")
Back to Journals » International Journal of Nanomedicine » Volume 11
Polarity-sensitive nanocarrier for oral delivery of Sb(V) and treatment of cutaneous leishmaniasis
Authors Lanza J, Fernandes F, Corrêa-Júnior JD, Vilela J, Magalhães-Paniago R, Ferreira L, Andrade M , Demicheli C, Melo M, Frezard F
Received 6 February 2016
Accepted for publication 23 March 2016
Published 25 May 2016 Volume 2016:11 Pages 2305—2318
DOI https://doi.org/10.2147/IJN.S105952
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Juliane S Lanza,1 Flaviana R Fernandes,1 José D Corrêa-Júnior,2 José MC Vilela,3 Rogério Magalhães-Paniago,4 Lucas AM Ferreira,5 Margareth S Andrade,3 Cynthia Demicheli,6 Maria N Melo,7 Frédéric Frézard1
1Department of Physiology and Biophysics, 2Department of Morphology, Instituto de Ciências Biológicas (ICB), Universidade Federal de Minas Gerais (UFMG), 3Innovation and Technology Center SENAI FIEMG – Campus CETEC, 4Department of Physics, Instituto de Ciências Exatas (ICEX), 5Department of Pharmaceutical Products, Faculty of Pharmacy, Universidade Federal de Minas Gerais (UFMG), 6Department of Chemistry, Instituto de Ciências Exatas (ICEX), 7Department of Parasitology, Instituto de Ciências Biológicas (ICB), Universidade Federal de Minas Gerais (UFMG), Belo Horizonte, Brazil
Abstract: There is a great need for orally active drugs for the treatment of the neglected tropical disease leishmaniasis. Amphiphilic Sb(V) complexes, such as 1:3 Sb–N-octanoyl-N-methylglucamide complex (SbL8), are promising drug candidates. It has been previously reported that SbL8 forms kinetically stabilized nanoassemblies in water and that this simple dispersion exhibits antileishmanial activity when given by oral route to a murine model of visceral leishmaniasis. The main objective of the present work was to interfere in the structural organization of these nanoassemblies so as to investigate their influence on the oral bioavailability of Sb, and ultimately, optimize an oral formulation of SbL8 for the treatment of cutaneous leishmaniasis. The structural organization of SbL8 nanoassemblies was manipulated through addition of propylene glycol (PG) to the aqueous dispersion of SbL8. The presence of 50% (v/v) PG resulted in the loss of hydrophobic microenvironment, as evidenced by fluorescence probing. However, nanostructures were still present, as demonstrated by dynamic light scattering, small-angle X-ray scattering, and atomic force microscopy (AFM). A remarkable property of these nanoassemblies, as revealed by AFM analysis, is the flexibility of their supramolecular organization, which showed changes as a function of the solvent and substrate polarities. The formulation of SbL8 in 1:1 water:PG given orally to mice promoted significantly higher and more sustained serum levels of Sb, when compared to SbL8 in water. The new formulation, when given as repeated doses (200 mg Sb/kg/day) to BALB/c mice infected with Leishmania amazonensis, was significantly more effective in reducing the lesion parasite burden, compared to SbL8 in water, and even, the conventional drug Glucantime® given intraperitoneally at the same dose. In conclusion, this work introduces a new concept of polarity-sensitive nanocarrier that was successfully applied to optimize an oral formulation of Sb(V) for treating cutaneous leishmaniasis.
Keywords: propylene glycol, antimony, Leishmania amazonensis, AFM, amphiphilic complex, SAXS
Introduction
Even though pentavalent antimonials, including meglumine antimoniate (Glucantime®) and sodium stibogluconate (Pentostam), are still the first-line drugs in most developing countries for treatment of all forms of leishmaniasis,1–5 they have several limitations. These drugs have to be given parenterally, daily, for at least 3 weeks. Antimony therapy is often accompanied by local pain during intramuscular injections and by systemic side effects, requiring very careful medical supervision.1 Typical side effects include nausea, myalgia, diarrhea, skin rashes, and renal and hepatotoxicity, together with the most severe pancreatitis and cardiotoxicity.1,2,5 All these factors contribute to compliance difficulties, and eventually, treatment failures.2,6 Depending on Leishmania parasite species, clinical manifestations of the disease can involve the viscera (liver, spleen, and bone marrow) or the skin, with local (cutaneous), diffuse (diffuse cutaneous), or disfiguring lesions (mucocutaneous).7–9
So far, miltefosine is the only oral drug available for leishmaniasis, but its use is restricted to the visceral form. Furthermore, teratogenicity restrains the use of this drug in women of childbearing age.10,11 Amphotericin B is another clinically important antileishmanial agent. It is effective when given parenterally, and liposomal amphotericin B (AmBisome®) has emerged as the safest injectable formulation.12 Previous studies have reported the efficacy of novel oral formulations of amphotericin B in experimental models of visceral leishmaniasis: a lipid-based formulation13 and amphotericin B attached to functionalized carbon nanotubes.14 However, these oral formulations required much higher doses of amphotericin B, compared to parenteral AmBisome®, and showed efficacy using multiple-dose regimen, in contrast to AmBisome® that is effective at a single dose.13,14 Furthermore, no report was found on the efficacy of these formulations in experimental models of cutaneous leishmaniasis (CL). Thus, there is a great need for orally or topically active drug for treatment of cutaneous and mucocutaneous leishmaniasis.5,15,16
Our group recently reported an effective oral delivery strategy for pentavalent antimonials, based on the formation of an amphiphilic Sb(V) complex.17 Such a complex was obtained from the reaction of Sb(V) with a nonionic surfactant from the N-alkyl-N-methylglucamide series, N-octanoyl-N-methylglucamide (L8), at a molar ratio of 1:3. The oral formulation of 1:3 Sb–N-octanoyl-N-methylglucamide complex (SbL8) in water exhibited an improved bioavailability of Sb in mice, when compared to Glucantime® given orally at the same Sb dose. This formulation showed antileishmanial activity by the oral route in a murine model of visceral leishmaniasis, but still to a lower extent than parenteral Glucantime®.17 The ability of the oral formulation to deliver Sb to the main site of infection, in this case, the liver, was further demonstrated. The formation of nanoassemblies in SbL8 aqueous dispersion and the identification of hydrophobic microenvironment in this system were also evidenced through transmission electron microscopy (TEM) analysis and fluorescence spectroscopy. Interestingly, these nanoassemblies showed improved thermodynamic and kinetic stabilities, compared to conventional L8 surfactant micelles.17 These data, together with the ability of Sb(V) to act as a polymerizing agent,18 led us to propose that Sb(V) may stabilize the L8 nanoassemblies through multiple Sb(−O−C)n bonds.
In the present work, our main objective was to manipulate the structural organization of these nanoassemblies so as to investigate their influence on the oral bioavailability of Sb and the efficacy of SbL8 in a murine model of CL. Such effect was achieved by reducing the solvent polarity through addition of propylene glycol (PG), resulting in changes in the supramolecular organization of SbL8 and the oral drug bioavailability.
Materials and methods
Materials
N-Octanoyl-N-methylglucamide (L8, 98%), potassium hexahydroxoantimoniate [KSb(OH)6], and 1,6-diphenylhexatriene (DPH) were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). PG was obtained from Labsynth (Diadema, Brazil). The reference drug, Glucantime® (Sanofi-Aventis, Bridgewater, NJ, USA), was a courtesy of health department of Sete Lagoas city (Minas Gerais, Brazil) for exclusive use in this research. Double-distilled deionized water was used throughout all the experiments.
Animals and parasites
Swiss female mice (4–6 weeks old, 20–25 g) were used in pharmacokinetic studies. BALB/c female mice (4–6 weeks old, 20–25 g) were used to establish a murine model of CL and perform efficacy assays. The animals were obtained from the CEBIO of the Instituto de Ciências Biológicas (ICB), Universidade Federal de Minas Gerais (UFMG). Free access to a standard diet was allowed, and tap water was supplied ad libitum. The studies involving animals were approved by the Ethical Committee for Animal Experimentation of the UFMG with protocol numbers 215/2009 and 318/2013.
