")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
PM2.5 downregulates miR-194-3p and accelerates apoptosis in cigarette-inflamed bronchial epithelium by targeting death-associated protein kinase 1
Authors Zhou TY, Zhong YJ, Hu Y, Sun C, Wang YX, Wang GF
Received 19 March 2018
Accepted for publication 17 May 2018
Published 1 August 2018 Volume 2018:13 Pages 2339—2349
DOI https://doi.org/10.2147/COPD.S168629
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Tianyu Zhou, Yijue Zhong, Yan Hu, Chao Sun, Yunxia Wang, Guangfa Wang
Department of Respiratory and Critical Care Medicine, Peking University First Hospital, Beijing, 100034, China
Background: Persistent exposure to cigarette smoke or biomass fuels induces oxidative stress and apoptosis in bronchial epithelium, which is one of the most important pathogenic mechanisms of chronic obstructive pulmonary disease (COPD). Fine particulate matter (PM2.5) is an aggravating risk factor of COPD exacerbation. Animal evidence showed PM2.5accelerated lung inflammation and oxidative stress in COPD mice, but the mechanism is still not clear. Recently, we found that miR-194-3p is a novel biomarker of both COPD and PM2.5 exposure, and miR-194 family has been reported to be involved in cell proliferation and apoptosis. Thus, we propose a hypothesis: PM2.5 can accelerate apoptotic response of airway epithelial cells in COPD and miR-194 is a potential involved regulator.
Materials and methods: Human bronchial epithelial cells (HBEpiCs) were treated with normal media, cigarette smoke solution (CSS) and PM2.5-CSS for 24 h. miR-194-3p mimics, inhibitors and scrambled controls were non-transfected or pre-transfected into HBEpiCs for 48 h. MircroRNAs and mRNA expression were quantified by qRT-PCR. Protein expression was analyzed by western blotting. Caspase activities, mitochondrial membrane potential and TUNEL-positive cells were detected to analyze apoptosis. Bioinformatics and luciferase analysis were used to identify the predicted binding site of miR-194-3p and potential targets.
Results: In our study, we found that PM2.5 significantly aggravated apoptosis in cigarette-inflamed HBEpiCs. miR-194-3p was dramatically downregulated in PM2.5-CSS-treated HBEpiCs. Bioinformatics and luciferase experiments reported that death-associated protein kinase 1 (DAPK1), regulating caspase 3 activities in apoptosis, was directly targeted by miR-194-3p. Inhibition of miR-194-3p increased DAPK1 expression and apoptosis in normal HBEpiCs. Importantly, overexpression of miR-194-3p suppressed apoptosis in PM2.5-CSS HBEpiCs.
Conclusion: These results suggested that miR-194-3p was a protective regulator involved in apoptosis pathway and a potential therapeutic target for treatment of bronchial epithelial injury aggravation induced by PM2.5.
Keywords: fine particulate matter, PM2.5, apoptosis, COPD, bronchial epithelial cells, miR-194-3p
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by persistent and aggressive airflow limitation and lung inflammation,1 and COPD has become the third highest cause of death in the world.2 One of the most important pathogenic mechanisms of COPD is inflammatory response and abnormal apoptosis in airway epithelium, due to long-term exposure to cigarette smoke or biomass fuels.3–6 Not only that, the inflammation and cell apoptosis induced by cigarette smoke remain in the lungs after quitting smoking.7,8 Thus, COPD is not easily managed and causes severe economic burden. Air pollution is one of the most challenging problems in COPD management. According to the World Health Organization, approximately 14% of 3.7 million deaths in 2012 were associated with COPD and respiratory infection.9
Particulate matter (PM) is a complex mixture of small particles and liquid droplets in the air, and fine particulate matter (PM2.5) is the PM with a diameter of 2.5 μm or less.10 Once inhaled, PM2.5 deposits in lung tissues and diffuses in blood inducing airway and systematic inflammation.11,12 In recent decades, epidemiological studies demonstrated that PM2.5 is an aggravating risk factor of COPD exacerbation. The latest systemic review showed that every 10 μg/m3 increase of PM2.5 was associated with a 2.5% increased risk of COPD-related emergency visits and hospital admissions.13 Animal studies have shown that PM2.5 accelerated the original lung inflammation in COPD mice,14 but the mechanism is still not clear.
MicroRNAs (miRNAs) are a class of endogenous small noncoding RNAs regulating cell development, proliferation, differentiation and death.15,16 miRNAs are confirmed to play an important role in the pathogenesis of COPD. For example, the downregulation of miR-146a17 and the upregulation of miR-22318 were associated with the inflammatory responses of COPD. Meanwhile, miRNAs are sensitive biomarkers to PM2.5. Expression of miRNAs such as miR-9, miR-10b and miR-21 were significantly altered after exposure to PM2.5,19 suggesting that miRNAs might be ideal tools for understanding the mechanism of PM2.5 in COPD.
In our unpublished data, we found that miR-194-3p was a novel biomarker of COPD, and miR-194-3p was also correlated with PM2.5-induced lung function decline (Zhou et al, unpublished material, 2018). miR-194 family participates in cell proliferation and senescence.20–23 Recently, miR-194 was confirmed to express in non-small lung cancer cells.24 However, miR-194 has not been discussed either in COPD or in PM2.5 exposure. Thus, we suggested a hypothesis that PM2.5 regulates the expression of miR-194-3p and accelerates apoptosis of cigarette-inflamed bronchial epithelial cells. In this study, we aimed to find out whether PM2.5 accelerated apoptosis and altered the expression of miR-194-3p in cigarette-inflamed human bronchial epithelial cells (HBEpiCs). If so, we aimed to further explore the underlying mechanism.
Materials and methods
Cigarette smoke solution (CSS)
According to the method of Blue and Janoff,25 smoke from one cigarette was pumped slowly into a negative pressure tube that contained 10 mL of sterile medium via a 50 mL syringe. After being filtered through 22 μm filters, the concentration of the original CSS was considered to be 100%. The concentration of final CSS was 5%.
PM2.5
The preparation of PM2.5 has been already described.26 Briefly, PM2.5 was gathered by a PM2.5 high volume sampler system (Staplex® No PM-2.5 SSI; USA) set on the roof of Peking University First Hospital, which was located very close to one of the busiest roads in central Beijing. Glass fiber filter paper (20.3×25.4 cm) was used to collect PM2.5 with an air flow rate of 1.13 m3/min. Then, the paper was sonicated three times with deionized water. Finally, the filtered products were lyophilized and refrigerated at −80°C. Final extracted PM2.5 was suspended and sonicated in culture media to ensure that the stimulus dose for cells was 500 ng/mL.
