Cosilencing of PLK2 and TAp73 in cells resulted in a weakening of PLK2 inhibition of cell cycle arrest and cell apoptosis. These results suggest that PLK2 inhibition sensitized tumor cells to DNA-damaging drugs by restoring TAp73 activity.
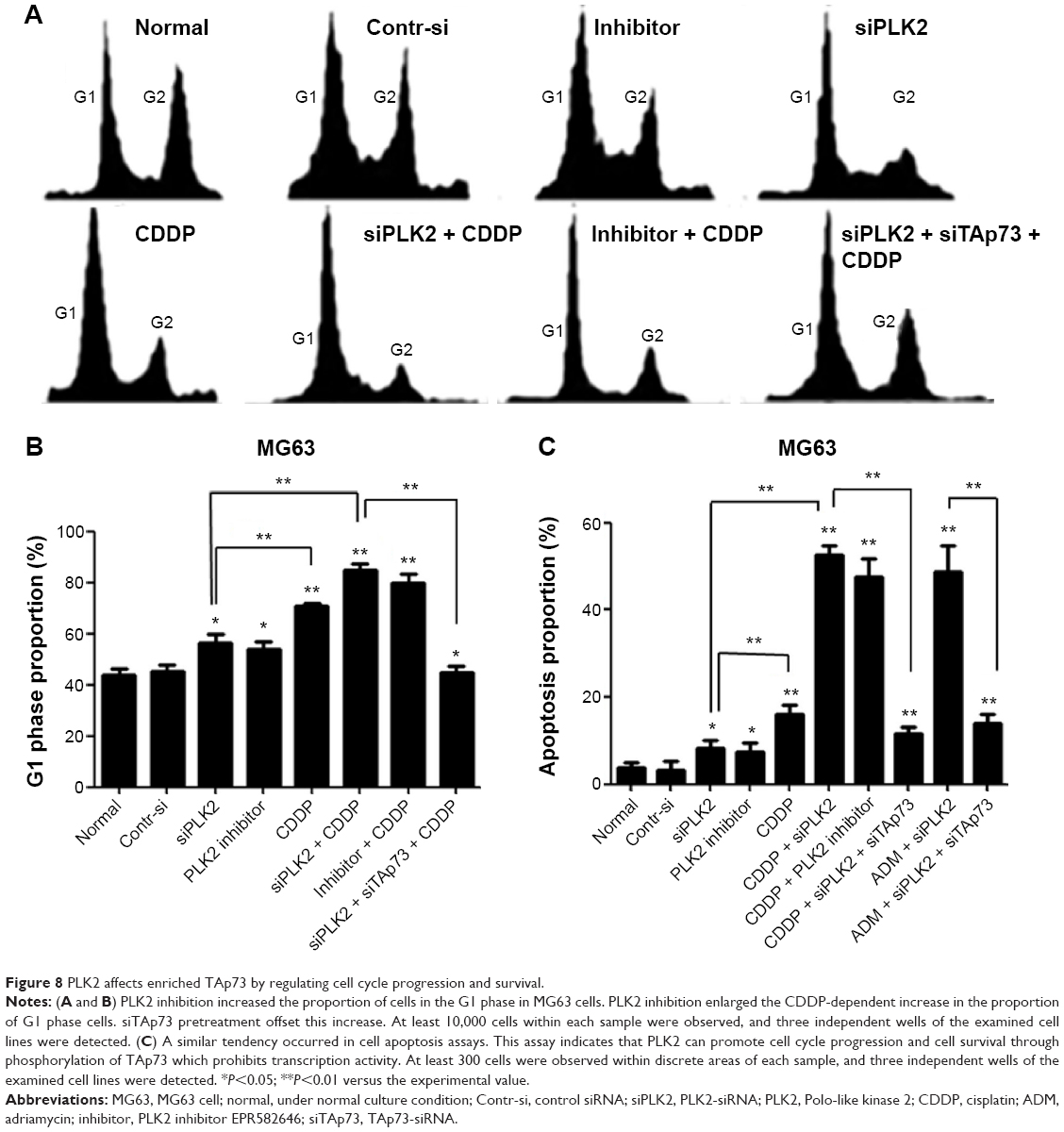 | Figure 8 PLK2 affects enriched TAp73 by regulating cell cycle progression and survival. |
Discussion
Variations in TAp73 abundance and phosphorylation of TAp73 can induce changes in tumor activity. In this study, we found that a range of DNA-damaging drugs stimulates the upregulation of TAp73 and PLK2 in several human tumor cell lines, indicating that their effect is broadly similar across these cell lines. Regarding TAp73 phosphorylation, we demonstrated that, like CK2, PLK2 and TAp73 directly bind to each other, and PLK2 phosphorylates TAp73 both in vivo and in vitro.