Promastigotes of Leishmania (Leishmania) amazonensis, strain MHOM/BR/1989/BA199, were obtained from the cryopreservation bank of the Leishmania Biology Laboratory at ICB, UFMG. The Leishmania isolate was obtained from a patient who presented diffuse form of CL in the state of Maranhão, Brazil.19 The cells were maintained in alpha minimal essential medium (αMEM; Gibco®; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% inactivated fetal bovine serum (Multicell; Wisent Inc., St Bruno, QC, Canada), 100 μg/mL kanamycin (Sigma-Aldrich Co.), and 50 μg/mL ampicillin (Sigma-Aldrich Co.), and incubated at 24°C±1°C and pH 7.0 in BOD greenhouse (Model: 2005; Johns-VWR Scientific, Toronto, ON, Canada). The promastigotes were grown in cell culture flasks of 25 mL volume (Corning Incorporated, Corning, NY, USA) with an initial inoculum of 106 cells/mL and transferred to a new medium after reaching the stationary growth phase, twice a week.
Synthesis of amphiphilic antimony complex and preparation of oral formulations
SbL8 was synthesized, as previously described,17 from reaction of KSb(OH)6 with L8 in water at 1:3 molar ratio. Reaction took place through heating at 70°C and solvent evaporation, leading to the formation of a film that was redispersed in water at room temperature. SbL8 dispersion was finally submitted to freeze-drying. To prepare oral formulations, freeze-dried SbL8 was dispersed in solvents of different polarities, water and binary (water:propylene glycol [W:PG], 1:1) mixture, at final L8 concentration of 534 mM. Macroscopic observations indicate that SbL8 formulation in 1:1 W:PG is clear and does not form precipitate. Homogeneous distribution of the compound was also supported by circular dichroism measurements showing the same spectrum characteristics, whether the sample to be analyzed was collected on the top or the bottom of the formulation.
Characterization of hydrophobic microenvironment
In all characterization studies, SbL8 was dispersed, either in water or in different mixtures of water and PG, at final L8 concentration of 30 mM. The presence of hydrophobic microenvironment in SbL8 dispersions was investigated using the lipophilic fluorescent probe DPH, as previously described.17 DPH aggregates and is essentially nonfluorescent in polar solvents and tends to partition, dissociate, and become fluorescent in hydrophobic microenvironments. DPH was added to SbL8 dispersions, either in water or different mixtures of water and PG, at a final concentration of 0.5 μM. After 24-hour incubation at 25°C and under light protection, fluorescence measurements were carried out using a Cary Eclipse fluorescence spectrometer (Varian Inc., Palo Alto, CA, USA) at excitation and emission wavelengths of 360 and 428 nm, respectively.
Particle size and zeta potential
The mean hydrodynamic diameter, polydispersity index, and zeta potential were determined by dynamic light scattering (DLS) using a Zetasizer (Nano ZS90; Malvern Instruments, Malvern, UK). The particle size was also investigated using Nanoparticle Tracking Analysis (NTA) (Nanosight; Malvern Instruments) and NTA 3.1 software to collect and analyze data. Measurements were carried out at 25°C and 1 day after complete solubilization of SbL8 in respective solvents. In order to validate measurements in the 1:1 W:PG solvent, a 100 nm standard polystyrene nanoparticle was used. Whereas DLS measurements were in agreement with the expected size in both solvent conditions, NTA data showed population with much smaller size than expected in the mixed solvent. Thus, only NTA results registered in water are presented.
Small-angle X-ray scattering
Small-angle X-ray scattering (SAXS) studies were conducted in line 2 of LNLS – Brazilian Synchrotron Light Laboratory/MCT (Campinas, Sao Paulo, Brazil). This line is equipped with a monochromator (λ=1.54 Å), an ionization chamber, and a 300k Pilatus detector placed at 1 m from the sample to record the intensity of the scattering. The scattering from the samples was subtracted from that of the system without the sample. The intensities of all samples were measured in relative units, but for the purpose of comparison, the measurements were standardized under the same experimental conditions. A volume of 50 μL of SbL8 dispersions in either water or W:PG was injected into the sample compartment maintained at 25°C. The I(q) vs q curves were obtained after subtraction of the signal of the samples from that of the corresponding blank (water or W:PG mixture). Data were fitted using the SASfit Version 0.93.5 by J Kohlbrecher and I Bressler (Paul Scherrer Institut, Villigen, Switzerland) available online,20 in order to identify the best model of particle size and shape.
Atomic force microscopy
A drop (20 μL) of the samples was deposited onto substrates of different polarities: previously cleaved mica or paraffin. The samples were disseminated and partially dried with a stream of argon. Topographic and phase contrast images were obtained under the tapping mode (intermittent contact) at a scan rate of 1 Hz and room temperature in Device Dimension 3000 monitored by a Nanoscope IIIa controller from Digital Instruments (Santa Barbara, CA, USA). The images were analyzed using the “section analysis” tool system. The diameters were measured at half-height of the particles. A minimum of ten images of each sample were analyzed to ensure the representativeness of the results.21 The dimensional analyses were then performed using the NanoScope Analysis (v1.40r1; Bruker Corporation, Munich, Germany) software.
Transmission electron microscopy
Samples of 1.5 mL volume were treated with 20 μL of a solution of OsO4 5% as a positive contrast agent for hydrophobic environments. A drop of bacitracin solution (0.1 mg/mL; Sigma-Aldrich, St Louis, MO, USA) was placed onto a 300-mesh Formvar copper grid (TABB Laboratories Equipment, Reading, UK) and allowed to adsorb, and the surplus was removed by filter paper (Whatman #44). After drying, the Formvar was covered with a drop of the samples and dried at room conditions before the nanostructures were imaged. The images were obtained using a TEM operating at an acceleration voltage of 120 kV, a line resolution of 0.34 nm, a point resolution of 0.49 nm, and a Cs of 6.3 (spherical aberration), implying high contrast (model Tecnai G2 Spirit, 2006; FEI, Hillsboro, OR, USA) equipped with digital cameras, of high resolution (11 megapixels) and high speed, located in the Electron Microscopy Center of UFMG. Representative electron micrographs were taken at magnifications of ×22,000–30,000.
Serum and liver pharmacokinetics of Sb in mice
Swiss mice received orally, by gavage, 200 μL of SbL8 dispersions at 205 mM of Sb, corresponding to a dose of 200 mg Sb/kg of body weight. Animals were divided into two groups that received SbL8 in different solvents (water or 1:1 W:PG). At different times after administration (15, 30, 60, 120, 180, 240, 360, and 480 minutes), mice were anesthetized with xylazine 8 mg/kg and ketamine 35 mg/kg (n=7/time), and blood samples from the brachial plexus and liver were collected. The serum was recovered and then stored at −20°C. The concentration of Sb was determined in serum and liver by graphite furnace atomic absorption spectroscopy, using Perkin Elmer AAnalyst 600 spectrometer (PerkinElmer Inc., Waltham, MA, USA) as described previously.17 Serum was submitted to 40-fold dilution in 0.2% nitric acid solution before measurements. The liver tissue was homogenized with two-thirds of the mass of phosphate-buffered saline and subjected to digestion with 65% nitric acid, using a heating block (Model 4004 Dry block; Marconi Equipamentos Para Laboratórios Ltda, Piracicaba, Brazil). The analytical methods were validated beforehand showing quantitation limits of 240 μg Sb/L and 0.93 μg Sb/g tissue, for the serum and liver, respectively. The Sb concentration vs time curves were obtained for both formulations and compared using two-way analysis of variance (ANOVA) and Bonferroni posttest. The area under the curve (AUC) was also determined for each dispersion on the 0–24-hour period. All the analyses were made through GraphPad Prism® 5.0 statistical packages software (GraphPad Software, Inc., La Jolla, CA, USA).
Antileishmanial activity in murine model of CL
BALB/c mice were shaved in the dorsal region and infected with 1×106 promastigotes/20 μL at stationary phase, by intradermal injection at the tail base. After 35 days of infection, corresponding to the first ulceration sign of the infection papule, animals were randomly divided into four groups (n=5/group): animals treated with SbL8 in water at 200 mg Sb/kg/day by oral route (200 μL by gavage), animals treated with SbL8 in 1:1 W:PG at 200 mg Sb/kg/day by oral route (200 μL by gavage), animals treated with standard therapy (Glucantime®) by intraperitoneal route at 200 mg Sb/kg/day, and nontreated animals. One animal in group treated with SbL8 in water and two animals in the group treated with SbL8 in W:PG died in the course of the treatment. Death was attributed to suffocation of animals after gavage, as a consequence of the formation of foam during administration of the formulation, its regurgitation, and transfer to the lungs. This undesired side effect was minimized by a slower administration.