Cell culture
HBEpiCs (ScienCell Research Laboratories, Inc., San Diego, CA, USA) were cultured in bronchial epithelial cell medium (ScienCell Research Laboratories, Inc.) at 37°C in a 5% CO2 atmosphere. In total, 30 nM miR-194-3p mimics, inhibitors or scrambled controls were non-transfected or pre-transfected into HBEpiCs with DMEM with 3% fetal calf serum (Thermo Fisher Scientific, Waltham, MA, USA) for 48 h. Transfection of each reagent was performed using Lipofectamine® 3000 (Thermo Fisher Scientific). The mimics and negative control of miR-194-3p were chemically synthesized as double-stranded RNAs (GenePharma Co., Ltd., Shanghai, China) using the following sequence (5′→3′): mimics sense CCAGUGGGGCUGCUGUUAUCUG and anti-sense GAUAACAGCAGCCCCACUGGUU; negative control sense UUCUCCGAACGUGUCACGUTT and anti-sense ACGUGACACGUUCGGAGAATT. The inhibitors and inhibitor-negative control were synthesized as single-stranded RNAs using the following sequence: inhibitor CAGAUAACAGCAGCCCCACUGG and inhibitor-negative control CAGUACUUUUGUGUAGUACAA. Before analysis, HBEpiCs were treated with normal media, CSS or PM2.5-CSS for 24 h.
Quantitative assessment of miRNA and mRNA
Total RNA was isolated from HBEpiCs via TRIzol® Reagent (Thermo Fisher Scientific). For mRNA analyzing, 1 ug of total RNA quantified by Nanodrop 2000 (Thermo Fisher Scientific) was reverse transcribed into 20 μL of complementary DNA using High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific), while for analysis of miRNA, 600 ng of total RNA was reverse transcribed by Mir-X miRNA First-Strand Synthesis Kit (TAKARA, Tokyo, Japan). qRT-PCR was performed on StepOnePlus Real-Time PCR System (Thermo Fisher Scientific) in a 20 μL reaction that contained 1 μL of cDNA and Power SYBR® Green PCR Master Mix (Thermo Fisher Scientific). Relative expressions of death-associated protein kinase 1 (DAPK1) and miR-194-3p were determined. β-Actin and U6 were used as the reference genes, respectively. The sequences of primers for miRNA analysis were as follows (5′→3′): U6 from Mir-X miRNA First-Strand Synthesis Kit; hsa-miR-194-3p ATTATTCCAGTGGGGCTGCT. Gene primers for mRNA analysis: hsa-β-actin forward CTGTGGCATCCACGAAACTA, reverse GTGTTGGCGTACAGGTCTT; hsa-DAPK1 forward GTGGATGGTCATTGCAGTTTAAG, reverse TACTGGAGGATGAGAGATGGAG.
Luciferase analysis
According to the binding site on DAPK1 mRNA 3′-untranslated region (3′-UTR), a wild-type (wt) DAPK1-3′-UTR gene or a mutated (mut) DAPK1-3′-UTR gene was constructed and cloned into the reporter vector; pMIR-REPORT miRNA expression reporter vector (Obio Technology Corp., Shanghai, China). The HEK293T cells (National Infrastructure of Cell Line Resource, Shanghai, China Infrastructure of Cell Line Resource, China) were transfected with empty vector, DAPK1-3′-UTR-wt vector and DAPK1-3′-UTR-mut vector with miR-194-3p mimic or scramble control. After 48 h, the transfected cells were analyzed by Dual-Luciferase Reporter Assay System (Promega Corporation, Fitchburg, WI, USA).
Immunoblotting analysis
Proteins were extracted from cell lysis. The expressions of caspase 3 (anti-pro-caspase 3 from Abcam plc.; anti-cleaved-caspase 3 from Cell Signaling Technology, Inc.) and caspase 9 (anti-caspase 9, from Abcam plc.) in cell lysis were analyzed using 12% SDS-PAGE, whereas DAPK1 (anti-DAPK1, from Sigma-Aldrich Co., St Louis, MO, USA.), AKT (anti-AKT, from Cell Signaling Technology, Inc.) and phosphorylated AKT (anti-phos-AKTser473, from Cell Signaling Technology, Inc.) were analyzed using 10% SDS-PAGE. β-Actin (anti-β-actin, from #TA-09, ZSGB-BIO, China) was the reference control. After being resolved by SDS-PAGE, proteins were transferred to Polyvinylidene Fluoride (PVDF) membranes and then blocked with 5% bovine serum albumin (Sigma-Aldrich Co.) for 1 h. Next, the membranes were separately incubated with primary antibodies at 4°C overnight, and then with appropriate HRP-conjugated secondary antibody (from #ZDR-5306 and −5307, ZSGB-BIO) at room temperature for 1 h. After detecting signals using ECL reagents (Merck Millipore, Billerica, MA, USA), gray value of different proteins was quantified with ImageJ v1.28 and normalized to β-actin.
TUNEL analysis
TUNEL (Roche Molecular Systems, Inc., Basel, Switzerland) staining was used for measuring DNA fragmentation. HBEpiCs were fixed before detection. The procedures were carried out according to the manufacturer’s instructions.
Caspase-3/7-positive cell detection and TMRM detection
The CellEvent™ Caspase-3/7 Green detection reagent (Thermo Fisher Scientific) labels nuclei of caspase 3/7-positive cells to report apoptosis. Tetramethylrhodamine, methyl ester reagent (TMRM; Thermo Fisher Scientific) was used to observe the mitochondrial membrane potential. HBEpiCs were loaded with 50 nM TMRM followed by 20 μM CellEvent™ detection reagent. Each reagent was incubated for 30 min, and fluorescence signals were observed in living cells.
Statistical analysis
All experiments were conducted independently at least 3 replicates. Continuous variables are presented as mean ± SD. Data were analyzed using SPSS 13.0 (SPSS Inc., Chicago, IL, USA), and bar graphs were protracted using Prism (version 5.0, GraphPad Software Ltd, San Diego, CA, USA). If the data distribution was normal, comparisons among three or more groups were estimated with an ANOVA test and comparisons between two groups were estimated by two-tailed Student’s t-test. If the data distribution was not normal, comparisons were estimated by Wilcoxon signed-rank test for nonparametric analysis. p<0.05 was considered statistically significant.
Results
PM2.5 increased the apoptotic response in cigarette-inflamed HBEpiCs
To verify the effect of PM2.5 in cigarette-inflamed bronchial epithelium, HBEpiCs were cultured with normal media, CSS or PM2.5-CSS for 24 h. Typical features of apoptosis were evaluated including mitochondrial membrane potential loss, DNA fragmentation and caspase activation. Caspase activities and mitochondrial membrane potential were detected in living cells (Figure 1A). Compared with normal HBEpiCs, CSS- and PM2.5-CSS-treated HBEpiCs showed a significant loss of mitochondrial membrane potential with an increase of caspase 3/7 activation. Compared with CSS-treated HBEpiCs, PM2.5-CSS cells showed a greater loss of mitochondrial membrane potential with a greater increase of caspase 3/7 activation. Meanwhile, DNA fragmentation was detected in fixed cells via TUNEL analysis (Figure 1C and D). Compared with normal HBEpiCs, the proportion of TUNEL-positive cells of CSS (18.6%±3.6% vs 1.4%±0.7%, p<0.001) and PM2.5-CSS-treated HBEpiCs (40.9%±3.1% vs 1.4%±0.7%, p<0.001) showed a significant increase. The difference between CSS- and PM2.5-CSS treated HBEpiCs was significant (p<0.001).
