")
Back to Journals » Cancer Management and Research » Volume 11
Plausibility of trophoblastic-like regulation of cancer tissue
Authors Piechowski J
Received 15 October 2018
Accepted for publication 30 April 2019
Published 31 May 2019 Volume 2019:11 Pages 5033—5046
DOI https://doi.org/10.2147/CMAR.S190932
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Rituraj Purohit
Jean Piechowski
Physician-Radiotoxicologist, Paris F-75011, France
Background: Thus far, a well-established logical pattern of malignancy does not exist. The current approach to cancer properties is primarily descriptive with usually, for each of them, extensive analyses of the underlying associated biomolecular mechanisms. However, this remains a catalog and it would be valuable to determine the organizational chart that could account for their implementation, hierarchical links and input into tumor regulation.
Hypothesis: Striking phenotypic similarities exist between trophoblast (invasive and expanding early placenta) and cancer regarding cell functions, logistics of development, means of protection and capacity to hold sway over the host organism. The concept of cancer cell trophoblastic-like transdifferentiation appears to be a rational proposal in an attempt to explain this analogy and provide a consistent insight into how cancer cells are functioning. Should this concept be validated, it could pave the way to promising research and therapeutic perspectives given that the trophoblastic properties are vital for the tumor while they are permanently epigenetically turned off in normal cells. Specifically targeting expression of the trophoblastic master genes could thereby be envisaged to jeopardize the tumor and its metastases without, in principle, inducing adverse side effects in the healthy tissues.
Conclusion: A wide set of functional features of cancer tissue regulation, including some apparently paradoxical facts, was reviewed. Cancer cell misuse of physiological trophoblastic functions can clearly account for them, which identifies trophoblastic-like transdifferentiation as a likely key component of malignancy and makes it a potential relevant anticancer target.
Keywords: cancer stem cells, trophoblast, transdifferentiation, epigenetics, immune system
Background
Cancer tissue is an ectopic tissue and its forced acceptance and thereafter support by the host organism cannot invariably occur by chance as a result of random mutations. Malignant cell survival and interactions with the host organism are based on a coordinated set of functions that all contribute to the tumor development with a systematic ascendency over the healthy tissues. Deregulated proliferation, constitutive genetic and/or epigenetic oncogenic alterations, ineffective tumor suppressor genes, and multifaceted genome anomalies and instability are at the root of the cancer phenotype. Cell survival with such disorders, a striking feature of cancer, is particularly challenging and no doubt necessitates a super efficient logistical support, even more so given the stress induced by the potential host tissue inhospitality.
Severe, nonrepairable DNA damage is a potential pitfall for cell functioning and progeny. It typically leads to senescence, apoptosis, mitotic catastrophe or necrosis. However, diverse gross chromosome aberrations or gene mutations slip through the safety net of the genes in charge of the quality control of the genome and may remain nonlethal provided that the cell logistics and vital properties are not jeopardized. Well-known examples are the chromosome abnormalities of Down syndrome (trisomy 21), Turner and Klinefelter sexual syndromes (respectively X0 and XXY aneuploidies), cat’s cry syndrome (5p deletion), etc, or the gene mutations causing hemophilia, cystic fibrosis, albinism, etc. Likewise, cells survive if they are able to compensate the malfunction by an appropriate epigenetic reprogramming. This is exactly what seems to happen in cancer. In fact, the oncogene-induced premalignant lesions present a “sluggish”, erratic development with commonly observed, scattered all over the place, occurrence of foci of cell blockage manifested as postmitotic-like state (senescence) or cell death (apoptosis). Besides an array of irreversible structural DNA damages, this precarious survival mainly reflects a background of cell functioning insufficiency resulting from corruption of signaling pathways and from cell inability to cover the extra vital needs imposed by the aberrant proliferation.1–4 Moreover, the microenvironment is a priori not suitable for, and counteracts, any ectopic cell growth. Quite similar situations exist in other fields of biology where various cell blockages may occur due to “over-activation or saturation” phenomena. This kind of physiological shock is, for instance, the case of thyroid function shutdown after administration of an acute high dosage of iodine (the “Wolff–Chaikoff effect”).5 In plant biology, addition of a limited number of gene copies may lead to a co-suppression of gene expression, eg, loss instead of increase of color in pigmented petunia petals.6
Coming back to the issue of cancer, certain cells are able to override the overgrowth-related and host-induced stresses, which implies a substantial functional reprogramming. They would form the actual malignant tissue that, unlike premalignant lesions, is virtually free of foci of senescence and apoptosis,3 although precancer and cancer cells are potentially mixed, especially during the initial stage of tumor growth. The cells engaged in the malignant progression have a high phenotypic plasticity and, in addition to this, a self-renewal ability to ensure a continuous development of the cancer tissue. They are typically stem-like cells and certainly comprise true stem cells, most likely progenitors, and perhaps certain de-differentiated cells. Concerning the resurgent program that determines the malignant evolution, it turns out that a physiological condition, involving a singular multifunctional tissue interrelation, is strikingly plagiarized by cancer tissues. It is the implantation of trophoblast, ie, the early placenta,7 in endometrium. Besides its invasive properties, trophoblast promotes angiogenesis and participates in uterine artery remodeling for oxygen and nutrient supply. In parallel, it determines a special local immune status involving immune tolerance to avoid any immune reaction from the mother against the embryo, while maintaining effective means of defense against pathogens.
The strong similarities between the mechanisms of trophoblast and cancer development were hypothetically attributed to re-expression of the trophoblastic master genes in cancer cells. This epigenetic reprogramming was called “malignant transdifferentiation” because, in parallel to save the unstable precancer cells it would constitute, in combination with the structural oncogenic genome alterations, the basis of malignancy.8 Expression of the trophoblastic program is physiologically strictly forbidden after birth because it would determine ectopic, nonhomeostatic tissue implantation. Hence, acquisition of the trophoblastic logistic properties, normally dormant, would explain the invasive behavior of cancer cells and the distinctive hijacking, to their advantage, of the host tissue functions.
Both trophoblastic and cancer cells have a comprehensive set of pro-survival functions devoted to fast growth of “semi-foreign” cells, including:8–12
- Autocrine growth factors enabling an autonomous growth inside the host organism;
- Metabolism with adaptive enzymatic pathways ensuring an optimal balance between synthesis of cell components and energy production whatever the oxygen tension. Aerobic glycolysis13 (the “Warburg effect”) is a distinctive option consisting in nonmitochondrial, oxygen-free ATP production despite normoxic conditions, when the glucose metabolites are mainly used for biosynthesis purposes. It leads to an excess of lactic acid release;
- Cell resistance and resilience, as shown by survival under poor and even adverse conditions, and by very efficient capacity to process xenobiotics;
- Antiapoptotic factors;
- Latent mesenchymal properties, eg, ability to achieve epithelial-mesenchymal transition (EMT)14–16 or get a vascular endothelial-like phenotype,17 a process called “vascular mimicry”. Through plagiarizing these trophoblastic phenotypic changes, malignant cells may develop advantageous adaptive properties for themselves or for the tumor;
- EMT-related invasion of the host tissues with extracellular matrix proteolysis. Cells may also migrate via amoeboid movement or transfer as in the fetomaternal microchimerism;
- Promotion of cell motility and metastases by neural (N) and placental (P) cadherins;18
- Giant cell formation by cell-cell fusion or acytokinetic DNA replication;
- Host tissue and local stroma conditioning, especially by implementation of angiogenesis and immune tolerance, in order to support the development of a “foreign” tissue;
- Secretion of the human chorionic gonadotropin or part (the beta chain) thereof.
However, due to various mutations, corruption of signaling pathways and dysfunctions in cancer cells, the symbiotic relationship between the tumor and the host stroma is not as perfect as in the case of the trophoblast and the decidua. Despite these obstacles, the basic trophoblastic pro-survival mechanisms are clearly working in cancer. To complement the properties discussed above, the purpose of this paper is to consider significant functional features and paradoxes resulting from the presumed trophoblastic-like regulation of cancer tissue, with the aim of assessing the soundness of this hypothesis. A unified pattern of cancer setting-up and development is then described.