The evolution of the lesions size was determined in each animal by averaging the horizontal and vertical diameters of the lesion, respectively, perpendicular and parallel to the spine of mice. Measurements were made with the aid of universal caliper, every 3 days during the entire treatment period. The graphs consisted in the time course of lesion progression ΔD (corresponding to the average diameter of the day subtracted by the average diameter obtained on day 1). Statistical analysis was made by two-way repeated measures ANOVA and Bonferroni posttest. After 30 days of treatment, mice were anesthetized and euthanatized by cervical dislocation. The lesions were excised, and parasite load was determined by the limiting dilution assay as described previously.22,23 Briefly, homogenates of tissues were collected in 1 mL of αMEM supplemented with 20% fetal bovine serum (pH 7.0). Each homogenate was serially diluted tenfold in 96-well flat-bottomed culture plates and then incubated at 26°C. The parasite load was determined by the highest dilution at which promastigotes grew after 10 days of incubation. Values are expressed as log(parasite load/mg of tissue).17 The data with normal distribution were analyzed statistically by one-way ANOVA, followed by Bonferroni posttest.
Results
Characterization of the nanostructures formed by SbL8 in solvents with different polarities
With the aim of manipulating the supramolecular organization of SbL8 in solution, SbL8 was dissolved in W:PG mixtures of varying proportions. The formulations were then characterized, for the presence of hydrophobic microenvironment using DPH lipophilic fluorescent probe and for the morphology of the nanoassemblies by DLS, NTA, SAXS, and atomic force microscopy (AFM). Figure 1 shows the fluorescence intensity of DPH in SbL8 or L8 dispersions as a function of PG proportion. The concentrations of L8 in the dispersions (30 mM for SbL8 and 75 mM for L8) were chosen so as to be higher than the critical micelle concentration (CMC), which were reported to be ~10 and 60 mM for SbL8 and L8, respectively.17
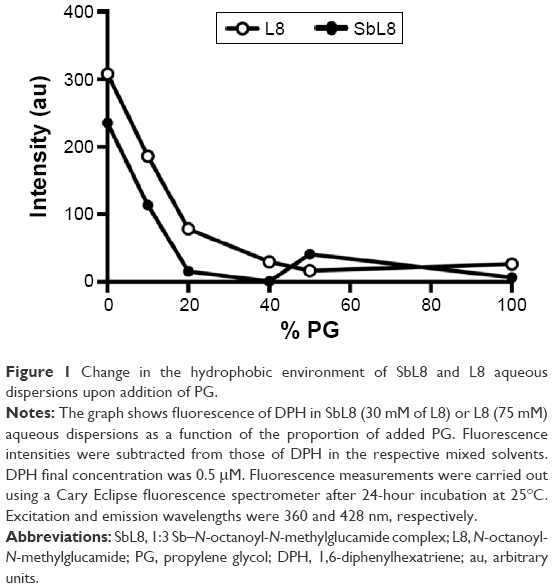 | Figure 1 Change in the hydrophobic environment of SbL8 and L8 aqueous dispersions upon addition of PG. |
As expected, in the absence of PG, the probe showed no fluorescence in water and maximum fluorescence in SbL8 and L8 dispersions. Upon increasing the proportion of PG in L8 and SbL8 solutions, DPH fluorescence decreased gradually and reached a plateau at 20% PG for SbL8 and 40% for L8. On the other hand, in the absence of L8 or SbL8, DPH fluorescence increased slightly with the amount of PG and reached a plateau at 20% PG, at a similar value as in L8 and SbL8 dispersions (data not shown), presumably because of PG-induced reorganization of DPH aggregates. As a control experiment, we checked if the probe could still recognize hydrophobic environment in the presence of PG, and found that the addition of phosphatidylcholine liposomes to a DPH solution in 1:1 W:PG resulted in a marked increase in fluorescence intensity (Figure S1). These data strongly support the loss of hydrophobic microenvironment and change in the supramolecular aggregation state of SbL8 upon addition of the less polar PG solvent. From this experiment, the 1:1 W:PG proportion was selected as “low-polarity” condition to be used in the subsequent studies. DLS analyses of SbL8 in water and 1:1 W:PG revealed polydisperse nanoparticles (polydispersity index between 0.3 and 0.5) with a mean hydrodynamic diameter in the range of 100–300 nm. The DLS multimodal analysis also suggested the coexistence of different particle populations: one with diameter in the 0.8–1.5 micrometer range, another with diameter ~60–90 nm, and an intermediate-sized particle population (~300 nm) in the mixed solvent only. These particles showed about the same negative values of zeta potential (~−52 mV) in both solvent conditions, which can be attributed to the negative charge of Sb complex. The results of NTA of SbL8 in water are consistent with DLS data, showing several particle populations with apparent mean diameter ranging from 45 to 385 nm (Figure S2).
The AFM study allowed the comparison of the morphology of the nanostructures (topography, size, and shape) formed by SbL8 in water and W:PG and their interaction with substrates of different polarities. Samples were applied and partially dried onto cleaved mica or paraffin, as hydrophilic and hydrophobic substrates, respectively. In the case of SbL8 in water, both layers with ~3 nm height and nanoassemblies with horizontal and vertical dimensions ranging from 20 to 250 nm and 2.8 to 15 nm, respectively, were observed. The largest nanoassemblies showed irregular surface and shape and seemed to arise from the aggregation of smaller discoidal nanoparticles of ~20 nm diameter (Figures 2A, B, and D and S3). L8 dispersion applied in the same conditions showed essentially 3 nm layers with very few discoidal nanoparticles of ~30 nm diameter and 4.5 nm height (data not shown). Conversely, neither nanoparticles nor layers were observed when the SbL8 aqueous dispersion was applied onto paraffin hydrophobic substrate (Figure 2C).
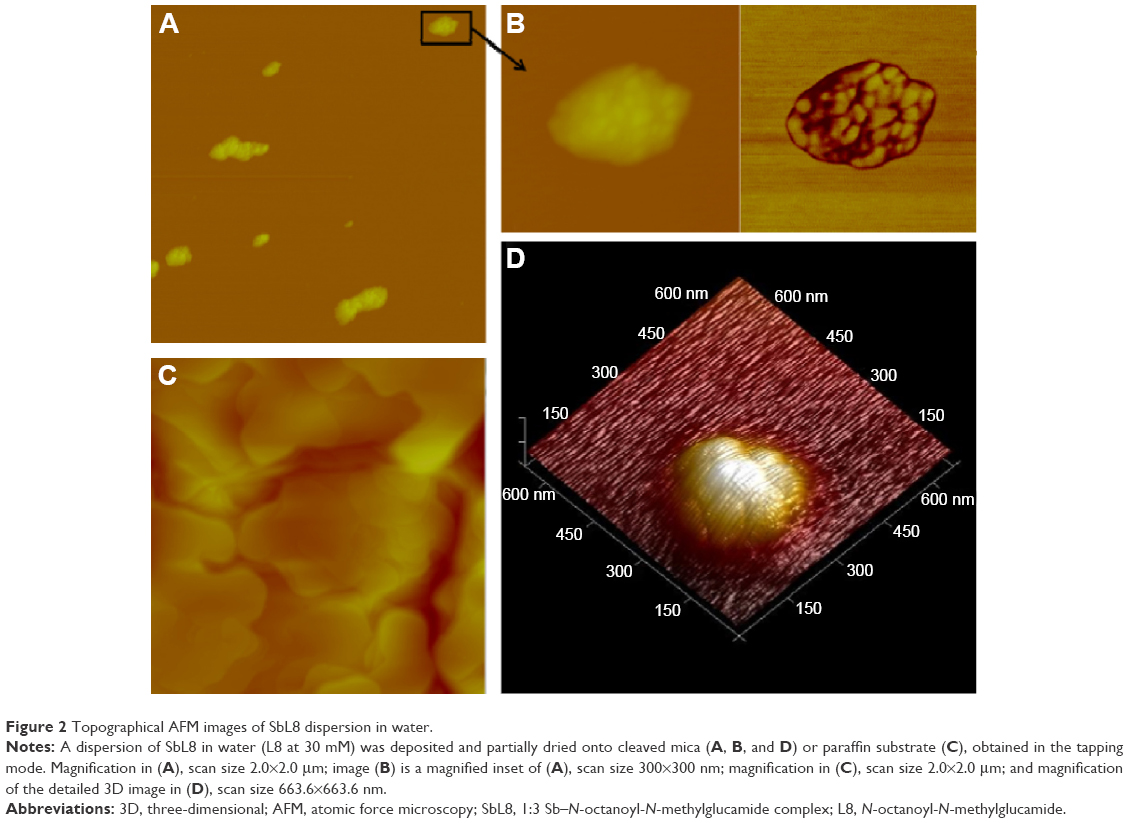 | Figure 2 Topographical AFM images of SbL8 dispersion in water. |
Interestingly, SbL8 applied in W:PG onto the mica substrate formed only layers with ~3 nm height (Figure 3A). Similar layers were observed with L8 dispersions (data not shown). On the other hand, when applied onto the hydrophobic surface (paraffin substrate), the SbL8 dispersion in W:PG showed the formation of discoidal nanoparticles with diameter and height of 2.2–100 and 2.6–19 nm, respectively (Figure 3B–D). Although the two formulations of SbL8 in water and W:PG showed particles with dimensions in the same range, the largest nanoparticles exhibited a more irregular surface in water than in W:PG presumably as a result of a more aggregated state. Taking into account that SbL8 particles in water and W:PG showed distinct structural organizations when applied onto the same substrate and that this organization was modified upon changing the substrate polarity, one can infer that the conformation of SbL8 nanoparticles is highly flexible and depends on the polarity of both the solvent and the substrate with which they interact.