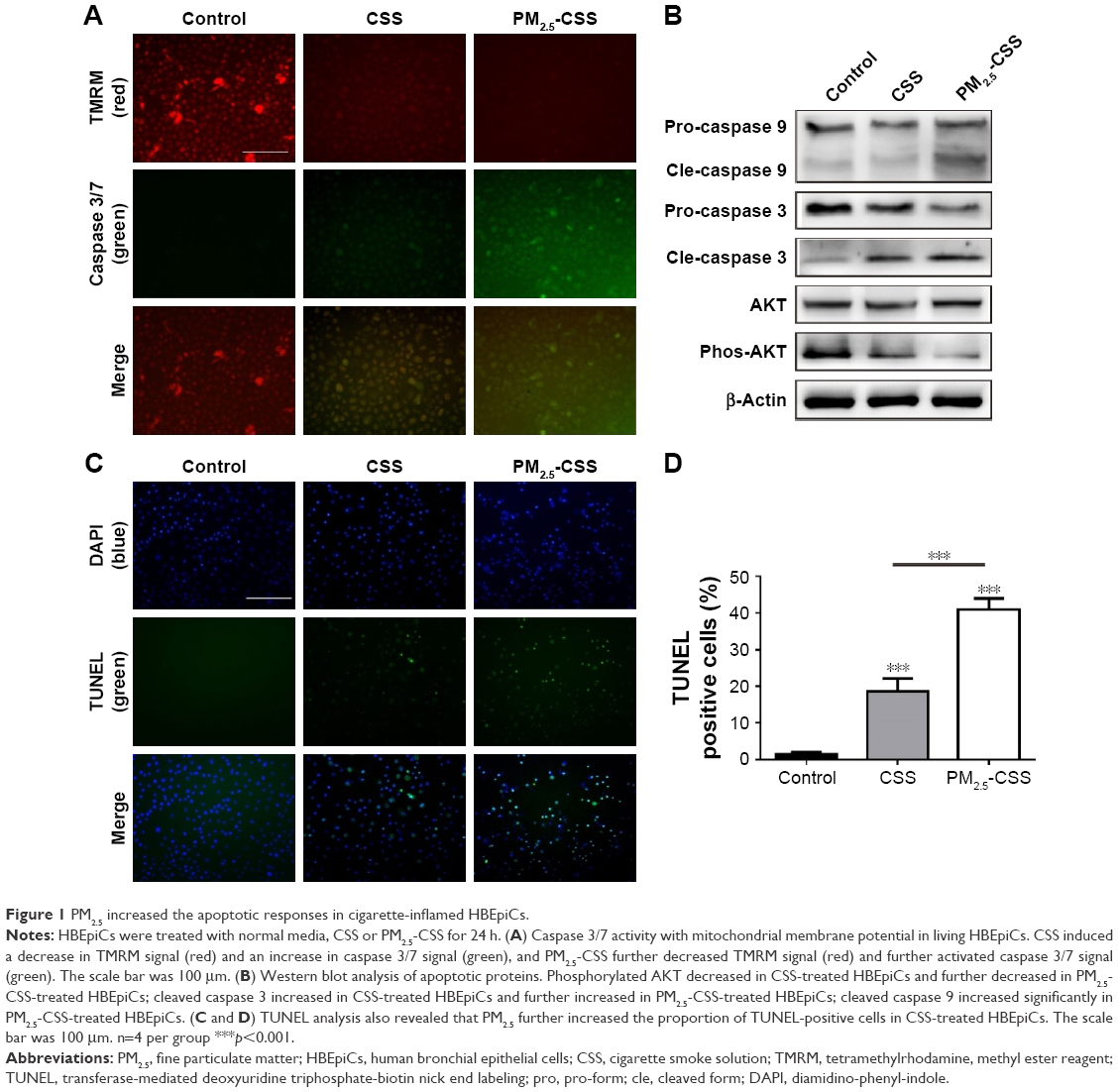 | Figure 1 PM2.5 increased the apoptotic responses in cigarette-inflamed HBEpiCs. |
AKT plays a critical role in cell survival and resistance,27 while caspases play an essential role in apoptosis activation.28,29 Expressions of these proteins were detected using western blot analysis (Figure 1B). The results showed that AKT phosphorylation decreased, while cleavage of caspase 3 increased in CSS-treated HBEpiCs. In PM2.5-CSS-treated cells, AKT phosphorylation further decreased and cleavage of caspase 3 further increased. Cleavage of caspase 9 increased significantly in PM2.5-CSS cells but showed no significant change in CSS cells. The abovementioned results demonstrated that PM2.5 aggravated apoptosis in CSS-treated HBEpiCs.
PM2.5 decreased the expression of miR-194-3p in cigarette-inflamed HBEpiCs
In order to observe the expression of miR-194-3p in HBEpiCs, we detected the expression of miR-194-3p by qRT-PCR (Figure 2A). Compared with control, the expression of miR-194-3p showed no statistical difference in CSS-treated HBEpiCs (0.75±0.26 vs 1.00±0.06, p=0.152), whereas the expression of miR-194-3p showed a significant decrease in PM2.5-CSS-treated cells (0.26±0.19 vs 1.00±0.13, p=0.019). The difference between CSS- and PM2.5-CSS treated HBEpiCs was significant (p=0.003).