Pro-survival self-sufficiency in growth factors
A key factor for cell life consists in stimulation by growth factors. Absence of growth factors ends up with senescence or apoptosis. A basic characteristic of trophoblastic cells is their self-sufficiency in growth factors thanks to autocrine loops, allowing the early trophoblast to survive even in the case of absent or weak extrinsic stimulation.19 This is also a biological feature of cancer cells involving the same range of growth factors,9–11 including receptor tyrosine kinase (RTK) ligands and TGF-β. It results in protumor effects, ie, growth autonomy, increase of the original set of growth factors and enhancement of the oncogenic signaling, all of them most likely contributing to the transition from precancer to real cancer. These growth factors promote tumor progression through boosting the proliferation and invasion processes,20 just as it happens in trophoblast. Another concurrent pro-survival and de facto promalignant action is that of inhibitor of apoptosis proteins (IAPs) such as survivin, expressed in both trophoblastic and cancer cells.
Tumor shaping by the TGF-β superfamily
The TGF-β superfamily is a large group of ubiquitous multifunctional cytokines, structurally similar to the originally discovered TGF-β1, that activate TGF-β cell-surface serine/threonine kinase receptors (TGFBRs). The signal propagates along the SMAD proteins while interfering in several non-SMAD signaling networks, eg, by stimulating the mitogen-activated protein kinase (MAPK) and protein kinase B (Akt) pathways downstream of RTKs.21 The outcome of the TGF-β action depends on the type and current biological status of the cell, and on the activating TGF-β ligand, thus determining context-dependent and possibly variable effects on cell fate and functions. Put simply, the canonical SMAD proteins are antiproliferative while the noncanonical pathways, for instance, MAPK and Akt, are pro-proliferative. Negative feedback regulation of TGF-β signaling is achieved by Lefty, a TGF-β-related cytokine, and by SMAD6 and SMAD7 proteins, all of them selectively neutralizing signal transmission along the SMAD pathways.22
Basically, TGF-β participates in the regulation of the cellular transition from stemness to differentiated state and contributes to the homeostatic balance between the parenchymal and stromal components of a tissue.23 TGF-β fosters the mesenchymal/fibroblastic phenotype24,25 in concert with fibroblast growth factor (FGF) signaling whereas it participates in the restrictive control of the other cell populations.26 Thus, TGF-β improves the development of fibroblasts and actively promotes EMT, while it inhibits growth and possibly favors apoptosis of parenchymal cells. TGF-β also restricts the development and activity of immune cells, and fosters the immune suppressive regulatory T cells. When excessive, TGF-β actions determine fibrosis and high immune tolerance.
It is currently admitted that the core signaling component involved in the contradictory effects of TGF-β on cell proliferation, in function of the cell type, is the SMAD cytostatic transcriptional program27 that prevents expression of c-myc and cyclin-dependent kinases (CDKs). It is operational in parenchymal cells but not in mesenchymal/fibroblastic cells where it is being silenced by activation of the MAPK and Akt pathways driven by TGF-β non-SMAD signaling, FGF and more broadly RTK-related growth factors.28–30 The silencing mechanism consists in specific impediment to the SMAD-driven transcription of the cytostatic genes. Owing to their mesenchymal potential, a similar process, with disruption of the SMAD cytostatic program, occurs in trophoblastic31–33 and malignant cells,30 strengthened by the impact of oncogenic abnormalities in the latter. Nevertheless, this is clearly not an easy issue to get to grips with since the TGF-β signaling is sensitive to numerous interfering factors that may determine an apparent versatility of its final action.
TGF-β expressed in the endometrium and the trophoblast contributes to embryo implantation, especially through promoting a hospitable stromal microenvironment inside the decidua and acquisition of immune tolerance at the maternal–fetal interface. TGF-β secreted from the blastocyst has also been proposed to foster apoptosis of endometrial epithelial cells at the site of implantation.34,35 Virtually similar effects concerning the local tissue conditioning are observed during tumor development. These effects can be explained, at least partly, by enhanced secretion of TGF-β both from growing cancer cells endowed with trophoblastic-like properties and from stromal cells. The implantation-related apoptosis of host cells is also boosted by trophinin, a marker of both trophoblastic and cancer cells.36,37
Antioncogenic in normal tissues and at the very beginning of the cancer process, the role of TGF-β seems to change, apparently becoming pro-oncogenic and promoting tumor progression once the cancer process really starts. Convergence of various events interfering with the regular TGF-β functions may explain this functional alteration.30 Thus, TGF-β may lose its antiproliferative efficiency due to inactivating mutations occurring in the TGF-β/TGFBR/SMAD axis, and/or oncogene-induced hindrance to the SMAD-driven transcription of the cytostatic genes, and/or nuclear factor kappa B (NF-κB)-induced expression of SMAD7. All this would lead TGF-β to signal primarily via the noncanonical pro-growth MAPK/Akt pathways in place of SMAD.
In addition, the putative cancer cell trophoblastic-like transdifferentiation may well largely participate in weakening the antiproliferative action of TGF-β and even explain its apparent switch from an antitumor towards a protumor role. This applies to:
- The constitutive loops of autocrine RTK-related growth factors, a typical feature of both malignant and trophoblastic cells, activating the MAPK/Akt/c-Myc/CDKs pathways and disrupting the TGF-β/SMAD cytostatic program;
- Acquisition of latent mesenchymal properties, a phenotypic condition facilitating cell development under the control of TGF-β;
- Autocrine production of the β-chain of the human chorionic gonadotropin (hCG), a typical trophoblastic hormone, in advanced cancers. It may structurally trick and block the TGF-β receptors of the tumor cells, thus preventing the normal TGF-β signaling.38
In sum, the physiological antiproliferative action of TGF-β does not work in cancer cells while it remains optimal in normal parenchymal cells. This leads to a development imbalance between the malignant and the normal tissues at the expense of the latter (Figure 1).
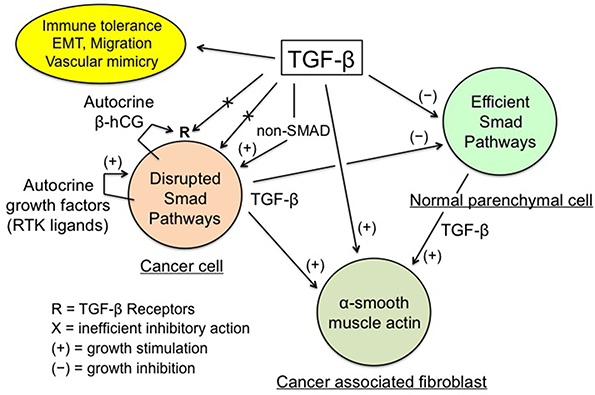 | Figure 1 TGF-β regulation of cancer tissue. Notes: Enhanced TGF-β secretion by cancer and stromal cells promotes the development of a dense myofibroblastic microenvironment with CAFs expressing α-smooth muscle actin, together with immune tolerance and EMT, thus plagiarizing the features of the placental trophoblast-decidua interface. Conversely, inhibition of cancer cell growth by the physiological TGF-β cytostatic program is virtually nonexistent, due to various obstacles: impairment of the TGF-β/SMAD axis and of the SMAD-driven gene transcription, high expression of SMAD7, and actions of autocrine growth factors and β-hCG. The β-chain of hCG, a hormone normally produced by trophoblast, may interfere with TGF-β receptors, thus preventing the normal action of TGF-β. The above figure is valid for trophoblastic cells (in place of cancer cells), with the SMAD cytostatic program being silenced by RTK ligands and β-hCG, and for decidual myofibroblasts (in place of CAFs). |
TGF-β production increases with tumor size and promotes the development of a tumor stroma caricaturing the myofibroblastic differentiation of the decidua and wounds. Its main features are presence of mesenchyme-related cancer-associated fibroblasts (CAFs) and marked imbalance of the connective tissue in favor of a myofibroblastic microenvironment with a high extracellular matrix deposition and fibrosis-like texture. The synergistic TGF-β/FGF profibroblastic action is the determining fact, which sends us back to the historical discovery of the “transforming factor” allowing fibroblasts to develop in very poor culture media39 and thus in any tissue and microenvironment. Aggressiveness of cancer cells correlates with high production of TGF-β, stiff stroma and high mechanotransduction signaling, all of them presenting a positive gradient towards the invasive front of the tumor.40 Increase of TGF-β also strengthens the local immune tolerance, this being true for cancer tissue and trophoblast. Another aspect of malignancy is the prometastatic action of TGF-β through its active promotion of EMT with type switching of cadherins and integrins, a condition conducive to cell migration, just as in the case of the invasive extravillous cytotrophoblast.