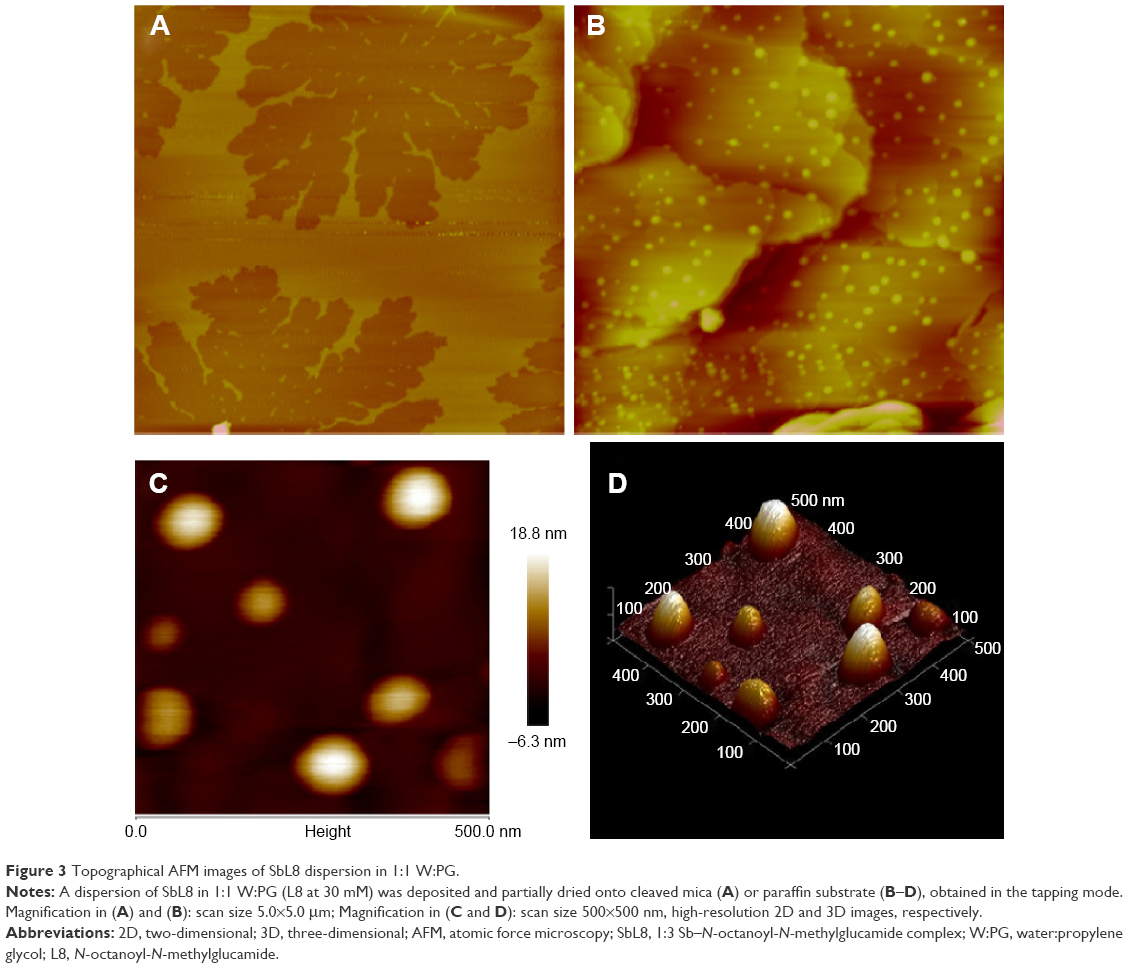 | Figure 3 Topographical AFM images of SbL8 dispersion in 1:1 W:PG. |
TEM analysis of SbL8 in water showed similar polydisperse nanostructures, with high-order structures as illustrated in Figure 4A. In the W:PG solvent, SbL8 organized as highly aggregated structures that seemed to result from the association of much smaller nanostructures (Figure 4B).
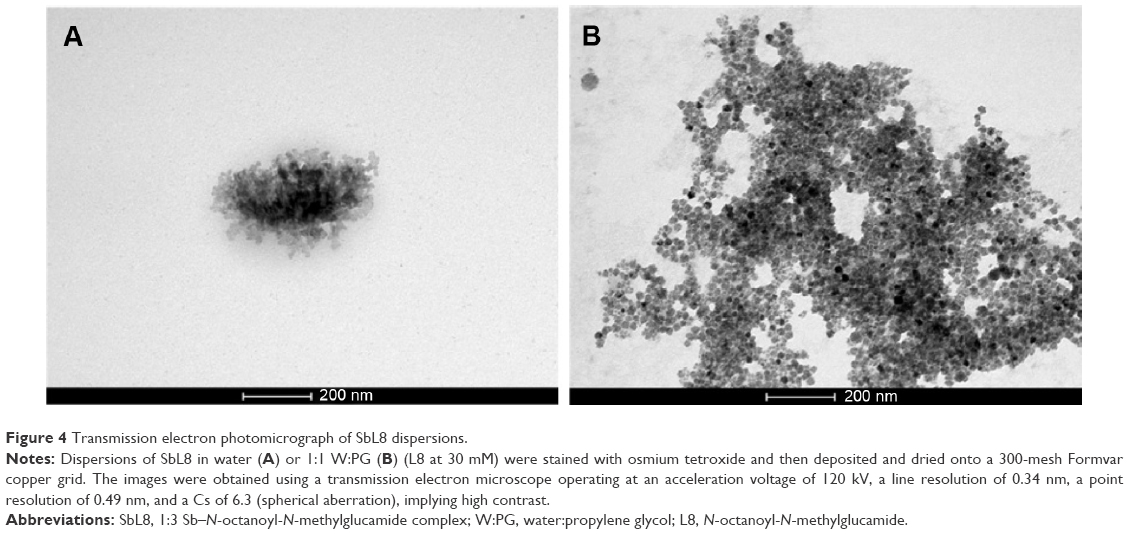 | Figure 4 Transmission electron photomicrograph of SbL8 dispersions. |
The microscopic data taken altogether suggest the formation of discoidal nanostructures in both water and 1:1 W:PG solvents, that seemed to associate to form larger nanoassemblies of varying sizes. Evidence was also obtained that these nanostructures suffer conformational changes upon reduction of the polarity of both the solvent and substrate.