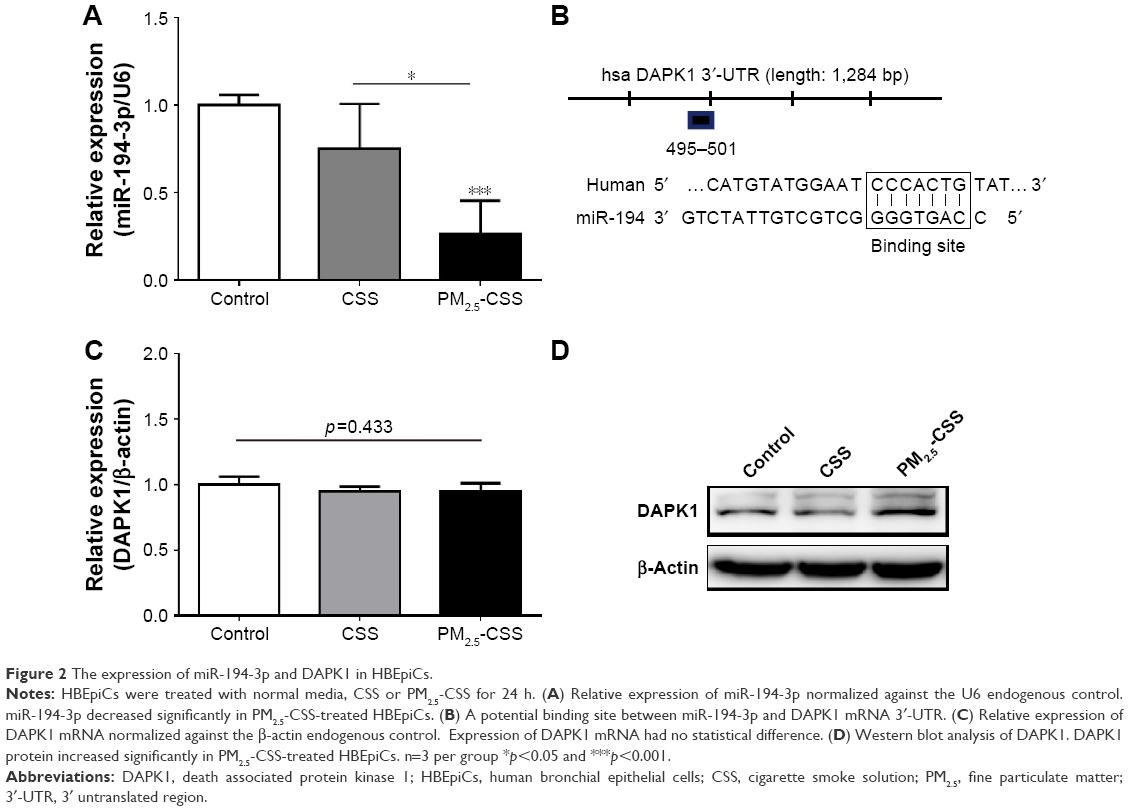 | Figure 2 The expression of miR-194-3p and DAPK1 in HBEpiCs. |
DAPK1 was a potential target of miR-194-3p and upregulated in PM2.5-CSS HBEpiCs
To explore the mechanism of miR-194-3p in PM2.5-CSS HBEpiCs, three bioinformatics algorithms, miRanda, TargetScan and miRWalk, were searched to find potential regulatory targets of miR-194-3p. All algorithms predicted DAPK1 as a target of miR-194-3p and the putative target sequence is located in 495-501nt of the 3′-UTR of human DAPK1 mRNA (Figure 2B). Then, expressions of DAPK1 mRNA and protein were detected in normal, CSS- and PM2.5-CSS-treated HBEpiCs. There was no significant difference in the expression of DAPK1 mRNA (normal 1.00±0.06 vs CSS 0.95±0.04 vs PM2.5-CSS 0.96±0.06, p=0.433) (Figure 2C). On the other hand, compared with control, the expression of DAPK1 protein showed a significant upregulation in PM2.5-CSS-treated HBEpiCs (normalized gray value: PM2.5-CSS 2.88±0.44 vs control 1.00±0.00, p<0.001), but it was not significantly different in CSS-treated HBEpiCs (normalized gray value: CSS 1.12±0.31 vs control 1.00±0.00, p=0.662). The difference in the expression of DAPK1 protein between CSS and PM2.5-CSS HBEpiCs was statistically significant (p<0.001) (Figure 2D).
DAPK1 mRNA was directly targeted by miR-194-3p
Next, luciferase analysis was used to identify the predicted binding site of miR-194-3p and DAPK1. The DAPK1 mRNA containing the wt putative binding site or mut binding site of miR-194-3p were constructed (Figure 3A) and cloned into the luciferase expressing pMIR vector. DAPK1-3′-UTR-wt vector or DAPK1-3′-UTR-mut vector was co-transfected with hsa-miR-194-3p mimics or scrambled control (miR-NC) into HEK293T cells, respectively. The results showed that the relative luciferase activity decreased significantly in cells transfected with DAPK1-3′-UTR-wt vector (0.78±0.04 vs 1.00±0.06, p<0.001), but luciferase activity showed no significant changes in cells transfected with DAPK1-3′-UTR-mut vector (0.92±0.08 vs 1.00±0.10, p=0.137) (Figure 3B). This result confirmed that miR-194-3p directly regulated DAPK1.
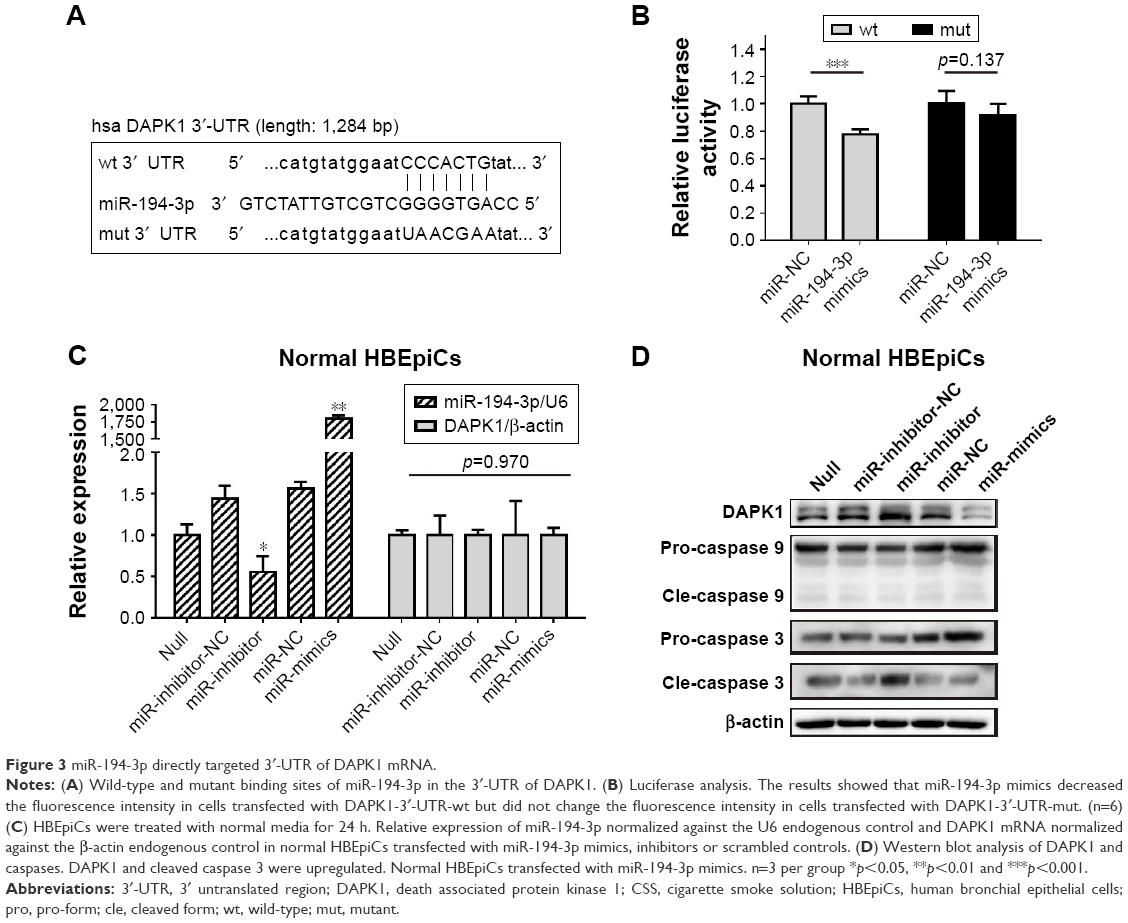 | Figure 3 miR-194-3p directly targeted 3′-UTR of DAPK1 mRNA. |
Decreased miR-194-3p directly upregulated the expression of DAPK1 protein and induced activation of caspase 3
To explore the potential regulation relationship between DAPK1 and miR-194-3p, normal HBEpiCs were transfected with miR-194-3p mimics, inhibitors and scrambled controls. The efficiency of transfection and the expression of DAPK1 mRNA were detected by qRT-PCR, and the expression of DAPK1 mRNA showed no significant difference among different HBEpiCs (Figure 3C). The expression of DAPK1 protein was detected using western blot analysis. The expression of DAPK1 protein increased significantly after inhibition of miR-194-3p (normalized value: inhibitors 3.12±0.92 vs null 