Ambivalent role of the immune system
Immune system participates in multifaceted protection of tissues. It ensures defense against a broad variety of pathogens and helps in maintaining tissue quality through cleaning and healing damaged tissues. Innate immune activity consists of immediate and nonspecific defense from pathogens. Cytotoxic T lymphocytes act directly against damaged, infected, and cancer cells, and microbial agents. Antigen presenting cells (dendritic cells, macrophages, B cells, granulocytes) respond in a short delay to toll-like receptor (TLR) detection of ligands related to potentially harmful situations, ie, pathogen-associated molecular patterns (PAMPs) and debris-associated molecular patterns (DAMPs), through engulfing and processing such materials, and then presenting the antigenic fragments to helper T cells in order to initiate an adaptive immune response, while activating a wide inflammatory signaling network.41 TLRs, NF-κB transcription factor and tumor necrosis factor-α (TNF-α) cytokine are key actors of the inflammatory process.42 The memory-based adaptive immune system, consisting of B cells producing antibodies and of antigen-specific cytotoxic T cells, provides long lasting, efficient specific defense against pathogens. Immunological activity is tightly controlled. Inhibitory immune checkpoint receptors (programmed cell death protein 1, PD-1) and their PD-L1/L2 ligands, all being transmembrane proteins present on the surface of immune cells, limit the immune response. The PD-1: PD-L1/L2 interaction antagonizes the co-stimulation of immune cells that are responding to antigenic signals.43 This immune activity mitigation, reinforced by the immunosuppressive action of regulatory T cells, prevents any excessive immune reaction.
Proper progress of pregnancy requires that decidua is a site enabling development of the “foreign” semi-allogeneic trophoblast while, at the same time, maintaining host defense against pathogens. This immunological duality is also part of the relation between the tumor and the host organism. The same multiple synergistic mechanisms as in pregnancy contribute to a discriminative immune tolerance towards the population of cancer cells.12 They act locally in the immediate vicinity of the trophoblastic or cancer cells. Hence, (a) HLA-G antigens and PD-L1/L2 ligands expressed on these cells activate the matching inhibitory immune checkpoint receptors on the immune cells in situ, thus antagonizing the antigen-induced co-stimulation, (b) myeloid-derived suppressive cells (MDSC)44,45 recruited by chemoattraction blunt the immune response, typically by inducing extra catabolism of tryptophan and arginine, and subsequent T cell depletion, (c) the complement-mediated cytolysis is locally impeded by action of specific cell-surface anticomplement factors, and (d) the trophoblastic C-terminal truncated helicase-like transcription factor, an alternative splicing isoform, would lessen the cytotoxicity of natural killer cells.46 Hence, the putative trophoblastic-like transdifferentiation of cancer cells and the related polyvalent mechanisms of local immune tolerance would explain why immune fighting against cancer has clearly a rather poor achievement. Fighting against infectious agents remains concurrently fully preserved through TLR activation on immune, stromal and trophoblastic or tumor cells (see the next section), and ensuing both release of chemokines and activation of the cytokine network.47
In addition to the flux of molecules regulating the inflammatory process aimed at fighting pathogens or other potentially harmful components, immune cells participate in tissue recovery through secretion of cytokines and growth-and-angiogenic factors promoting healing. The dual function results in a contradictory effect on cancer tissue, this one being both attacked and, at the same time, supported by the immune system. The tumor progrowth effectiveness comes from the fact that cancer cells take advantage of the physiological inflammation-related means being used to heal wounded or damaged healthy tissues.48 Interestingly, this also applies to invasion of endometrium (or of another tissue in the case of ectopic pregnancy) by trophoblast. Indeed, DAMPs-induced inflammation improves embryo implantation and subsequent pregnancy progress (Figure 2).49
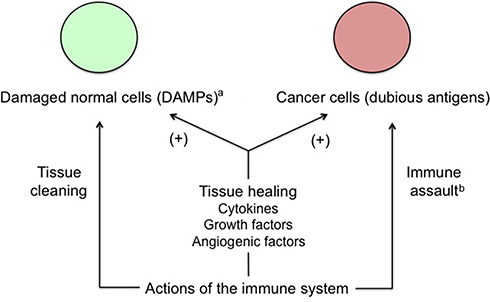 | Figure 2 Apparent conflicting actions of the immune system on cancer cells. Notes: Cancer tissue conveniently takes advantage of the physiological, immune-related healing mechanisms and improves its growth potential this way. Moreover, cancer cells induce a trophoblastic-like immune tolerance limiting the immune destructive action against them. aFor example, wounds or invasion of endometrium by trophoblast. bLimited by trophoblastic-like immune tolerance. |
A clearly dual role characterizes the group of tumor-associated macrophages (TAMs).50 They schematically fall into two subpopulations: (a) pro-inflammatory M1 macrophages that sustain the immune reaction and fight pathogens and undesirable materials, and (b) anti-inflammatory M2 macrophages that lower the immune reaction and help in cleaning and regenerating the damaged tissues. M2 macrophages support wound healing. M1/M2 polarization is parallel and related to the Th1, Th17/Th2 polarization of helper T cells, and is modulated by specific cytokines and microenvironment events. Singularly, besides the common M1 subpopulation, a significant and very active M2 subpopulation is present in the decidua to support trophoblast development,51,52 and in the tumor stroma where it exerts a protumor activity.53 There was a metaphorical suggestion that “trophoblast cells ‘educate’ monocytes/macrophages to create an adequate environment that promotes trophoblast survival”. As a consequence of the putative trophoblastic-like transdifferentiation, this aphorism could apply to cancer cells as well.
Signaling maze of TLRs, TNF-α and NF-κB
NF-κB protein dimer is a transcription factor having a central role in inflammation. It is, in particular, activated downstream of the TLRs, TNF-α and TGF-β signaling pathways42,54 (Figure 3) and has multifaceted functions, including modulation of secretion of inflammatory cytokines, notably TNF-α, but also support to cell growth and survival through limitation of apoptosis, increase of angiogenesis and activation of cell-cycle promoting genes.55 Moreover, NF-κB upregulates SMAD7 that exerts a negative feedback control on SMAD signaling.56
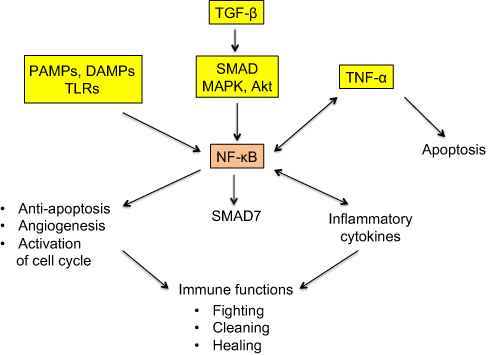 | Figure 3 Simplified representation of the signaling maze of TLRs, TNF-α and NF-κB. Notes: NF-κB is a pleiotropic transcription factor with ambivalent, pro- and antitumor actions. PAMPs and DAMPs activate NF-κB via TLRs. TNF-α is an inflammatory cytokine present both downstream and upstream of NF-κB. TGF-β activates NF-κB via the interlinked SMAD and MAPK/Akt signaling cascades, and NF-κB induces a SMAD7-driven negative feedback loop. All these facts clearly mean that the final action of the represented system, that works, among others, in cancer and trophoblast, is not unequivocal and that it strongly depends on a lot of interfering factors. Striking similarities exist between cancer and trophoblast as regards the immune-related fighting, cleaning, and healing actions. |
TLR4 receptors are engaged in innate immunity against endotoxin/lipopolysaccharide (LPS). In pregnancy, their activation in placenta and uterine stroma triggers TNF-α-induced placental inflammation and hemorrhage.57 LPS causes the same TNF-α-related effects inside tumors.58 This analogy is consistent with the fact that, besides the regular immune cells, TLRs are also expressed on decidual and trophoblastic cells at the placental-maternal interface,59 and on stromal and cancer cells in cancer tissue.60 TLR presence on trophoblastic and cancer cells provides evidence of an immune-like activity shared by these “canonically nonimmune cells”, concurrently with “canonically immune cells”. PAMPs, DAMPs and antigenic tumor debris activate TLRs on all these cells, which, not only induces both innate and adaptive immune reactions against the pathogens and the tumor, but also inherently results in activation of NF-κB that triggers production of inflammatory cytokines and promotes cell growth and survival through its previously mentioned multifunctional actions. This potential support to tumor expansion is in line with the dual fighting/healing role of the immune system discussed in the previous section. The conflicting roles of TLRs in cancer progression are pretty difficult to evaluate. This matter is somewhat akin to the below TNF-α paradox.