It has already been established that proteins such as TAp73 contain many functional domains.28,29 An analysis of previous studies indicated that changes to the phosphorylation activity of the TA1 domain in TAp73 often impair the transcriptional activity of TAp73.30–39 On the other hand, phosphorylation of other domains usually results in an increment in TAp73 antitumor activity. Thus, the identification of the PLK2 phosphorylation site within TAp73 domains is a vital clue to TAp73 function. In this research, in agreement with our computational analysis, we found that PLK2 phosphorylates Ser48, which is located in the TA1 domain. Furthermore, RT-PCR and WB results also demonstrated that PLK2 inhibition significantly increases the activity of TAp73 even where it is at high levels. A previous study40 has demonstrated that the TAp73 isoform ΔNp73, without the TA domain, has the opposite function to TAp73. This manifestation may, in part, explain variations in functionality after TAp73 phosphorylation in the TA1 domain.
Accumulating evidence, such as gene knockout research,41–43 DNA damaging response in human tumor cells,26,44 and up- and downregulation in tumors adapting to cellular contexts strongly suggest a close correlation between PLK2 and TAp73.45–48 In this study, we found that PLK2 phosphorylates TAp73 even at high levels of the latter, suppresses TAp73 transcriptional activity, and consequently causes cell cycle arrest and cell apoptosis. TAp73 may also stabilize PLK2 by prolonging the half-life of PLK2, which implies that TAp73 may potentiate PLK2 to regulate cell cycle progression.
As mentioned earlier, whether PLK2 plays a promotional or suppressant role in human tumors remains unclear. Previous research has demonstrated that deregulation of PLK2 results in a malignant phenotype.49,50 Recently, Li et al7 demonstrated that PLK2 is essential for the survival of tumor cells. In addition, Matthew et al51 observed that PLK2 inhibition sensitizes tumor cells to DNA-damaging drugs, a finding mirrored in our research. Furthermore, PLK2 is able to activate PLK1, suppress p53,52 and promote the oncogenic effects of mutant p53 in tumor cells.53 The aforementioned studies demonstrate that PLK2 has a tumor-promoting role. Thus, our results are consistent with the finding that PLK2 promotes tumors by prohibiting TAp73 activity, even where there is a rich abundance of TAp73.
Because numerous malignant tumors exhibited TAp73 overexpression in clinical patients,54,55 high levels of TAp73 are usually regarded as a marker of malignant tumors. One possible explanation for this is that TAp73 expression in tumors is upregulated under certain conditions, but its activity is inhibited. To confirm this observation, researchers have used several methods to restore TAp73 activity in tumors.55 In our study, we found that PLK2 inhibition is capable of restoring TAp73 activity, a finding that provides extra insight into this phenomenon.
Conclusion
In summary, we reveal here a novel mechanism of PLK2 as a prosurvival agent in tumor cells with a rich abundance of TAp73, where PLK2 phosphorylates the Ser48 residue of TAp73 and blocks TAp73 transcriptional activity (Figure 9). PLK2 inhibition may thus sensitize tumor cells to the effects of DNA-damaging drugs by rescuing TAp73 activity. Our research may aid the quest for antitumor therapies in tumors characterized by TAp73 overexpression.
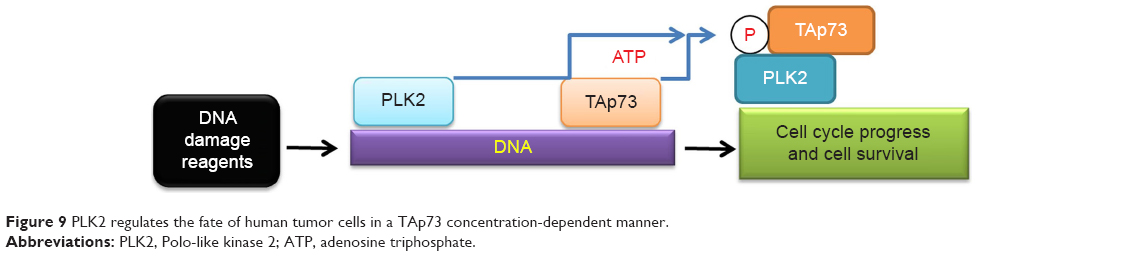 | Figure 9 PLK2 regulates the fate of human tumor cells in a TAp73 concentration-dependent manner. |
Acknowledgments
This work was supported by National Natural Science Foundation of China (grant numbers 81372869, 81172555, and 81572628 to ZBH, AMJ, and HL) and National Natural Science Foundation of Guangdong Province (grant number S2013010016052 to HL). The authors thank the State Key Laboratory of Respiratory Diseases and Professor Rong Zhou, National Clinical Research Center for Respiratory Disease, Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China, and they also thank the Dongguan Institute of Pediatrics and Director Xiaomei Lu, People’s Republic of China.