SAXS measurements of the dispersions in water at 30 mM showed a scattering curve for SbL8 but not for L8 dispersion, in agreement with the higher CMC value of L8. Interestingly, SbL8 at 30 mM showed a structured SAXS curve profile very similar to that of L8 surfactant at 140 mM (Figure 5A), except for the lowest angle region where the sharp increase of intensity for SbL8 may be attributed to particle aggregation. After exclusion of the low-angle part of the SAXS curves, it was possible to fit the data using the SASfit software and identify the best model of particle size and shape. The best adjustment was obtained with the spherical core–shell model, as illustrated in Figure 5B, and the following parameters: R1 =2.34 nm and R2 =23.47±0.29 nm for L8; and R1 =1.38 nm and R2 =23.85±0.44 nm for SbL8. The presence of PG in the SbL8 dispersion resulted in a much less structured curve profile at higher angle (Figure 5C, top), indicating a structural change of the nanoassemblies, but preventing adjustment to any structural model. This is in contrast with the L8 dispersion that showed complete loss of scattering (Figure 5C, bottom), supporting the disappearance of nanoparticles.
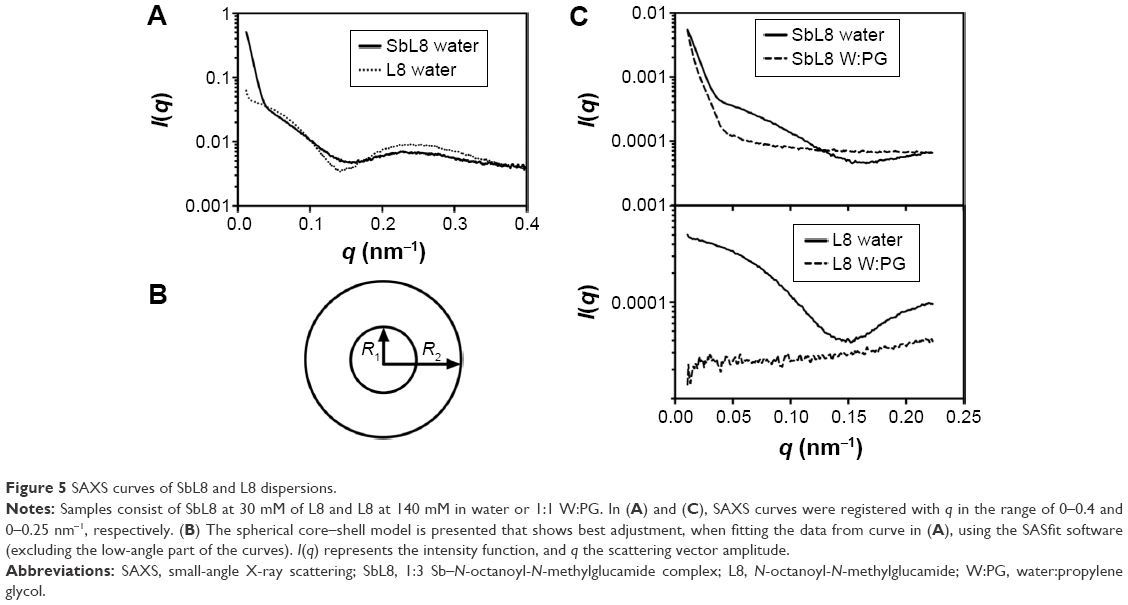 | Figure 5 SAXS curves of SbL8 and L8 dispersions. |
Pharmacokinetic study
To evaluate the impact of addition of PG to the SbL8 aqueous dispersion on the absorption of Sb through the gastrointestinal tract, comparative pharmacokinetic studies were performed in Swiss mice after administration by gavage of SbL8 in either water or 1:1 W:PG mixture. Figure 6 shows the concentration of Sb in the serum of animals, at different times after oral administration of SbL8 formulations at 200 mg Sb/kg. The formulation of SbL8 in W:PG promoted significantly greater serum levels of Sb at the earliest (0.25 hour) and longest (24 hours) time points, compared to the formulation in water (two-way ANOVA, P<0.001). These data suggest a higher efficiency and more sustained intestinal absorption of Sb from SbL8 in W:PG. The ability of the formulation of SbL8 in W:PG to promote more sustained serum levels of Sb is also evidenced by the fact that there was no significant variation in the concentration of Sb over the 0.25–24-hour interval (one-way ANOVA, P>0.05). Additionally, the AUC0–24 h for SbL8 in W:PG was approximately twice as high as AUC0–24 h for SbL8 in water (2,741 and 1,633 μg·h/L), indicating a higher bioavailability of Sb from oral administration of the drug in the binary solvent.
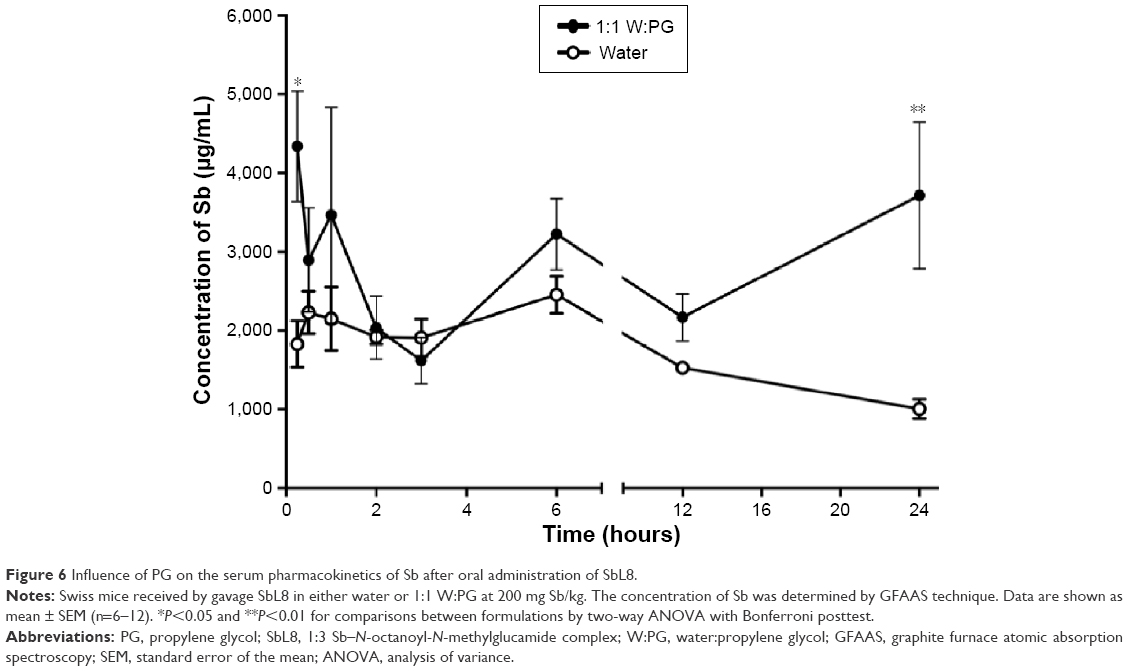 | Figure 6 Influence of PG on the serum pharmacokinetics of Sb after oral administration of SbL8. |
In vivo efficacy
The formulations of SbL8 in water and W:PG were compared for their antileishmanial efficacy in a murine model of CL. BALB/c mice were first infected by inoculation of L. amazonensis at the tail base. Treatment started just after ulceration of the lesions through administration by gavage of SbL8 dispersions at 200 mg Sb/kg/day. Comparisons were also made with a group treated intraperitoneally with Glucantime® at the same dose of Sb and with a negative control group that received saline orally. Figure 7 shows the variation of lesions size over the 30-day period of treatment. The formulation of SbL8 in 1:1 W:PG promoted a significant stabilization of the lesions size, when compared to saline, to a similar level as Glucantime® given intraperitoneally. On the other hand, the formulation of SbL8 in water, despite being effective in reducing the lesion, was significantly less effective than the standard parenteral drug. It is also noteworthy that some animals from the SbL8 W:PG group presented regression in ulcer characteristics after 30 days of treatment (Figure 7, inset).