Besides being an antitumor inflammatory cytokine, TNF-α appears to have a protumor effect at low concentration. Indeed, in this context, chemoattracted myeloid cells undergo a TNF-α-induced endothelial differentiation, which contributes to an increase of the microvascular density.61 However, TNF-α most likely remains primarily an antitumor agent and the effect in question probably mainly results from TNF-α-induced activation of the NF-κB transcription factor whose protective, proangiogenic and progrowth actions overcome the TNF-α regular cytotoxicity and apoptotic effects (Figure 3).62 Interestingly, a sustained activation of NF-κB is observed in both trophoblastic and cancer cells,63,64 and trophoblastic-like transdifferentiation may well be, among others, a potential explanation of this similarity.
TNF-α (cachectin) and NF-κB also participate in an inflammation-related, systemic alteration of metabolism characterized by a pronounced, unstoppable wasting of both skeletal muscle and fat storage, and an increase of brown adipose tissue where the energy produced by mitochondria is directly released as heat instead of being stored in ATP.65 The mechanism and pathophysiological interpretation of this organism collapse remain to be deciphered. Far from fostering the immune host defenses and hindering tumor development, the inflammatory status of the cachectic syndrome paradoxically reinforces tumor ascendancy over the healthy tissues. The systemic runaway of the host inflammatory machinery (Figure 3) caused by cancer invasion thus appears to have a procancer effect caricaturing the improving effect of inflammation on trophoblastic implantation.49
Puzzling expression of the trophoblast cell surface antigen 2 (Trop-2)
Intronless TACSTD2 gene encodes the Trop-2 glycoprotein, a cell surface transmembrane receptor interacting with several ligands, in particular insulin-like growth factor-1 (IGF-1). It induces intracellular calcium release and activates the MAPK and Akt signaling pathways, thus implementing cell growth and EMT.66,67 Its anti-adhesive activity boosts the migratory phenotype.68 Trop-2 is a marker of cell stemness and is present in many normal tissues. It was originally identified with a high expression level on human trophoblast cells. The putative cancer cell trophoblastic-like reprograming would lead to overexpression of the different trophoblastic markers and, in particular, Trop-2 (Figure 4). Trop-2 is typically associated with tumor progression. It is often overexpressed in various types of human carcinomas, compared with the corresponding normal tissue, and correlates with a poor patient prognosis.69 In the case of Trop-2 failure due to either TACSTD2 gene defect, loss of heterozygosity (LOH), or epigenetic silencing, there would yet be a lot of other trophoblastic biomarkers still expressed. Lack of just Trop-2 would most likely have no significant negative impact on the tumor fate because it is indeed one cancer-sustaining factor, probably subsidiary, among many others.
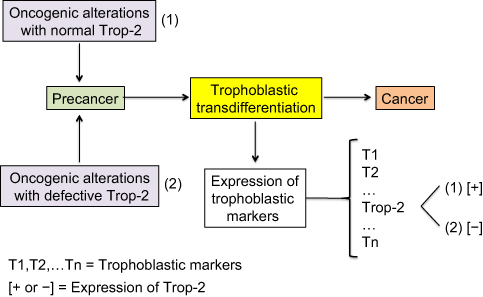 | Figure 4 Expression of the trophoblastic Trop-2 biomarker in cancer cells. Notes: Both overexpression and, in a few cases, nonexpression are observed in carcinogenesis. Overexpression of Trop-2 is assumed to result from the putative trophoblastic-like transdifferentiation and subsequent activation of the TACSTD2 gene. Nonexpression simply means that the TACSTD2 gene is defective or epigenetically silenced, while the other trophoblastic biomarkers should logically be expressed. Trop-2 typically promotes and less frequently restricts cancer progression, depending on the histotype. |
Less frequently, in a few histotypes, Trop-2 plays an opposite role, ie, high Trop-2 expression mitigates, while low expression promotes, cancer development.70 One tentative explanation is that Trop-2 would compete with the IGF-1 receptor (IGF-1R) for the shared IGF-1 ligand, in a specific context where IGF-1R is supposed to activate a strategic oncogenic pathway. The hypothesis is that Trop-2 is intrinsically a tumor promoter, although of weak effectiveness, and that its apparent ambivalent role results from the fact that, if it is highly expressed, it may outcompete and functionally take the place of the pro-oncogenic IGF-1R, thus shifting the cell towards a weaker malignant status. Conversely, in the case of low expression of Trop-2, the strategic IGF-1R pro-oncogenic pathway is not overcome and determines a high cell malignancy. Nevertheless, all this should yet be further clarified.
Modulation of tumor propagation by gap junction intercellular communication (GJIC)
Connexins (Cx) are short-lived transmembrane proteins able to form communication channels enabling ionic and molecular exchanges between adjacent cells. Their family includes 21 genes identified in humans. Connexins and cadherins are components of a tightly regulated dynamic system that governs intercellular relations, ensuring coordinated tissue functioning. Cx43, the most widespread of them, is virtually present in all cell types, whereas Cx40 is mainly expressed in cardiovascular structures. Cx43 has clearly conflicting roles in cancer, preventing early stages and conversely promoting late stages of cancer development.71–73
This duality may be explained by reference to the role of connexins (Cx) in the trophoblast where, while Cx43 fosters cell-cell adhesion, communication and fusion with formation of syncytiotrophoblast and giant cells, Cx40-expressing extravillous cells actively proliferate to form compact outgrowths, and connexin-depleted cells detach therefrom, migrate, colonize the decidua and invade the spiral arteries.74–76 Cx40 likely generates weaker gap junctions and cell-cell adhesion than Cx43, which facilitates cell proliferation and prevents cell-cell fusion. By analogy with the above, the regular Cx43-type gap junctions would mediate efficient cell-cell adhesion in the beginning tumor, thus limiting multiplication and dissemination of cancer cells. In this case, Cx43 would act as tumor suppressor, which is its typical role in early primary tumors. Then, while weaker gap junctions mimicking the Cx40-type ones mitigate cell-cell adhesion and facilitate the tumor growth, connexin-depleted cells gradually appear and detach, migrate and can enter blood and lymph vessels. Subsequent re-expression of Cx43 would support attachment of these cells to endothelial lining, thus enabling their diapedesis and extravasation, which paves the way to production of metastases. Once there, cancer cells would thrive, using the Cx43 gap junctions to implement vital exchanges with stromal cells.77,78 Cx43 would thus become a tumor promoter involved in the viability of metastases. In sum, weak gap junctions and connexin downregulation would be specific to proliferative and invasive cancer cells, whereas Cx43 would slow their proliferation but also promote their metastatic implantation and stromal support.
Hence, cancer cells misuse trophoblastic mechanisms of intercellular communication to grow and boost metastasis development. Connexins, cadherins and integrins are determining factors of the mode of migration (mesenchymal or amoeboid) of invasive cells.79 The regulation and quality of connexins are probably disrupted in cancer, with an uncertain functional impact.80
Growth pattern of cancer cell population and ascendancy over healthy tissues
Despite being impacted by an array of genetic and epigenetic anomalies, cancer cells overcome the host organism. They survive poor local conditions, colonize the healthy tissues away from the primary tumor and the malignant process often restarts after a therapeutic eradication. A model of cancer development and resilience, matching these features, has been proposed recently.8 The perennial, necessary element is a stem-like cell (true stem cell, progenitor or uncommonly de-differentiated cell) with self-renewal capacity. This type of cell can both continuously generate differentiated daughter cells through asymmetric divisions and participate to an extensive increase of the cell population through symmetric divisions. Phenotypic plasticity is another key feature allowing certain rare stem-like cells to acquire a malignant profile through trophoblastic-like transdifferentiation, this being constitutive and transmissible to the cell progeny.