1.00±0.00, p<0.001), whereas the expression decreased significantly after overexpression of miR-194-3p (normalized value: mimics 0.40±0.16 vs null 1.00±0.00, p=0.006) (Figure 3D). Previous research has reported that DAPK1 is an upstream regulator of caspase 3, which activates apoptosis.30–32 Cleavage of caspases was detected by western blot analysis. Inhibition of miR-194-3p significantly increased the cleavage of caspase 3 in normal HBEpiCs (normalized value: inhibitors 3.12±0.92 vs null 1.00±0.00, p<0.001) (Figure 3B). However, inhibition of miR-194-3p had little influence on the cleavage of caspase 3 in normal HBEpiCs. Also, there was no significant difference in the cleavage of caspase 9. These results showed that inhibition of miR-194-3p upregulated the expression of DAPK1 as well as cleavage of caspase 3 in normal HBEpiCs.
Overexpression of miR-194-3p downregulated DAPK1 protein directly and suppressed apoptosis in PM2.5-CSS HBEpiCs
Lastly, to observe whether miR-194-3p could regulate apoptosis in PM2.5-CSS-treated HBEpiCs, PM2.5-CSS HBEpiCs were pre-transfected with miR-194-3p mimics, inhibitors and scrambled controls. The efficiency of transfection and the expression of DAPK1 mRNA were detected by qRT-PCR (Figure 4A), and the expression of DAPK1 mRNA showed no significant difference. However, western blot results showed the DAPK1 protein expression decreased significantly in cells treated with mimics (normalized value: 0.60±0.15 vs 1.00±0.00, p=0.009). The DAPK1 protein expression significantly increased in cells treated with inhibitors (normalized value: 1.46±0.11 vs 1.00±0.00, p=0.020) (Figure 4B).
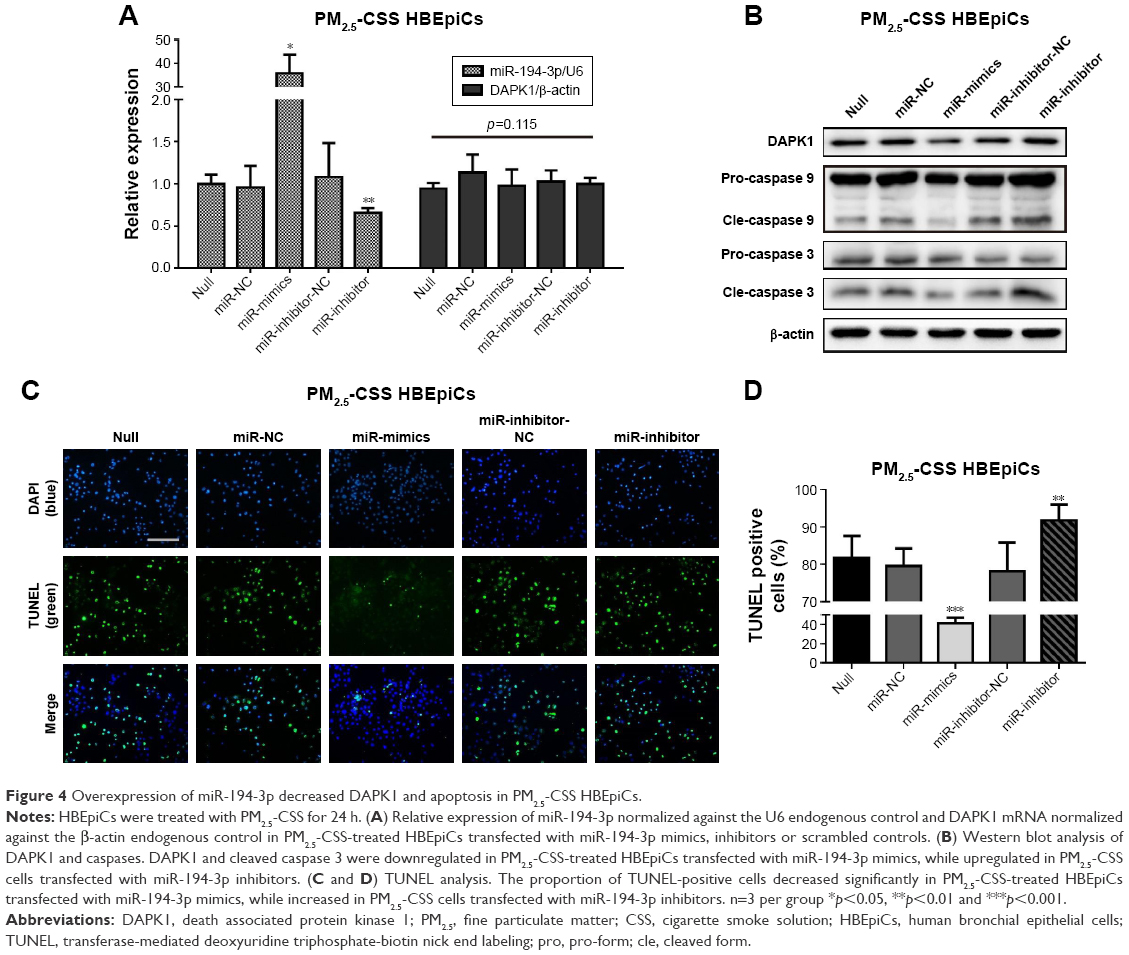 | Figure 4 Overexpression of miR-194-3p decreased DAPK1 and apoptosis in PM2.5-CSS HBEpiCs. |
Apoptosis was firstly verified using western blot analysis (Figure 4B). Overexpression of miR-194-3p significantly decreased cleavage of caspase 3 in PM2.5-CSS HBEpiCs (normalized value: 0.53±0.25 vs 1.00±0.00, p=0.024), but there was just a marginal significance in cleavage of caspase 9 (normalized value: 0.79±0.09 vs 1.00±0.00, p=0.060). To the contrary, inhibition of miR-194-3p significantly increased both cleavage of caspase 3 (normalized value: 1.46±0.11 vs 1.00±0.00, p<0.001) and cleavage of caspase 9 (normalized value: 1.41±0.12 vs 1.00±0.00, p=0.002) in PM2.5-CSS HBEpiCs. TUNEL analysis is shown in Figure 4C and D. Overexpression of miR-194-3p significantly decreased the proportion of TUNEL-positive cells in PM2.5-CSS HBEpiCs (mimics 41.2%±5.8% vs null 81.7%±5.9%, p<0.001; mimics 41.2%±5.8% vs NC 79.6%±4.7%, p<0.001), whereas inhibition of miR-194-3p significantly increased the proportion (inhibitors 91.7%±4.3% vs null 81.7%±5.9%, p=0.028; inhibitors 91.7%±4.3% vs inhibitor-NC 78.1%±4.2%, p=0.005). These results demonstrated that overexpression of miR-194-3p inhibited the production of DAPK1 and suppressed apoptosis in PM2.5-CSS HBEpiCs.