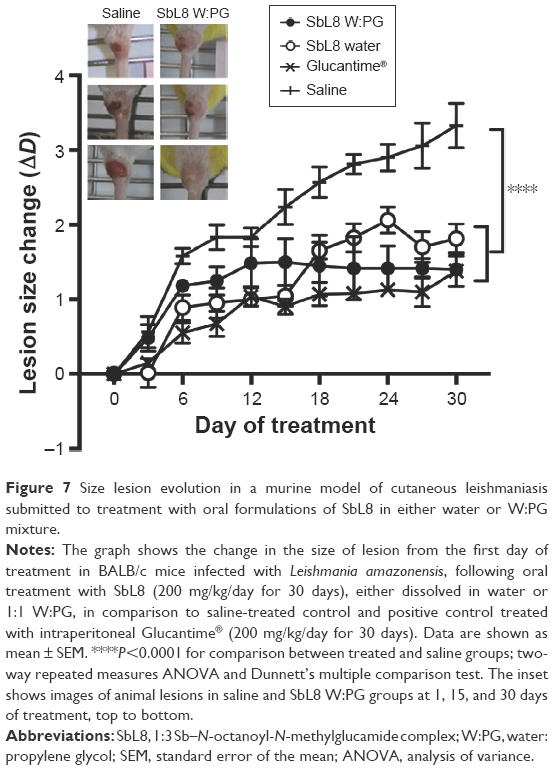 | Figure 7 Size lesion evolution in a murine model of cutaneous leishmaniasis submitted to treatment with oral formulations of SbL8 in either water or W:PG mixture. |
Figure 8 shows the parasite load in the lesions of animals. All the treated groups showed a significant reduction in parasite burden, in comparison to the control saline group. The oral formulation of SbL8 in water and intraperitoneal Glucantime® were equally effective in promoting the reduction of parasite burden. Strikingly, oral treatment with SbL8 in 1:1 W:PG was significantly more effective than all other treatments in the experiment (P<0.001, one-way ANOVA with Tukey’s posttest). The reduction of the parasitic load also showed a good correlation with the stabilization of the lesion size evolution in animals treated with intraperitoneal Glucantime® and oral SbL8 in either water or W:PG.
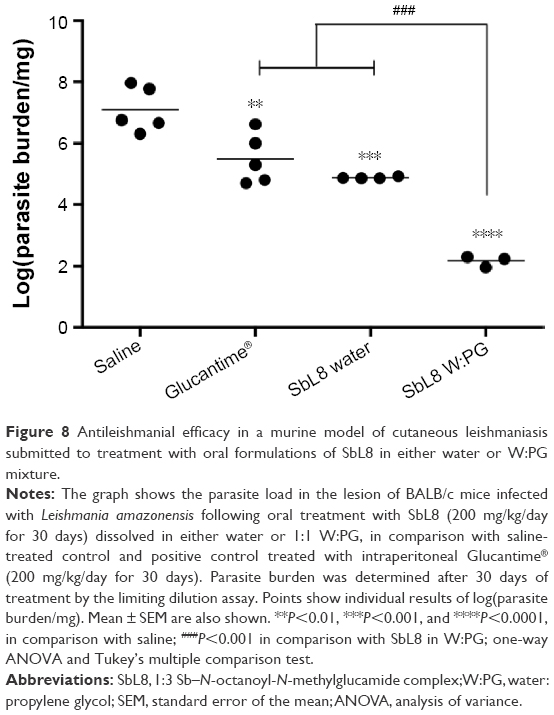 | Figure 8 Antileishmanial efficacy in a murine model of cutaneous leishmaniasis submitted to treatment with oral formulations of SbL8 in either water or W:PG mixture. |
Discussion
In a previous study conducted by our group, the antileishmanial activity of SbL8 in water given by oral route was studied in a murine model of visceral leishmaniasis.17 The formation of nanoassemblies in SbL8 aqueous dispersion was also evidenced through TEM analysis and the identification of hydrophobic microenvironments using the DPH probe. In the present work, our main objective was to interfere in the structural organization of these nanoassemblies so as to investigate their influence on the oral bioavailability of Sb and the efficacy of SbL8 in a murine model of CL. The primary idea behind the use of PG as a solvent was to exploit its weaker hydrogen bond donor character and its lower dielectric constant, when compared to water.24 By adding PG to a surfactant solution in water, the dielectric constant of medium and also the solvophobic effect in the system are reduced, causing an increase in CMC of the surfactant.25 In the case of SbL8, even though the hydrophobic microenvironment was lost upon addition of 50% PG, nanostructures were still present, as evidenced by DLS, SAXS, AFM, and TEM analyses.
To understand the precise effect of PG, one should first consider the supramolecular organization of SbL8 in water. SAXS analysis and NTA support the formation of spherical core–shell nanoparticles with diameter in the range of 40–50 nm. Such nanostructures, also observed in concentrated solution of L8, may consist in nanoaggregates of micelles. On the other hand, AFM data indicate the formation of discoidal nanoparticles with ~20 nm diameter that further aggregate to form larger (200–300 nm) nanoassemblies. However, the formation of bicelle-type (Figure S3) rather than micelle-type nanoparticles, as revealed by AFM, may have been induced by the drying process (during preparation of the sample) and subsequent interaction of nanostructures with the hydrophilic substrate. The small and larger nanoassemblies observed with SbL8 are most probably stabilized by Sb(V)-induced polymerization through formation of multiple Sb(−O−C)n intermolecular bonds. Indeed, previous electrospray ionization mass spectrometry analysis of SbL8 dispersion in water revealed not only 1:3 Sb–L8 complexes but also higher order species, such as 2:5 Sb–L8 complex.17 The fact that electrostatic repulsion between negatively charged SbL8 nanoparticles apparently did not prevent their aggregation also supports the model of polymerization.
Regarding the effect of PG on the organization of SbL8 nanostructures, one can propose that PG weakened the hydrophobic effect and hydrophilic interactions, but preserved the covalent Sb–O–C bonds which stabilize the nanostructures. The preservation of Sb–L8 complexation in the mixed solvent is also supported by circular dichroism analysis of SbL8 in 1:1 W:PG (7.5 mM of L8), showing that SbL8 spectrum differed significantly from that of L8 (Figure S4). Such effects of PG are consistent with a previous report that increasing amount of this solvent in aqueous solution of serum albumin induced noncooperative conformational change of the protein, by weakening its tertiary structure through hydrophobic interaction with nonpolar side chains and promoting α-helix formation.26
Another interesting property of SbL8 nanoassemblies, as suggested by the AFM and SAXS studies, is the flexibility of their supramolecular organization that suffers modifications as a function of the polarity of the solvent and the substrate with which they interact. The results of in vivo studies suggest that this flexibility and polarity sensitivity can be exploited to improve the oral bioavailability of Sb and the efficacy of SbL8 against leishmaniasis caused by L. amazonensis or by other Leishmania species. On the basis of thermodynamics, nanoassemblies in the presence of PG are expected to adopt a more “open” conformation, which may facilitate their interaction with hydrophobic substrates, such as membranes. The higher serum concentration of Sb promoted by the formulation of SbL8 in W:PG during the initial phase of the pharmacokinetics may be due to either a more permeable conformational state of the nanoassemblies or a possible permeation enhancement of PG. In addition, the more sustained serum level of Sb from the W:PG formulation suggests a specific supramolecular organization of SbL8 in the intestine which may favor the formation of a drug depot or the absorption of a specific antimonial species with a lower blood clearance.
From the pharmacological point of view, it is expected that higher and more sustained serum levels of Sb, as seen from SbL8 formulation in W:PG, may facilitate the distribution of Sb to the skin and its action in the area of the infection caused by Leishmania parasites. Therefore, the significantly higher antileishmanial efficacy of the formulation of SbL8 in W:PG is consistent with its distinct pharmacokinetic profile. In order to rule out the possibility of the higher efficacy of SbL8 in W:PG being due to a nonspecific permeation enhancer effect of PG, both solvents were compared for their ability to promote the oral antileishmanial efficacy of Glucantime® (200 mg/kg/day for 30 days) in the same model of CL. Glucantime®, in either water or 1:1 W:PG solvent, did not induce significant reduction in the lesion parasite load in comparison to saline-treated group, suggesting PG does not act through nonspecific membrane permeation enhancer effect (Figure S5). Interestingly, the higher efficacy of SbL8 in W:PG seems to be specific of the CL model. Indeed, in a mice model of VL, SbL8 showed similar efficacies when given in water or W:PG (Figure S6).