It is important to notice that the transdifferentiation considered here is not strictly conventional since the resurgent, vital and growth properties are simply added to—and do not supersede—the original set of cell properties, especially those concerning the histological type. This means, for instance, that a skin cell does not become a trophoblast cell but that it acquires the latter’s logistic functions, and in particular the pro-survival and pro-invasive attributes, through reexpression of the respective master genes. The growth-restricting regulation that applies during pregnancy, ie, the time-and-space limitation of invasion of the endometrium by the trophoblast, is not operating here, so that the cell invasive potential is fully preserved and may be reactivated anytime to meet nutrient or oxygen needs or growth related space requirement. Secretion of the β-chain of the placental hCG hormone, a subsidiary effect of the epigenetic reprogramming of the trophoblastic logistical properties, is observed in certain advanced cancers. All this is the reason why the process was called “trophoblastic-like transdifferentiation” and not simply “trophoblastic transdifferentiation”.
This phenotypic evolution makes cells malignant because it confers them life supremacy on the model of trophoblast, a semi-allogeneic tissue having elaborated vital properties, able to hold sway over the host tissues, induce angiogenesis and immune tolerance, and invade healthy tissues. Typical features of normal stem cells are dormancy and high resistance to stress and xenobiotics. The reprogrammed trophoblastic properties certainly boost the resilience and resistance of malignant stem-like cells since trophoblast derives directly from totipotent cells, ie, the cells that possess the highest stem potential after the zygote.81
In this model, tumor development is based on stem-like cells producing an increasingly differentiated progeny that will constitute the bulk of the cancer tissue (Figure 5).82 The collection of differentiated cells produced by any given stem-like cell represents an increment to the malignant tissue and may be seen as an elementary malignant module (MM). Its growth and size depend on cell cycle kinetics, cell lifetime, microenvironment reaction, nutrient and oxygen supply, and a lot of intrinsic and extrinsic interfering factors. Such a model is fully compatible with formation of “spheres” in vitro,83 assuming that spheres play in vitro the role of MMs in vivo. As stem-like cells present symmetrical mitoses, the tumor is supposed to concurrently grow through progressive build-up of newly formed MMs. All the cells may potentially migrate but only stem-like cells can generate metastases because the differentiated cells lose self-renewal capability and have a decreasing proliferative potential. This approach stresses the pivotal role of cancer stem-like cells in both tumor growth and formation of metastases. Those being in a quiescent state may explain posttreatment relapses.
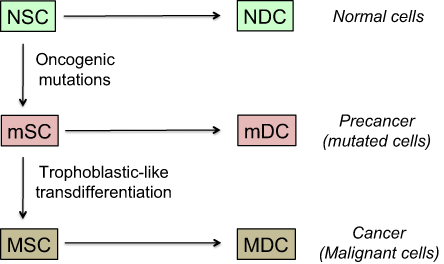 | Figure 5 Model of cancer tissue development. Notes: The general pattern and hierarchy of tissue structure, based on stem cells (SC) with self-renewal capabilities and differentiated daughter cells (DC) with decreased proliferation potential, is supposed to be the same for normal, mutated, or malignant cells. Stem cells are rare but certain of them have a metastatic potential and those being in a quiescent state may explain posttreatment relapses. They are hence key actors of the malignant process. Differentiated cells form the bulk of the tumor, but cannot create metastases nor induce posttreatment relapses because their lifetime is limited. The probability of trophoblastic-like transdifferentiation, that provides certain precancer cells with the vital logistical support leading to malignancy, is infinitesimal. More precisely, the transdifferentiation component that interests us here is essentially that which drives the extravillous phenotype. |
There is a degree of similarity between the metastatic process and invasion of endometrium by trophoblast, since both trophoblastic and tumor cells implement the same basic mechanisms and migrate individually as single cells or collectively as clusters of cells.84,85 Metastasis is a multistep and highly inefficient process.86 As mentioned above, only stem-like cells are able to initiate metastases and their rarity is de facto a strong limiting factor regardless of the other numerous potential pitfalls. However, progressive impairment of cancer cell differentiation over time leads to expansion of the stem-like cell population and thus boosts formation of metastases.
In practice, tumor development is not really well defined because, besides fluctuations in the microenvironment, tumor cells have neither a constant frequency of division nor a regular cell cycle kinetics and may be in an active or inactive quiescent state. Mobility, fusion and death of certain cells also interfere. Moreover, in the mixture of cancer and precancer cells inside a tumor certain precancer stem-like cells may achieve a trophoblastic-like transdifferentiation too and become malignant, thus inaugurating development of extra malignant populations.
Another point concerns mutated cells displaying an apparently normal phenotype despite typical oncogenic genome alterations.87–92 Actual unambiguous precancer lesions may appear after a long latent period or may possibly never appear. Then, transition from indolent precancer to cancer, a likely infinitesimal probability event involving a trophoblastic-like reprogramming, would yet again be lasting a long time (Figure 5). Therefore, the entire carcinogenesis process is usually extremely long and has a very low chance of success.
Information was given on the transcription factors implicated in trophoblast development and function.93 Even if several of them, eg, TEAD, Ets1, TFAP2C, CREB, may definitely act as tumor promoters,94–97 further data are still needed on their role in tumorigenesis. Whatever the transcription factors, all are the product of secondary genes present in every cell. Their expression, depending on the cell type, is governed and regulated by upstream master genes (Figure 6). Here, our key issue relates to the trophoblastic master genes. Identifying them, especially those of the extravillous phenotype, should become the actual challenge.
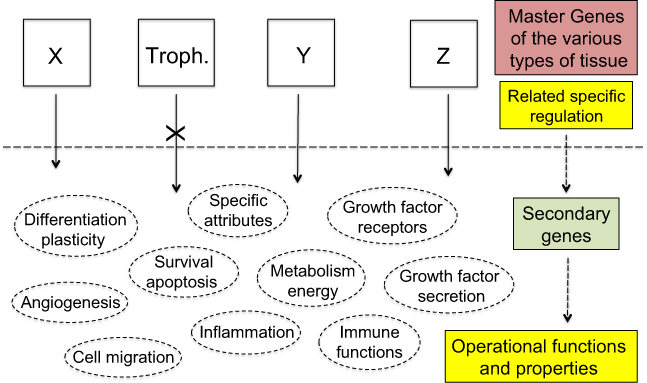 | Figure 6 Therapeutic strategies. Notes: The current anti-cancer therapies target operational functions and properties common to all cells and thus have adverse side effects against normal tissues for which use of certain of these functions and properties is potentially vital. Exclusively targeting the specific products of the trophoblastic master genes would de facto make cancer cells lack a suitable logistical support to survive whereas normal tissues would not be impacted as every operational function and property of normal cells would remain unaltered. X, Y, Z represent the master genes that determine the various types of tissue, eg, X may be the group of epidermis master genes, alike Y for kidney epithelium, or Z for thyroid epithelium, etc. “Troph.” represents the trophoblastic master genes. |
In sum, a lot of unpredictable cellular events occur during the tumorous process but constitutive expression of trophoblastic properties is certainly a constant feature that most likely accounts for the ascendancy of cancer cells over healthy tissues. The functional equivalence between the two pairs, (trophoblast + endometrium decidua) and (tumor + host stroma), that both are aimed at developing a “semi-foreign or ectopic tissue”, could explain the paradoxical host organism support to cancer tissue development.