Disclosure
ZH, ZX, and XL are equal contributors in this work and should be considered as first co-authors. AJ and HL contributed equally to this work as senior authors. The authors report no conflicts of interest in this work.
References
Du W, Jiang P, Mancuso A, et al. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat Cell Biol. 2013;15(8):991–1000. | ||
Llamazares S, Moreira A, Tavares A, et al. Polo encodes a protein kinase homolog required for mitosis in Drosophila. Genes Dev. 1991;5(12A):2153–2165. | ||
Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt-Dias M. Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol. 2014;15(7):433–452. | ||
Raab M, Krämer A, Hehlgans S, et al. Mitotic arrest and slippage induced by pharmacological inhibition of Polo-like kinase 1. Mol Oncol. 2015;9(1):140–154. | ||
Sanhaji M, Ritter A, Belsham HR, et al. Polo-like kinase 1 regulates the stability of the mitotic centromere-associated kinesin in mitosis. Oncotarget. 2014;5(10):3130–3144. | ||
Bowers S, Truong AP, Ye M, et al. Design and synthesis of highly selective, orally active Polo-like kinase-2 (Plk-2) inhibitors. Bioorg Med Chem Lett. 2013;23(9):2743–2749. | ||
Li J, Ma W, Wang PY, Hurley PJ, Bunz F, Hwang PM. Polo-like kinase 2 activates an antioxidant pathway to promote the survival of cells with mitochondrial dysfunction. Free Radic Biol Med. 2014;73:270–277. | ||
Seet BT, Dikic I, Zhou MM, Pawson T. Reading protein modifications with interaction domains. Nat Rev Mol Cell Biol. 2006;7(7):473–483. | ||
Ubersax JA, Ferrell JE. Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8(7):530–541. | ||
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. | ||
Lu H, Yan C, Quan XX, et al. CK2 phosphorylates and inhibits TAp73 tumor suppressor function to promote expression of cancer stem cell genes and phenotype in head and neck cancer. Neoplasia. 2014;16(10):789–800. | ||
Songyang Z, Lu KP, Kwon YT, et al. A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol Cell Biol. 1996;16(11):6486–6493. | ||
Mbefo MK, Paleologou KE, Boucharaba A, et al. Phosphorylation of synucleins by members of the Polo-like kinase family. J Biol Chem. 2010;285(4):2807–2822. | ||
Schreurs S, Gerard M, Derua R, et al. In vitro phosphorylation does not influence the aggregation kinetics of WT α-synuclein in contrast to its phosphorylation mutants. Int J Mol Sci. 2014;15(1):1040–1067. | ||
Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31(13):3635–3641. | ||
Liu Z, Ren J, Cao J, et al. Systematic analysis of the Plk-mediated phosphoregulation in eukaryotes. Brief Bioinform. 2013;14(3):344–360. | ||
Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43(Database issue):D512–D520. | ||
Lu H, Yang X, Duggal P, et al. TNF-alpha promotes c-REL/DeltaNp63alpha interaction and TAp73 dissociation from key genes that mediate growth arrest and apoptosis in head and neck cancer. Cancer Res. 2011;71(21):6867–6877. | ||
Thakur A, Sun Y, Bollig A, et al. Anti-invasive and antimetastatic activities of ribosomal protein S6 kinase 4 in breast cancer cells. Clin Cancer Res. 2008;14(14):4427–4436. | ||
Fitzgerald K, Bergeron M, Willits C, et al. Pharmacological inhibition of Polo like kinase 2 (PLK2) does not cause chromosomal damage or result in the formation of micronuclei. Toxicol Appl Pharmacol. 2013;269(1):1–7. | ||