Although SbL8 in W:PG promoted lower average lesion dimensions (from day 18) and parasite burden (day 30) when compared to SbL8 in water, only the parasite burden showed significant difference among the groups. From the literature, one would expect correlation between lesion development and Leishmania parasite burden in BALB/c mice.22 However, our previous experience with the L. amazonensis BALB/c mice model showed that oral Glucantime® can promote small but significant reduction in parasite burden, without affecting the lesion growth.27 This suggests that, when evaluating the efficacy of antimonial drugs in this model, the determination of parasite burden may be more sensitive than the measurement of size lesion. Another possible explanation for this apparent lack of correlation is that evolution of the lesion depends on both the parasite burden and the inflammatory response28 and that inflammatory response in this model may compromise the effect of pentavalent antimonials on the lesion development. It is also noteworthy that BALB/c mice have Th2-biased immune responses that make this model extremely susceptible to Leishmania infection and explains why BALB/c mice do not show self-healing lesions, contrary to immunocompetent humans infected with L. amazonensis. This feature also compromises the measurement of lesion development as a predictor of potential clinical efficacy. Thus, in our experimental model, the parasite burden together with the drug pharmacokinetic profile can be considered as the most reliable parameters to evaluate the potential clinical antileishmanial efficacy of the formulation.
A major finding of the present study is the higher antileishmanial efficacy of SbL8 in W:PG given by oral route, when compared to conventional drug Glucantime® given intraperitoneally at the same Sb dosage. The differential pharmacokinetic profile of the amphiphilic Sb(V) complex, that is, its ability to promote prolonged plasma level of Sb, most probably contributes to its greater effectiveness. As mentioned earlier, this sustained plasma level of Sb may be due to the formation of a drug depot in the intestine or to the amphiphilic character of absorbed antimonial species, which may favor its interaction with plasma protein and thereby delay the Sb clearance. In contrast, the hydrophilic drug Glucantime® has a high plasma clearance with average residence time of only 20 minutes.17 On the other hand, one cannot rule out the possibility that the two antimonial compounds may present differential cell capture efficiencies and metabolisms, which could lead to differences in their antileishmanial activity.
Conclusion
This work reports for the first time the oral efficacy of SbL8 nanoassemblies against CL and shows that decreasing the vehicle polarity promotes conformational change of the nanostructures that contributes to enhanced drug bioavailability and efficacy. A new concept of polarity-sensitive nanocarrier for oral drug delivery is introduced.
Acknowledgments
The authors would like to specially thank Diógenes de Sousa Neto, Mateus Borba Cardoso, Nayara Kesia Lima Mendes Moura, and Larissa Procópio Carvalho for technical support. This work was supported by the following Brazilian agencies: Conselho Nacional de Desenvolvimento Científico e Tecnológico (303227/2013-3, 472468/2013-8), Fundação de Amparo a Pesquisa do Estado de Minas Gerais (RED-00007-14, APQ-01373-14 PRONEX, APQ-01542-13), Coordenação de Aperfeicoamento de Pessoal de Nível Superior (studentship), and the LNLS – Brazilian Synchrotron Light Laboratory/MCT (SAXS1-14258).
Author contributions
FRF, JSL, LAMF, CD, and MNM conceived and designed the study. JSL carried out the experiments. FRF, JSL, and FF carried out the SAXS measurements. RM-P contributed to the analysis and interpretation of SAXS curves. JDC-J carried out TEM analysis. JMCV carried out AFM analysis. JMCV and MSA contributed to the analysis of AFM images. FF and JSL drafted the manuscript. All authors have contributed to writing and the critical revision of the manuscript during all stages of submission. All authors read and approved the final manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Marsden PD. Pentavalent antimonials: old drugs for new diseases. Rev Soc Bras Med Trop. 1985;18:187–198. | ||
Berman JD. Human leishmaniasis: clinical, diagnostic, and chemotherapeutic developments in the last 10 years. Clin Infect Dis. 1997;24:684–703. | ||
Ridley RG. The need for new approaches to tropical disease drug discovery and development for improved control strategies. In: Failamb AH, Ridley RG, Vial HJ, editors. Drugs Against Parasitic Diseases: R&D Methodologies and Issues. Geneva: UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases (TDR); 2003:13–21. | ||
Organización Panamericana de la Salud. Leishmaniasis en las Américas: recomendaciones para el tratamiento [Leishmaniasis in the Americas: Recommendations for Treatment]. Washington, DC: OPS; 2013. | ||