Uniqueness of the malignant profile for whatever type of cell
In spite of various modulations in their development, malignant tumors do consistently present certain “canonical traits” concerning the parenchymal and stromal constituent parts, interactions with the host tissues, and continuous expansion in the host organism. Such a systematic pattern of cancer formation and evolution cannot only result from accumulation of random genetic and epigenetic alterations. Interestingly, although the landscape of leukemia apparently seems to be different from that of tumors, many basic features are indeed virtually similar:98
- Various triggering mutations, long duration and low chance of success of the process of leukemogenesis;
- Progression from preleukemia or chronic leukemia (equivalent to a precancer) to aggressive or acute leukemia (real cancer);
- Pyramidal structure of the cell population, with leukemic stem cells at the top, proliferative cells in the middle and lowly or nonproliferative cells at the bottom. The group of stem cells increases through symmetrical divisions. As described above for tumors, the differentiated cell progeny of a given stem cell would represent a MM and the leukemic cell population, that is like a “fluid tumor”, would result from the build up of MMs;
- Residence of leukemic stem cells inside supportive niches, in a context of bone marrow fibrosis. Leukemic stem cells also move away from the niches and form metastases in various tissues outside the bone marrow;
- Distinctive “trophoblastic-like” pro-survival and pro-expanding properties:99–104 autocrine growth factors, metabolism shift of leukemic cells towards aerobic glycolysis (the “Warburg effect”), microenvironment alteration promoting angiogenesis, active downregulation of immune surveillance, expression of antiapoptotic factors (survivin), cell migration and invasion with expression of EMT-related genes;
- Rarely, possible secretion of the β-chain of hCG, a typical trophoblastic hormone.105
It thus appears that the malignant process in leukemia comprises the same typical components as in tumors, ie, structural oncogenic genome alterations combined with an epigenetic reprogramming of cell logistics that results in implementation of trophoblastic-like functions. The same is true for lymphomas.106–111 Development of multiple myeloma is also supported by essentially the same cell logistics,112–117 and this seems to apply to sarcomas too.118–123 The shared group of functional properties at issue here is in fact a subset of the currently recognized hallmarks of cancer.4
Conclusion
Both cancer and early embryo are faced with the same problematic, ie, that of a semi-foreign or ectopic tissue with a high development rate and huge vital needs, engaged in colonization of a host organism. Trophoblast, the early placental structure, is designed to address the logistical challenges of embryo implantation in an optimized way. The trophoblastic program is found in a strictly dormant state in every cell of the body, as evidenced through reproductive cloning by transfer of the nucleus of a somatic cell into a denucleated oocyte. It seems to be reactivated in cancer cells, endowing them with pro-survival and latent mesenchymal properties, and to play a key functional role in cancer tissue regulation, in particular in expansion of mesenchyme-related myofibroblastic CAFs. A notable point is that in tumorous tissue both parenchyma and stroma present a high mesenchymal potential. The concept, in which the trophoblastic master genes act as master regulators of malignancy, would represent a unified mechanistic framework for the different types of cancer.
The distinctive properties of cancer tissue result from misuse of physiological trophoblastic functions. Obvious examples are: (a) boosting of tumor development by action of synergistic autocrine growth factors (RTK ligands, TGF-β), and by the vital support of angiogenesis and vascular mimicry; (b) involvement of EMT and specific connexin expression in the metastatic process; (c) unsetting of immune cytotoxic mechanisms to achieve immune tolerance. Such a functional redesign can be explained by a trophoblastic-like epigenetic reprogramming, a process at the root of the “malignant transdifferentiation” turning precancer into cancer. It would allow tumor cells to survive, expand and control the host organism so that this one actively supports tumor progression, with angiogenesis and immune tolerance being essential facts. It is hence proposed that future research and therapeutic choices should focus on the master genes at the root of the close similarities between trophoblastic and cancer properties.
The interesting point is that trophoblastic properties are vital for the malignant cells while they are epigenetically tightly silenced and never expressed in normal cells after birth. Hence, specifically targeting the master gene products governing the trophoblastic program would jeopardize the tumor with, in principle, no adverse side effects on healthy tissues (Figure 6). This would mean developing innovative therapies against the distinctive trophoblastic signaling cascades, targeting the upstream transcription factors and/or the downstream trophoblastic biomarkers. Finally, given that the trophoblastic program could be not only a major component of cancer cell functioning but also the target of specific therapies with, in theory, no adverse side effects, then the trophoblastic-like transdifferentiation would appear to be both the power and the Achilles’ heel of cancer.
Disclosure
The author reports no conflict of interest in this work.
References
1. Courtois-Cox S, Jones SL, Cichowski K. Many roads lead to oncogene-induced senescence. Oncogene. 2008;27:2801–2809. doi:10.1038/sj.onc.1210950
2. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi:10.1126/science.1140735
3. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. doi:10.1038/nrc2772
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi:10.1016/j.cell.2011.02.013
5. Leung AM, Braverman LE. Consequences of excess iodine. Nat Rev Endocrinol. 2014;10:136–142.
6. van der Krol AR, Mur LA, Beld M, Mol JN, Stuitje AR. Flavonoid genes in petunia: addition of a limited number of gene copies may lead to a suppression of gene expression. Plant Cell. 1990;2:291–299.
7. Burton GJ, Fowden AL. The placenta: a multifaceted, transient organ. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140066.
8. Piechowski J. Hypothesis about transdifferentiation as backbone of malignancy. Front Oncol. 2017;7:126.
9. Ferretti C, Bruni L, Dangles-Marie V, Pecking AP, Bellet D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum Reprod Update. 2007;13:121–141.
10. Holtan SG, Creedon DJ, Haluska P, Markovic SN. Cancer and pregnancy: parallels in growth, invasion, and immune modulation and implications for cancer therapeutic agents. Mayo Clin Proc. 2009;84:985–1000. doi:10.1016/S0025-6196(11)60669-1
11. Soundararajan R, Rao AJ. Trophoblast ‘pseudo-tumorigenesis’: significance and contributory factors. Reprod Biol Endocrinol. 2004;2:15. doi:10.1186/1477-7827-2-15
12. Piechowski J. Trophoblastic-like transdifferentiation: a key to oncogenesis. Crit Rev Oncol Hematol. 2016;101:1–11. doi:10.1016/j.critrevonc.2016.01.019
13. Smith DG, Sturmey RG. Parallels between embryo and cancer cell metabolism. Biochem Soc Trans. 2013;41:664–669. doi:10.1042/BST20120352
14. Vincent-Jordan N, Johnson GL, Abell AN. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle. 2011;10:2865–2873. doi:10.4161/cc.10.17.17188
15. DaSilva-Arnold S, James JL, Al-Khan A, Zamudio S, Illsley NP. Differentiation of first trimester cytotrophoblast to extravillous trophoblast involves an epithelial-mesenchymal transition. Placenta. 2015;36:1412–1418. doi:10.1016/j.placenta.2015.10.013
16. Davies JE, Pollheimer J, Yong HE, et al. Epithelial-mesenchymal transition during extravillous trophoblast differentiation. Cell Adh Migr. 2016;10:310–321. doi:10.1080/19336918.2016.1170258
17. Sun B, Zhang D, Zhao N, Zhao X. Epithelial-to-endothelial transition and cancer stem cells: two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget. 2017;8:30502–30510. doi:10.18632/oncotarget.8461
18. Vieira AF, Paredes J. P-cadherin and the journey to cancer metastasis. Mol Cancer. 2015;14:178. doi:10.1186/s12943-014-0278-9
19. Adamson ED. Activities of growth factors in preimplantation embryos. J Cell Biochem. 1993;53:280–287. doi:10.1002/jcb.240530403
20. Knöfler M, Pollheimer J. IFPA award in placentology lecture: molecular regulation of human trophoblast invasion. Placenta. 2012;33(Suppl):S55–S62. doi:10.1016/j.placenta.2011.09.019
21. Chaudhury A, Howe PH. The tale of transforming growth factor-beta (TGFbeta) signaling: a soigné enigma. IUBMB Life. 2009;61:929–939. doi:10.1002/iub.239
22. Ulloa L, Tabibzadeh S. Lefty inhibits receptor-regulated Smad phosphorylation induced by the activated transforming growth factor-beta receptor. J Biol Chem. 2001;276:21397–21404. doi:10.1074/jbc.M010783200
23. Xu X, Zheng L, Yuan Q, et al. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018;6:2.
24. Denu RA, Nemcek S, Bloom DD, et al. Fibroblasts and mesenchymal stromal/stem cells are phenotypically indistinguishable. Acta Haematol. 2016;136:85–97. doi:10.1159/000445096
25. Clark RA, McCoy GA, Folkvord JM, McPherson JM. TGF-beta 1 stimulates cultured human fibroblasts to proliferate and produce tissue-like fibroplasia: a fibronectin matrix-dependent event. J Cell Physiol. 1997;170:69–80. doi:10.1002/(SICI)1097-4652(199701)170:1<69::AID-JCP8>3.0.CO;2-J
26. Siegel PM, Massagué J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–821. doi:10.1038/nrc1208
27. Massagué J, Gomis RR. The logic of TGFbeta signaling. FEBS Lett. 2006;580:2811–2820. doi:10.1016/j.febslet.2006.04.033
28.