Aubele DL, Hom RK, Adler M, et al. Selective and brain-permeable Polo-like kinase-2 (Plk-2) inhibitors that reduce α-synuclein phosphorylation in rat brain. ChemMedChem. 2013;8(8):1295–1313. | ||
Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics. 2006;5(4):749–757. | ||
Goto H, Kosako H, Tanabe K, et al. Phosphorylation of vimentin by rho-associated kinase at a unique amino-terminal site that is specifically phosphorylated during cytokinesis. J Biol Chem. 1998;273(19):11728–11736. | ||
Ma S, Liu MA, Yuan YL, Erikson RL. The serum-inducible protein kinase Snk is a G1 phase polo-like kinase that is inhibited by the calcium- and integrin-binding protein CIB. Mol Cancer Res. 2003;1(5):376–384. | ||
Fu L, Liu N, Han Y, Xie C, Li Q, Wang E. ADAM10 regulates proliferation, invasion, and chemoresistance of bladder cancer cells. Tumour Biol. 2014;35(9):9263–9268. | ||
Flores ER, Tsai KY, Crowley D, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416(6880):560–564. | ||
Shimizu-Yoshida Y, Sugiyama K, Rogounovitch T, et al. Radiation-inducible hSNK gene is transcriptionally regulated by p53 binding homology element in human thyroid cells. Biochem Biophys Res Commun. 2001;289(2):491–498. | ||
Okada K, Kanaya S, Asai K. Accurate extraction of functional associations between proteins based on common interaction partners and common domains. Bioinformatics. 2005;21(9):2043–2048. | ||
Logotheti S, Pavlopoulou A, Galtsidis S, Vojtesek B, Zoumpourlis V. Functions, divergence and clinical value of TAp73 isoforms in cancer. Cancer Metastasis Rev. 2013;32(3–4):511–534. | ||
Burge S, Teufel DP, Townsley FM, Freund SM, Bycroft M, Fersht AR. Molecular basis of the interactions between the p73 N terminus and p300: effects on transactivation and modulation by phosphorylation. Proc Natl Acad Sci U S A. 2009;106(9):3142–3147. | ||
Koida N, Ozaki T, Yamamoto H, et al. Inhibitory role of Plk1 in the regulation of p73-dependent apoptosis through physical interaction and phosphorylation. J Biol Chem. 2008;283(13):8555–8563. | ||
Paliwal P, Radha V, Swarup G. Regulation of p73 by Hck through kinase-dependent and independent mechanisms. BMC Mol Biol. 2007;8:45. | ||
Gonzalez S, Prives C, Cordon-Cardo C. p73alpha regulation by Chk1 in response to DNA damage. Mol Cell Biol. 2003;23(22):8161–8171. | ||
Benosman S, Meng X, Von Grabowiecki Y, et al. Complex regulation of p73 isoforms after alteration of amyloid precursor polypeptide (APP) function and DNA damage in neurons. J Biol Chem. 2011;286(50):43013–43025. | ||
Merlo P, Fulco M, Costanzo A, et al. A role of p73 in mitotic exit. J Biol Chem. 2005;280(34):30354–30360. | ||
Gaiddon C, Lokshin M, Gross I, et al. Cyclin-dependent kinases phosphorylate p73 at threonine 86 in a cell cycle-dependent manner and negatively regulate p73. J Biol Chem. 2003;278(30):27421–27431. | ||
Niikura Y, Ogi H, Kikuchi K, Kitagawa K. BUB3 that dissociates from BUB1 activates caspase-independent mitotic death (CIMD). Cell Death Differ. 2010;17(6):1011–1024. | ||
Yuan ZM, Shioya H, Ishiko T, et al. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature. 1999;399(6738):814–817. | ||
Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature. 1999;399(6738):809–813. | ||