Discussion
miRNAs are acknowledged as key regulators in post-transcriptional modification. After constituting RNA-induced silencing complex, miRNAs bind directly to the 3′-UTR of target mRNA leading to mRNA degradation (complete complementary pairing) or translation suppression (incomplete complementary pairing). The stable expression of miRNAs in specific tissues and the conservative role of their mechanism in different species indicate their importance, with an enormous research and therapeutic potential.15,16 Several miRNA therapeutics such as mimics and inhibitors have been used in clinical trials or basic animal research. The most famous clinical appliance is miravirsen, an miR-122 inhibitor for the treatment of hepatitis C virus.33 So far, several miRNAs have been identified as potential therapeutic targets of lung diseases. For example, miR-16 mimics induce apoptosis and suppress proliferation of non-small cell lung cancer cells.34 miR-15535 and miR-19a36 are considered to be therapeutic targets of asthma. Nowadays, many experts have claimed that miRNAs are ideal therapeutic targets for COPD in the present and future.37,38 Hundreds of miRNAs in serum, plasma, sputum and lung tissues have been profiled to be associated with COPD. Several miRNAs have been identified to have therapeutic effects on COPD in vitro. Osei et al17 reported that miR-146-5p mimics reduced the release of IL-8 in COPD pulmonary fibroblasts. Ezzie et al18 found that miR-15b was significantly upregulated in stage IV COPD, and overexpression of miR-15b directly targets SMAD7 and then downregulates TGF-β in human bronchial epithelial cell line BEAS-2B. Hassan et al39 found that miR-199a-5p mimics effectively downregulated the unfolded protein response and decreased the release of cytokines in a1-antitrypsin-deficient monocytes.
In the meantime, epidemiological evidence has clarified that COPD patients are more vulnerable to PM2.5.40 Once PM2.5 is inhaled, COPD patients suffer from significant dyspnea and lung function decrease.41 Meta-analysis demonstrated that a 10 μg/m3 increase in PM2.5 was associated with a 2.36% (95% CI 1.00%–3.73%) increased risk of COPD-associated hospital admissions.42 Short-term exposure to outdoor air pollution increased the mortality rate of COPD by 1%, 1% and 6% in China, the European Union and the USA, respectively. Long-term exposure increased the mortality rate by 10%.43 Oxidative stress is the main effect of PM2.5. Researchers suggest that the inflammatory response involved in PM2.5-induced lung injury is very similar to the inflammation involved in COPD.9,44 For example, PM2.5 activated NF-κB-dependent pathway inducing release of IL-6 and monocyte adhesion,45 just like the reaction in COPD. Ye et al46 found that PM2.5 promoted bronchial smooth muscle cell migration and airway remodeling, just as in COPD. So, this suggests that the mechanism of PM2.5 in COPD is that PM2.5 aggravates the original lung inflammation of COPD. In an animal model, Gu et al14 found that PM2.5 accelerated the original Th1 and Th17 inflammation in lung tissues of COPD mice via Notch signal pathways.
miRNAs are also sensitive to air pollution,19 and so miRNAs might be regulators in the mechanism of PM2.5 in COPD. Previously, we found that 10 miRNAs (miR-691, miR-181a, miR-146a, miR-146b, miR-21a-5p, miR-129, miR-155, miR-139-5p, miR-21a-3p and miR-340) were upregulated in lung tissues of mice exposed to PM2.5.47 However, because the design of these air pollution-related miRNA studies were different, there are still no consensus from different researches. Our unpublished preliminary study firstly identified that blood miR-194-3p was decreased significantly in untreated COPD patients. Then, we verified that miR-194-3p was decreased in healthy adults after PM2.5 exposure, and miR-194-3p showed statistically positive correlation with lung function at lag1 and lag0. This inspired us to conclude that miR-194-3p might be an important regulator participating in the mechanism of PM2.5 on COPD.
Members of the miR-194 family are novel markers that were first reported in the intestine and kidney. Previous evidence from tumor research showed that miR-194 was able to inhibit cell proliferation20–23 and suppress inflammation48,49 and epithelial mesenchymal transition.21,50,51 In the miR-194 family, miR-194-3p is reported less than miR-194-5p. Alteration of miR-194-3p has been reported in chronic hepatitis B infection52 and necrotizing enterocolitis.53 Jung et al54 reported that histone deacetylase inhibitor upregulated the expression of miR-194-3p and induced anti-tumor effects in cholangiocarcinoma cells. Recently, miR-194 was confirmed to be expressed in lung tissues. Zhu et al24 reported that miR-194 was decreased in non-small lung cancer samples and inhibited proliferation of non-small lung cancer cells. However, miR-194 has not been discussed in COPD or PM2.5 exposure.
In the present study, we found that PM2.5 aggravated apoptosis of cigarette-inflamed HBEpiCs, and we verified that miR-194-3p declined significantly in PM2.5-CSS-treated HBEpiCs. Furthermore, we found that miR-194-3p directly targeted DAPK1. DAPK1, belonging to a family of Ser/Thr Kinase, is a regulator of cell apoptosis, and DAPK1 acts as a mediator in apoptosis.55 Previous research found that DAPK1 mediates the pro-apoptotic activity via TGF-β death signals,56 p53 pathway57 and NF-κB signaling pathways.58 There is sufficient evidence showing that DAPK1 is the upstream regulator of caspase 3 in apoptosis. Apoptosis in cells transfected with DAPK1 increased with activation of the caspase 3-dependent pathway,30 while caspase 3 activation decreased in DAPK1 knockdown cells compared to control siRNA-transfected cells.59 Our study found that inhibition of miR-194-3p upregulated the expression of DAPK1 and cleaved caspase 3 in normal HBEpiCs. Not only that, overexpression of miR-194-3p decreased DAPK1 and caspase 3 cleavage, resulting in the suppression of apoptosis in PM2.5-cigarette smoke exposed HBEpiCs. This suggested that miR-194-3p might be a protective miRNA and a potential therapeutic target.