29. Horowitz JC, Lee DY, Waghray M, et al. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem. 2004;279:1359–1367. doi:10.1074/jbc.M306248200
30. Seoane J. Escaping from the TGFbeta anti-proliferative control. Carcinogenesis. 2006;27:2148–2156. doi:10.1093/carcin/bgl068
31. Erlebacher A, Price KA, Glimcher LH. Maintenance of mouse trophoblast stem cell proliferation by TGF-beta/activin. Dev Biol. 2004;275:158–169. doi:10.1016/j.ydbio.2004.07.032
32. Lash GE, Otun HA, Innes BA, Bulmer JN, Searle RF, Robson SC. Inhibition of trophoblast cell invasion by TGFB1, 2, and 3 is associated with a decrease in active proteases. Biol Reprod. 2005;73:374–381. doi:10.1095/biolreprod.105.040337
33. Forbes K, Westwood M. Maternal growth factor regulation of human placental development and fetal growth. J Endocrinol. 2010;207:1–16. doi:10.1677/JOE-10-0174
34. Jones RL, Stoikos C, Findlay JK, Salamonsen LA. TGF-beta superfamily expression and actions in the endometrium and placenta. Reproduction. 2006;132:217–232. doi:10.1530/rep.1.01076
35. Staun-Ram E, Shalev E. Human trophoblast function during the implantation process. Reprod Biol Endocrinol. 2005;3:56. doi:10.1186/1477-7827-3-23
36. Tamura N, Sugihara K, Akama TO, Fukuda MN. Trophinin-mediated cell adhesion induces apoptosis of human endometrial epithelial cells through PKC-δ. Cell Cycle. 2011;10:135–143. doi:10.4161/cc.10.1.14448
37. Kim SW, Yang HG, Kang MC, et al. KIAA1114, a full-length protein encoded by the trophinin gene, is a novel surface marker for isolating tumor-initiating cells of multiple hepatocellular carcinoma subtypes. Oncotarget. 2014;5:1226–1240. doi:10.18632/oncotarget.1677
38. Cole LA. hCG, the wonder of today’s science. Reprod Biol Endocrinol. 2012;10:24. doi:10.1186/1477-7827-10-24
39. Sporn MB. The early history of TGF-beta, and a brief glimpse of its future. Cytokine Growth Factor Rev. 2006;17:3–7. doi:10.1016/j.cytogfr.2005.09.012
40. Acerbi I, Cassereau L, Dean I, et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol (Camb). 2015;7:1120–1134. doi:10.1039/c5ib00040h
41. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–175. doi:10.1111/j.1600-065X.2012.01146.x
42. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. doi:10.1038/sigtrans.2017.23
43. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi:10.1038/nrc3239
44. Köstlin N, Hofstädter K, Ostermeir AL, et al. Granulocytic myeloid-derived suppressor cells accumulate in human placenta and polarize toward a Th2 phenotype. J Immunol. 2016;196:1132–1145. doi:10.4049/jimmunol.1500340
45. Pyzer AR, Cole L, Rosenblatt J, Avigan DE. Myeloid-derived suppressor cells as effectors of immune suppression in cancer. Int J Cancer. 2016;139:1915–1926.
46. Kaur G, Helmer RA, Smith LA, Martinez-Zaguilan R, Dufour JM, Chilton BS. Alternative splicing of helicase-like transcription factor (Hltf): intron retention-dependent activation of immune tolerance at the feto-maternal interface. PLoS One. 2018;13:e0200211.
47. Barber EM, Fazzari M, Pollard JW. Th1 cytokines are essential for placental immunity to Listeria monocytogenes. Infect Immun. 2005;73:6322–6331.
48. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867.
49. Granot I, Gnainsky Y, Dekel N. Endometrial inflammation and effect on implantation improvement and pregnancy outcome. Reproduction. 2012;144:661–668.
50. Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res. 2012;4:376–389.
51. Zhang YH, He M, Wang Y, Liao AH. Modulators of the balance between M1 and M2 macrophages during pregnancy. Front Immunol. 2017;8:120.
52. Fest S, Aldo PB, Abrahams VM, et al. Trophoblast-macrophage interactions: a regulatory network for the protection of pregnancy. Am J Reprod Immunol. 2007;57:55–66.
53. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). 2014;6:1670–1690.
54. Grau AM, Datta PK, Zi J, Halder SK, Beauchamp RD. Role of Smad proteins in the regulation of NF-kappaB by TGF-beta in colon cancer cells. Cell Signal. 2006;18:1041–1050.
55. Wong ET, Tergaonkar V. Roles of NF-kappaB in health and disease: mechanisms and therapeutic potential. Clin Sci (Lond). 2009;116:451–465.
56. Freudlsperger C, Bian Y, Contag Wise S, et al. TGF-β and NF-κB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene. 2013;32:1549–1559.
57. Carpentier PA, Dingman AL, Palmer TD. Placental TNF-α signaling in illness-induced complications of pregnancy. Am J Pathol. 2011;178:2802–2810.
58. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975;72:3666–3670.
59. Koga K, Aldo PB, Mor G. Toll-like receptors and pregnancy: trophoblast as modulators of the immune response. J Obstet Gynaecol Res. 2009;35:191–202.
60. Li TT, Ogino S, Qian ZR. Toll-like receptor signaling in colorectal cancer: carcinogenesis to cancer therapy. World J Gastroenterol. 2014;20:17699–17708.
61. Li B, Vincent A, Cates J, Brantley-Sieders DM, Polk DB, Young PP. Low levels of tumor necrosis factor alpha increase tumor growth by inducing an endothelial phenotype of monocytes recruited to the tumor site. Cancer Res. 2009;69:338–348.
62. Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466.
63. Callejas NA, Casado M, Boscá L, Martín-Sanz P. Requirement of nuclear factor kappaB for the constitutive expression of nitric oxide synthase-2 and cyclooxygenase-2 in rat trophoblasts. J Cell Sci. 1999;112:3147–3155.
64. Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12:86.
65. Porporato PE. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis. 2016;5:e200.
66. Shvartsur A, Bonavida B. Trop2 and its overexpression in cancers: regulation and clinical/therapeutic implications. Genes Cancer. 2015;6:84–105.
67. Guerra E, Trerotola M, Tripaldi R, et al. Trop-2 induces tumor growth through AKT and determines sensitivity to AKT inhibitors. Clin Cancer Res. 2016;22:4197–4205.
68. Trerotola M, Jernigan DL, Liu Q, Siddiqui J, Fatatis A, Languino LR. Trop-2 promotes prostate cancer metastasis by modulating β(1) integrin functions. Cancer Res. 2013;73:3155–3167.
69. Zeng P, Chen MB, Zhou LN, Tang M, Liu CY, Lu PH. Impact of TROP2 expression on prognosis in solid tumors: a systematic review and meta-analysis. Sci Rep. 2016;6:33658.
70. Lin JC, Wu YY, Wu JY, et al. TROP2 is epigenetically inactivated and modulates IGF-1R signalling in lung adenocarcinoma. EMBO Mol Med. 2012;4:472–485.
71. Aasen T, Mesnil M, Naus CC, Lampe PD, Laird DW. Gap junctions and cancer: communicating for 50 years. Nat Rev Cancer. 2016;16:775–788.
72. Jiang JX, Penuela S. Connexin and pannexin channels in cancer. BMC Cell Biol. 2016;17(Suppl 1):12.
73. Wu JI, Wang LH. Emerging roles of gap junction proteins connexins in cancer metastasis, chemoresistance and clinical application. J Biomed Sci. 2019;26:8.
74. Cronier L, Bastide B, Defamie N, et al. Involvement of gap junctional communication and connexin expression in trophoblast differentiation of the human placenta. Histol Histopathol. 2001;16:285–295.
75. Cronier L, Defamie N, Dupays L, et al. Connexin expression and gap junctional intercellular communication in human first trimester trophoblast. Mol Hum Reprod. 2002;8:1005–1013.
76. Winterhager E, Kidder GM. Gap junction connexins in female reproductive organs: implications for women’s reproductive health. Hum Reprod Update. 2015;21:340–352.
77. Banerjee D. Connexin’s connection in breast cancer growth and progression. Int J Cell Biol. 2016;2016:9025905.
78. Chen Q, Boire A, Jin X, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493–498.
79. Defamie N, Chepied A, Mesnil M. Connexins, gap junctions and tissue invasion. FEBS Lett. 2014;588:1331–1338.
80. Dubina MV, Iatckii NA, Popov DE, Vasil’ev SV, Krutovskikh VA. Connexin 43, but not connexin 32, is mutated at advanced stages of human sporadic colon cancer. Oncogene. 2002;21:4992–4996.