Bunjobpol W, Dulloo I, Igarashi K, Concin N, Matsuo K, Sabapathy K. Suppression of acetylpolyamine oxidase by selected AP-1 members regulates DNp73 abundance: mechanistic insights for overcoming DNp73-mediated resistance to chemotherapeutic drugs. Cell Death Differ. 2014;21(8):1240–1249. | ||
Yang A, Walker N, Bronson R, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404(6773):99–103. | ||
Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289(5477):304–306. | ||
Niklison-Chirou MV, Steinert JR, Agostini M, et al. TAp73 knockout mice show morphological and functional nervous system defects associated with loss of p75 neurotrophin receptor. Proc Natl Acad Sci U S A. 2013;110(47):18952–18957. | ||
Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol. 2011;12(4):259–265. | ||
Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res. 2004;2(7):371–386. | ||
Syed N, Coley HM, Sehouli J, et al. Polo-like kinase Plk2 is an epigenetic determinant of chemosensitivity and clinical outcomes in ovarian cancer. Cancer Res. 2011;71(9):3317–3327. | ||
Tan LB, Chen KT, Yuan YC, Liao PC, Guo HR. Identification of urine PLK2 as a marker of bladder tumors by proteomic analysis. World J Urol. 2010;28(1):117–122. | ||
Syed N, Smith P, Sullivan A, et al. Transcriptional silencing of Polo-like kinase 2 (SNK/PLK2) is a frequent event in B-cell malignancies. Blood. 2006;107(1):250–256. | ||
Eckerdt F, Yuan J, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. 2005;24(2):267–276. | ||
Matsumoto T, Wang PY, Ma W, Sung HJ, Matoba S, Hwang PM. Polo-like kinases mediate cell survival in mitochondrial dysfunction. Proc Natl Acad Sci U S A. 2009;106(34):14542–14546. | ||
Matthew EM, Yen TJ, Dicker DT, et al. Replication stress, defective S-phase checkpoint and increased death in Plk2-deficient human cancer cells. Cell Cycle. 2007;6(20):2571–2578. | ||
Liu X, Erikson RL. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci U S A. 2003;100(10):5789–5794. | ||
Shen T, Li Y, Yang L, et al. Upregulation of Polo-like kinase 2 gene expression by GATA-1 acetylation in human osteosarcoma MG-63 cells. Int J Biochem Cell Biol. 2012;44(2):423–429. | ||
Zhao W, Zhang B, Guo X, et al. Expression of Ki-67, Bax and p73 in patients with hilar cholangiocarcinoma. Cancer Biomark. 2014;14(4):197–202. | ||
Moll UM. The role of p63 and p73 in tumor formation and progression: coming of age toward clinical usefulness. Commentary re: Koga F et al, Impaired p63 expression associates with poor prognosis and uroplakin III expression in invasive urothelial carcinoma of the bladder. Clin. Cancer Res. 9: 5501–5507, 2003, and Puig P et al, p73 Expression in human normal and tumor tissues: loss of p73alpha expression is associated with tumor progression in bladder Cancer. Clin. Cancer Res. 9: 5642–5651, 2003. Clin Cancer Res. 2003;9(15):5437–5441. | ||
PhosphoSitePlus: a protein modification resource [webpage on the Internet]. Cell Signaling Technology, Inc.; 2013. Available from: www.PhosphoSitePlus.org. Accessed November 10, 2015. |
Supplementary material
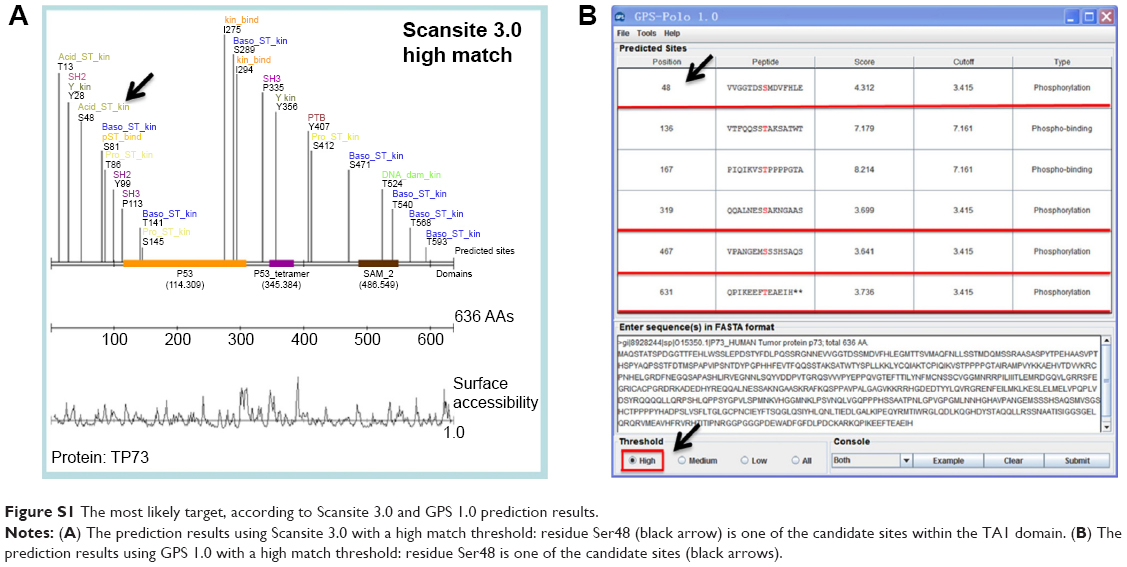 | Figure S1 The most likely target, according to Scansite 3.0 and GPS 1.0 prediction results. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.