There are some problems that need to be discussed in the future. First, as miR-194-3p decreased significantly only in PM2.5-CSS HBEpiCs not in CSS HBEpiCs, we did not discuss the role of miR-194-3p in CSS HBEpiCs. Whether miR-194-3p is a regulator in COPD should be verified in future using more appropriate cells from patients or animal models. Secondly, we found that miR-194-3p targeted DAPK1 directly and activated caspase 3 downstream; whether there were different apoptotic pathways involved was not analyzed in our study. Lastly, our study illustrated that miR-194-3p is a very promising therapeutic target of bronchial epithelial cells. Next, transfected animal studies should be prepared to find a reasonable dose and measurement of treatment. It is necessary to find the most appropriate way to deliver miR-194-3p mimics to bronchial epithelial cells and to reduce the nonspecific toxicity at the same time.
Conclusion
Our experiments proved that PM2.5 downregulates miR-194-3p and accelerates apoptosis in cigarette-inflamed bronchial epithelium by directly targeting DAPK1. Reduction of miR-194-3p upregulated expression of DAPK1 and cleavage of caspase 3. Overexpression of miR-194-3p could suppress apoptosis in PM2.5-cigarette-exposed bronchial epithelial cells. These findings suggest that miR-194-3p could be a potential therapeutic target for the treatment of PM2.5-induced bronchial epithelial injury aggravation.
Acknowledgments
The authors would like to acknowledge Obio Technology Corp. for their support on luciferase analysis. This work was supported by the National Natural Science Foundation of China (grant nos 81370106 and 8150010304), Beijing Municipal Natural Science Foundation (grant no 7161013) and National key research and development plan (grant no 2017YFC1309500). There was an oral presentation of this study at 2017 Congress of Asian Pacific Society of Respirology, named “THE ROLE OF PM2.5 PLAYED IN CIGARETTE SMOKEINFLAMED PULMONARY EPITHELIUM”.
Disclosure
The authors report no conflicts of interest in this work.
References
GOLD. Global Strategy for the Diagnosis, Management, and Prevention of COPD: Full Repo (2017), November 16, 2016. Available from: http://goldcopd.org | ||
Burney PG, Patel J, Newson R, Minelli C, Naghavi M. Global and regional trends in COPD mortality, 1990–2010. Eur Respir J. 2015;45(5):1239–1247. | ||
Jayes L, Haslam PL, Gratziou CG, et al; Tobacco Control Committee of the European Respiratory Society. SmokeHaz: systematic reviews and meta-analyses of the effects of smoking on respiratory health. Chest. 2016;150(1):164–179. | ||
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445–2454. | ||
Salvi S, Barnes PJ. Is exposure to biomass smoke the biggest risk factor for COPD globally? Chest. 2010;138(1):3–6. | ||
Hu G, Zhou Y, Tian J, et al. Risk of COPD from exposure to biomass smoke: a metaanalysis. Chest. 2010;138(1):20–31. | ||
Hogg JC. Why does airway inflammation persist after the smoking stops? Thorax. 2006;61(2):96–97. | ||
Lapperre TS, Postma DS, Gosman MM, et al. Relation between duration of smoking cessation and bronchial inflammation in COPD. Thorax. 2006;61(2):115–121. | ||
World Health Organization. Ambient (outdoor) air quality and health. [Fact sheet]. Updated May 2, 2018. Available from: http://www.who.int/mediacentre/factsheets/fs313/en/ | ||
Agency USEP. Particulate Matter (PM) Basics. Updated September 12, 2016. Available from: https://www.epa.gov/pm-pollution/particulate-matter-pm-basics#PM | ||
Bernstein JA, Alexis N, Barnes C, et al. Health effects of air pollution. J Allergy Clin Immunol. 2004;114(5):1116–1123. | ||
Brunekreef B, Holgate ST. Air pollution and health. Lancet. 2002;360(9341):1233–1242. | ||
DeVries R, Kriebel D, Sama S. Outdoor air pollution and COPD-related emergency department visits, hospital admissions, and mortality: a meta-analysis. COPD. 2017;14(1):113–121. | ||
Gu XY, Chu X, Zeng XL, Bao HR, Liu XJ. Effects of PM2.5 exposure on the Notch signaling pathway and immune imbalance in chronic obstructive pulmonary disease. Environ Pollut. 2017;226:163–173. | ||
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. | ||
Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16(3):203–222. | ||
Osei ET, Florez-Sampedro L, Tasena H, et al. miR-146a-5p plays an essential role in the aberrant epithelial-fibroblast cross-talk in COPD. Eur Respir J. 2017;49(5). pii: 1602538. | ||
Ezzie ME, Crawford M, Cho JH, et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax. 2012;67(2):122–131. | ||
Vrijens K, Bollati V, Nawrot TS. MicroRNAs as potential signatures of environmental exposure or effect: a systematic review. Environ Health Perspect. 2015;123(5):399–411. | ||
Pichiorri F, Suh SS, Rocci A, et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell. 2010;18(4):367–381. | ||
Meng Z, Fu X, Chen X, et al. miR-194 is a marker of hepatic epithelial cells and suppresses metastasis of liver cancer cells in mice. Hepatology. 2010;52(6):2148–2157. | ||
Xu S, Zhang B, Zhu Y, et al. miR-194 functions as a novel modulator of cellular senescence in mouse embryonic fibroblasts. Cell Biol Int. 2017;41(3):249–257. | ||
Han K, Zhao T, Chen X, et al. microRNA-194 suppresses osteosarcoma cell proliferation and metastasis in vitro and in vivo by targeting CDH2 and IGF1R. Int J Oncol. 2014;45(4):1437–1449. | ||
Zhu X, Li D, Yu F, et al. miR-194 inhibits the proliferation, invasion, migration, and enhances the chemosensitivity of non-small cell lung cancer cells by targeting forkhead box A1 protein. Oncotarget. 2016;7(11):13139–13152. | ||
Blue ML, Janoff A. Possible mechanisms of emphysema in cigarette smokers. Release of elastase from human polymorphonuclear leukocytes by cigarette smoke condensate in vitro. Am Rev Respir Dis. 1978;117(2):317–325. | ||
Zhao C, Liao J, Chu W, et al. Involvement of TLR2 and TLR4 and Th1/Th2 shift in inflammatory responses induced by fine ambient particulate matter in mice. Inhal Toxicol. 2012;24(13):918–927. | ||
Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal. 2011;23(10):1515–1527. | ||
Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81(4):505–512. | ||
Nagata S, Tanaka M. Programmed cell death and the immune system. Nat Rev Immunol. 2017;17(5):333–340. | ||
Lee JH, Rho SB, Chun T. Programmed cell death 6 (PDCD6) protein interacts with death-associated protein kinase 1 (DAPk1): additive effect on apoptosis via caspase-3 dependent pathway. Biotechnol Lett. 2005;27(14):1011–1015. | ||
Bialik S, Kimchi A. The death-associated protein kinases: structure, function, and beyond. Annu Rev Biochem. 2006;75:189–210. | ||
Singh P, Ravanan P, Talwar P. Death associated protein kinase 1 (DAPK1): a regulator of apoptosis and autophagy. Front Mol Neurosci. 2016;9:46. | ||
Janssen HL, Reesink HW, Lawitz EJ, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368(18):1685–1694. | ||
Wang W, Chen J, Dai J, Zhang B, Wang F, Sun Y. MicroRNA-16-1 inhibits tumor cell proliferation and induces apoptosis in A549 non-small cell lung carcinoma cells. Oncol Res. 2016;24(5):345–351. | ||
Zhou H, Li J, Gao P, Wang Q, Zhang J. miR-155: a novel target in allergic asthma. Int J Mol Sci. 2016;17(10). pii: E1773. | ||
Moheimani F, Hsu AC, Reid AT, et al. The genetic and epigenetic landscapes of the epithelium in asthma. Respir Res. 2016;17(1):119. | ||
Bracke KR, Mestdagh P. MicroRNAs as future therapeutic targets in COPD? Eur Respir J. 2017;49(5). pii: 1700431. | ||
Osei ET, Florez-Sampedro L, Timens W, Postma DS, Heijink IH, Brandsma CA. Unravelling the complexity of COPD by microRNAs: it’s a small world after all. Eur Respir J. 2015;46(3):807–818. | ||
Hassan T, Carroll TP, Buckley PG, et al. miR-199a-5p silencing regulates the unfolded protein response in chronic obstructive pulmonary disease and α1-antitrypsin deficiency. Am J Respir Crit Care Med. 2014;189(3):263–273. | ||
Heinrich J, Schikowski T. COPD patients as vulnerable subpopulation for exposure to ambient air pollution. Curr Environ Health Rep. 2018;5(1):70–76. | ||
Peacock JL, Anderson HR, Bremner SA, et al. Outdoor air pollution and respiratory health in patients with COPD. Thorax. 2011;66(7):591–596. | ||
Atkinson RW, Kang S, Anderson HR, Mills IC, Walton HA. Epidemiological time series studies of PM2.5 and daily mortality and hospital admissions: a systematic review and meta-analysis. Thorax. 2014;69(7):660–665. | ||
Song Q, Christiani DC, Wang X, Ren J. The global contribution of outdoor air pollution to the incidence, prevalence, mortality and hospital admission for chronic obstructive pulmonary disease: a systematic review and meta-analysis. Int J Environ Res Public Health. 2014;11(11):11822–11832. | ||
Ling SH, van Eeden SF. Particulate matter air pollution exposure: role in the development and exacerbation of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2009;4:233–243. | ||
Liu CW, Lee TL, Chen YC, et al. PM2.5-induced oxidative stress increases intercellular adhesion molecule-1 expression in lung epithelial cells through the IL-6/AKT/STAT3/NF-κB-dependent pathway. Part Fibre Toxicol. 2018;15(1):4. | ||
Ye X, Hong W, Hao B, et al. PM2.5 promotes human bronchial smooth muscle cell migration via the sonic hedgehog signaling pathway. Respir Res. 2018;19(1):37. | ||
Hou T, Liao J, Zhang C, Sun C, Li X, Wang G. Elevated expression of miR-146, miR-139 and miR-340 involved in regulating Th1/Th2 balance with acute exposure of fine particulate matter in mice. Int Immunopharmacol. 2018;54:68–77. | ||
Tian H, Liu C, Zou X, Wu W, Zhang C, Yuan D. MiRNA-194 regulates palmitic acid-induced toll-like receptor 4 inflammatory responses in THP-1 cells. Nutrients. 2015;7(5):3483–3496. | ||
Zhao HJ, Ren LL, Wang ZH, et al. MiR-194 deregulation contributes to colorectal carcinogenesis via targeting AKT2 pathway. Theranostics. 2014;4(12):1193–1208. | ||
Dong P, Kaneuchi M, Watari H, et al. MicroRNA-194 inhibits epithelial to mesenchymal transition of endometrial cancer cells by targeting oncogene BMI-1. Mol Cancer. 2011;10:99. | ||
Li Z, Ying X, Chen H, et al. MicroRNA-194 inhibits the epithelial-mesenchymal transition in gastric cancer cells by targeting FoxM1. Dig Dis Sci. 2014;59(9):2145–2152. | ||
Ninomiya M, Kondo Y, Kimura O, et al. The expression of miR-125b-5p is increased in the serum of patients with chronic hepatitis B infection and inhibits the detection of hepatitis B virus surface antigen. J Viral Hepat. 2016;23(5):330–339. | ||
Ng PC, Chan KY, Leung KT, et al. Comparative MiRNA expressional profiles and molecular networks in human small bowel tissues of necrotizing enterocolitis and spontaneous intestinal perforation. PLoS One. 2015;10(8):e0135737. | ||
Jung DE, Park SB, Kim K, Kim C, Song SY. CG200745, an HDAC inhibitor, induces anti-tumour effects in cholangiocarcinoma cell lines via miRNAs targeting the Hippo pathway. Sci Rep. 2017;7(1):10921. | ||
Shohat G, Spivak-Kroizman T, Cohen O, et al. The pro-apoptotic function of death-associated protein kinase is controlled by a unique inhibitory autophosphorylation-based mechanism. J Biol Chem. 2001;276(50):47460–47467. | ||
Jang CW, Chen CH, Chen CC, Chen JY, Su YH, Chen RH. TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase. Nat Cell Biol. 2002;4(1):51–58. | ||
Raveh T, Droguett G, Horwitz MS, DePinho RA, Kimchi A. DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat Cell Biol. 2001;3(1):1–7. | ||
Yoo HJ, Byun HJ, Kim BR, Lee KH, Park SY, Rho SB. DAPk1 inhibits NF-κB activation through TNF-α and INF-γ-induced apoptosis. Cell Signal. 2012;24(7):1471–1477. | ||
Wu B, Yao H, Wang S, Xu R. DAPK1 modulates a curcumin-induced G2/M arrest and apoptosis by regulating STAT3, NF-κB, and caspase-3 activation. Biochem Biophys Res Commun. 2013;434(1):75–80. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.