81. Morgani SM, Brickman JM. The molecular underpinnings of totipotency. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130549.
82. Gu G, Yuan J, Wills M, Kasper S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res. 2007;67:4807–4815.
83. Chen YC, Ingram PN, Fouladdel S, et al. High-throughput single-cell derived sphere formation for cancer stem-like cell identification and analysis. Sci Rep. 2016;6:27301.
84. Clark AG, Vignjevic DM. Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol. 2015;36:13–22.
85. Hannon T, Innes BA, Lash GE, Bulmer JN, Robson SC. Effects of local decidua on trophoblast invasion and spiral artery remodeling in focal placenta creta - an immunohistochemical study. Placenta. 2012;33:998–1004.
86. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292.
87. Martincorena I, Roshan A, Gerstung M, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886.
88. Brash DE. Preprocancer. Normal skin harbors cancer-causing mutations. Science. 2015;348:867–868.
89. Santarpia L, Bottai G, Kelly CM, Győrffy B, Székely B, Pusztai L. Deciphering and targeting oncogenic mutations and pathways in breast cancer. Oncologist. 2016;21:1063–1078.
90. Conconi D, Redaelli S, Bovo G, et al. Unexpected frequency of genomic alterations in histologically normal colonic tissue from colon cancer patients. Tumor Biol. 2016;37:13831–13842.
91. Martincorena I, Fowler JC, Wabik A, et al. Somatic mutant clones colonize the human esophagus with age. Science. 2018;362:911–917.
92. Yokoyama A, Kakiuchi N, Yoshizato T, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. 2019;565:312–317.
93. Baines KJ, Renaud SJ. Transcription factors that regulate trophoblast development and function. Prog Mol Biol Transl Sci. 2017;145:39–88.
94. Zhou Y, Huang T, Cheng AS, Yu J, Kang W, To KF. The TEAD family and its oncogenic role in promoting tumorigenesis. Int J Mol Sci. 2016;17:138.
95. Dittmer J. The biology of the Ets1 proto-oncogene. Mol Cancer. 2003;2:29.
96. Wang X, Sun D, Tai J, et al. TFAP2C promotes stemness and chemotherapeutic resistance in colorectal cancer via inactivating hippo signaling pathway. J Exp Clin Cancer Res. 2018;37:27.
97. Shankar DB, Cheng JC, Kinjo K, et al. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell. 2005;7:351–362.
98. Löwenberg B. Introduction to the review series on leukemic stem cells. Blood. 2017;129:1567.
99. Bieker R, Padró T, Kramer J, et al. Overexpression of basic fibroblast growth factor and autocrine stimulation in acute myeloid leukemia. Cancer Res. 2003;63:7241–7246.
100. Samudio I, Fiegl M, Andreeff M. Mitochondrial uncoupling and the Warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009;69:2163–2166. doi:10.1158/0008-5472.CAN-08-3722
101. Schmidt T, Carmeliet P. Angiogenesis: a target in solid tumors, also in leukemia? Hematology Am Soc Hematol Educ Program. 2011;2011:1–8. doi:10.1182/asheducation-2011.1.1
102. Knaus HA, Kanakry CG, Luznik L, Gojo I. Immunomodulatory drugs: immune checkpoint agents in acute leukemia. Curr Drug Targets. 2017;18:315–331. doi:10.2174/1389450116666150518095346
103. Carter BZ, Qiu Y, Huang X, et al. Survivin is highly expressed in CD34(+)38(-) leukemic stem/progenitor cells and predicts poor clinical outcomes in AML. Blood. 2012;120:173–180. doi:10.1182/blood-2012-02-409888
104. Stavropoulou V, Kaspar S, Brault L, et al. MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell. 2016;30:43–58. doi:10.1016/j.ccell.2016.05.011
105. Fraternali-Orcioni G, Falini B, Quaini F, et al. Beta-HCG aberrant expression in primary mediastinal large B-cell lymphoma. Am J Surg Pathol. 1999;23:717–721.
106. Renné C, Willenbrock K, Küppers R, Hansmann ML, Bräuninger A. Autocrine- and paracrine-activated receptor tyrosine kinases in classic Hodgkin lymphoma. Blood. 2005;105:4051–4059. doi:10.1182/blood-2004-10-4008
107. Mushtaq M, Darekar S, Klein G, Kashuba E. Different mechanisms of regulation of the warburg effect in lymphoblastoid and burkitt lymphoma cells. PLoS One. 2015;10:e0136142. doi:10.1371/journal.pone.0136142
108. Ribatti D, Nico B, Ranieri G, Specchia G, Vacca A. The role of angiogenesis in human non-Hodgkin lymphomas. Neoplasia. 2013;15:231–238.
109. Tanaka Y, Maeshima AM, Nomoto J, et al. Expression pattern of PD-L1 and PD-L2 in classical Hodgkin lymphoma, primary mediastinal large B-cell lymphoma, and gray zone lymphoma. Eur J Haematol. 2018;100:511–517. doi:10.1111/ejh.13033
110. Ansell SM, Arendt BK, Grote DM, et al. Inhibition of survivin expression suppresses the growth of aggressive non-Hodgkin’s lymphoma. Leukemia. 2004;18:616–623. doi:10.1038/sj.leu.2403281
111. Lemma S, Karihtala P, Haapasaari KM, et al. Biological roles and prognostic values of the epithelial-mesenchymal transition-mediating transcription factors Twist, ZEB1 and Slug in diffuse large B-cell lymphoma. Histopathology. 2013;62:326–333. doi:10.1111/his.12000
112. Chiron D, Maïga S, Surget S, et al. Autocrine insulin-like growth factor 1 and stem cell factor but not interleukin 6 support self-renewal of human myeloma cells. Blood Cancer J. 2013;3:e120. doi:10.1038/bcj.2013.18
113. Sanchez WY, McGee SL, Connor T, et al. Dichloroacetate inhibits aerobic glycolysis in multiple myeloma cells and increases sensitivity to bortezomib. Br J Cancer. 2013;108:1624–1633. doi:10.1038/bjc.2013.120
114. Giuliani N, Storti P, Bolzoni M, Palma BD, Bonomini S. Angiogenesis and multiple myeloma. Cancer Microenviron. 2011;4:325–337. doi:10.1007/s12307-011-0072-9
115. Rosenblatt J, Avigan D. Targeting the PD-1/PD-L1 axis in multiple myeloma: a dream or a reality? Blood. 2017;129:275–279. doi:10.1182/blood-2016-08-731885
116. Romagnoli M, Trichet V, David C, et al. Significant impact of survivin on myeloma cell growth. Leukemia. 2007;21:1070–1078. doi:10.1038/sj.leu.2404602
117. Azab AK, Hu J, Quang P, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. 2012;119:5782–5794. doi:10.1182/blood-2011-09-380410
118. Cassinelli G, Favini E, Bo L D, et al. Antitumor efficacy of the heparan sulfate mimic roneparstat (SST0001) against sarcoma models involves multi-target inhibition of receptor tyrosine kinases. Oncotarget. 2016;7:47848–47863. doi:10.18632/oncotarget.10292
119. Issaq SH, Teicher BA, Monks A. Bioenergetic properties of human sarcoma cells help define sensitivity to metabolic inhibitors. Cell Cycle. 2014;13:1152–1161. doi:10.4161/cc.28010
120. Rocchi L, Caraffi S, Perris R, Mangieri D. The angiogenic asset of soft tissue sarcomas: a new tool to discover new therapeutic targets. Biosci Rep. 2014;34:e00147. doi:10.1042/BSR20140075
121. Bertucci F, Finetti P, Perrot D, et al. PDL1 expression is a poor-prognosis factor in soft-tissue sarcomas. Oncoimmunology. 2017;6:e1278100. doi:10.1080/2162402X.2016.1278100
122. Kappler M, Kotzsch M, Bartel F, et al. Elevated expression level of survivin protein in soft-tissue sarcomas is a strong independent predictor of survival. Clin Cancer Res. 2003;9:1098–1104.
123. Sannino G, Marchetto A, Kirchner T, Grünewald TGP. Epithelial-to-mesenchymal and mesenchymal-to-epithelial transition in mesenchymal tumors: a paradox in sarcomas? Cancer Res. 2017;77:4556–4561. doi:10.1158/0008-5472.CAN-17-0032
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.