Frézard F, Demicheli C, Ribeiro RR. Pentavalent antimonials: new perspectives for old drugs. Molecules. 2009;14:2317–2336. | ||
Ouellette M, Drummelsmith J, Papadopoulou B. Leishmaniasis: drugs in the clinic, resistance and new developments. Drug Resist Updat. 2004;7(4–5):257–266. | ||
Lainson R. The Neotropical Leishmania species: a brief historical review of their discovery, ecology and taxonomy. Rev Pan-Amaz Saude. 2010;1:13–32. | ||
Silveira FT, Lainson R, Corbett CEP. Clinical and immunopathological spectrum of American cutaneous leishmaniasis with special reference to the disease in Amazonian Brazil: a review. Mem Inst Oswaldo Cruz. 2004;99(3):239–251. | ||
Harhay MO, Olliaro PL, Costa DL, Costa CHN. Urban parasitology: visceral leishmanisis in Brazil. Trends Parasitol. 2011;27(9):403–409. | ||
Jha TK, Sundar S, Thakur CP, et al. Miltefosine, an oral agent, for the treatment of Indian visceral leishmaniasis. N Engl J Med. 1999;341(24):1795–1800. | ||
Sundar S, Jha TK, Thakur CP, et al. Oral miltefosine for Indian visceral leishmaniasis. New Engl J Med. 2002;347(22):1739–1746. | ||
Bern C, Adler-Moore J, Berenguer J, et al. Liposomal amphotericin B for the treatment of visceral leishmaniasis. Clin Infect Dis. 2006;43:917–924. | ||
Wasan KM, Wasan EK, Gershkovich P, et al. Highly effective oral Amphotericin B formulation against murine visceral leishmaniasis. J Infect Dis. 2009;200:357–360. | ||
Prajapati VK, Awasthi K, Yadav PT, et al. An oral formulation of Amphotericin B attached to functionalized carbon nanotubes is an effective treatment for experimental visceral leishmaniasis. J Infect Dis. 2012;205:333–336. | ||
Chappuis F, Sundar S, Hailu A, et al. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat Rev Microbiol. 2007;5(11):873–882. | ||
Frézard F, Demicheli C. Novel delivery strategies for the old pentavalent antimonial drugs. Expert Opin Drug Deliv. 2010;7:1343–1358. | ||
Fernandes FR, Ferreira WA, Campos MA, et al. Amphiphilic antimony(V) complexes for oral treatment of visceral leishmaniasis. Antimicrob Agents Chemother. 2013;57(9):4229–4236. | ||
Frézard F, Demicheli C, Kato KC, Reis PG, Lizarazo-Jaimes EH. Chemistry of antimony-based drugs in biological systems and studies of their mechanism of action. Rev Inorg Chem. 2013;33:1–12. | ||
De Oliveira JP, Fernandes F, Cruz AK, et al. Genetic diversity of Leishmania amazonensis strains isolated in northeastern Brazil as revealed by DNA sequencing, PCR-based analyses and molecular karyotyping. Kinetoplastid Biol Dis. 2007;6:5–8. | ||
Breßler I, Kohlbrecher J, Thünemann AF. SASfit: a tool for small-angle scattering data analysis using a library of analytical expressions. J Appl Crystallogr. 2015;48:1587–1598. | ||
Sitterberg J, Ozcetin A, Ehrhardt C, Bakowsky U. Utilising atomic force microscopy for the characterisation of nanoscale drug delivery systems. Eur J Pharm Biopharm. 2010;74(1):2–13. | ||
Titus RG, Marchand M, Boon T, Louis JA. A limiting dilution assay for quantifying Leishmania major in tissues of infected mice. Parasite Immunol. 1985;7(5):545–555. | ||
Afonso LC, Scott P. Immune responses associated with susceptibility of C57BL/10 mice to Leishmania amazonensis. Infect Immun. 1993;61(7):2952–2959. | ||
Camesano TA, Nagarajan R. Micelle formation and CMC of Gemini surfactants: a thermodynamic model. Colloids Surf A Physicochem Eng Aspects. 2000;167:165–177. | ||
Sohrabi B, Bazyari A, Hashemianzadeh M. Effect of ethylene gycol on micellization and surface properties of Gemini surfactant solutions. Colloids Surf A Physicochem Eng Aspects. 2010;364:87–93. | ||
Gekko K, Koga S. The stability of protein structure in aqueous propylene glycol: amino acid solubility and preferential solvation of protein. Biochim Biophys Acta. 1984;786:151–160. | ||
Demicheli C, Ochoa R, Silva JBB, et al. Oral delivery of meglumine antimoniate-beta-cyclodextrin complex for treatment of leishmaniasis. Antimicrob Agents Chemother. 2004;48:100–103. | ||
Almeida RP, Barral-Netto M, De Jesus AM, et al. Biological behavior of Leishmania amazonensis isolated from humans with cutaneous, mucosal, or visceral leishmaniasis in BALB/C mice. Am J Trop Med Hyg. 1996;54:178–184. |
Supplementary materials
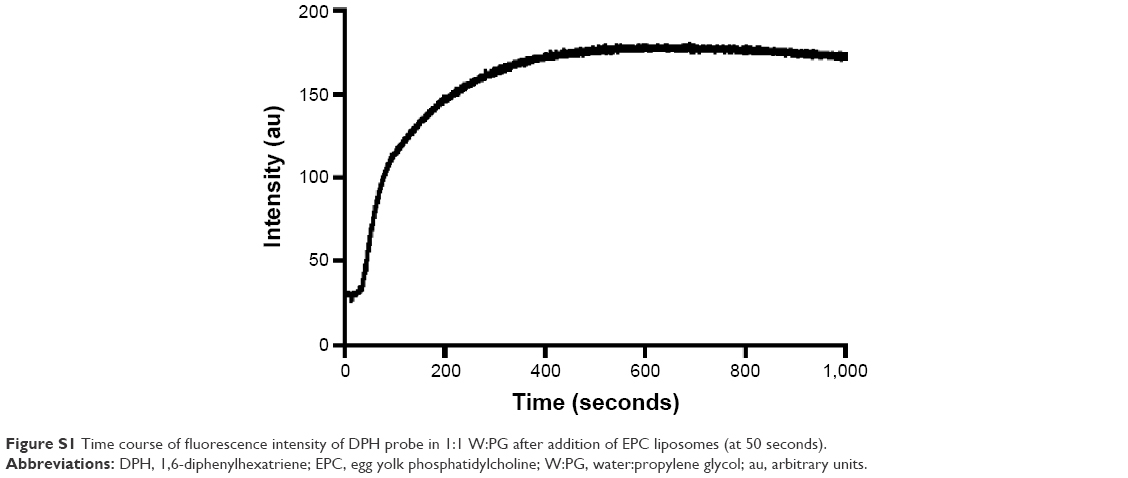 | Figure S1 Time course of fluorescence intensity of DPH probe in 1:1 W:PG after addition of EPC liposomes (at 50 seconds). |
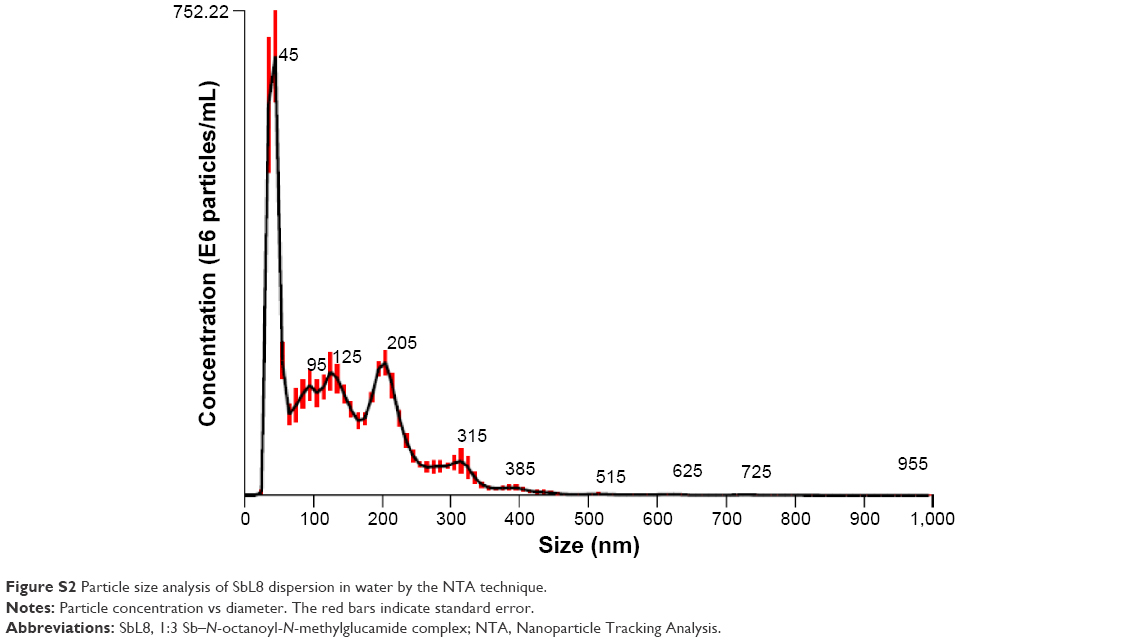 | Figure S2 Particle size analysis of SbL8 dispersion in water by the NTA technique. |
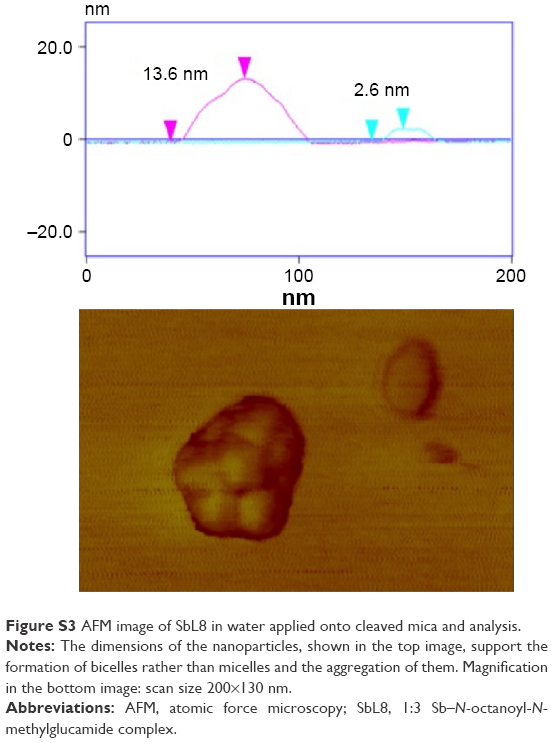 | Figure S3 AFM image of SbL8 in water applied onto cleaved mica and analysis. |
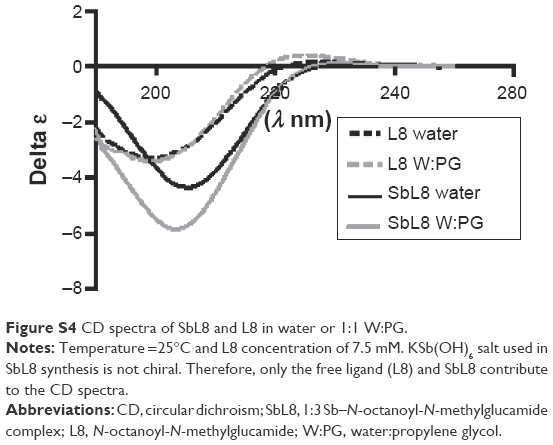 | Figure S4 CD spectra of SbL8 and L8 in water or 1:1 W:PG. |
 | Figure S5 Effect of the presence of 50% PG in Glucantime® on its antileishmanial efficacy, given by oral route, in a murine model of cutaneous leishmaniasis. |
 | Figure S6 Effect of the presence of PG in SbL8 oral formulation on its antileishmanial efficacy in a murine model of visceral leishmaniasis. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.