")
Back to Journals » Biologics: Targets and Therapy » Volume 8
Plasma membrane coenzyme Q: evidence for a role in autism
Authors Crane F, Löw H, Sun I, Navas P, Gvozdjáková A
Received 21 August 2013
Accepted for publication 8 October 2013
Published 29 May 2014 Volume 2014:8 Pages 199—205
DOI https://doi.org/10.2147/BTT.S53375
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Frederick L Crane,1 Hans Löw,2 Iris Sun,1 Placido Navas,3 Anna Gvozdjáková4
1Department of Biological Sciences, Purdue University, West Lafayette, IN, USA; 2Department of Molecular Medicine, Karolinska Institute, Stockholm, Sweden; 3Centro Andaluz de Biología del Desarrollo, Universidad Pablo de Olavide, Sevilla, Spain; 4Pharmacobiochemical Laboratory of Third Medical Department, Medical Faculty, Comenius University in Bratislava, Bratislava, Slovakia
Background: The Voltage Dependent Anion Channel (VDAC) is involved in control of autism. Treatments, including coenzyme Q, have had some success on autism control.
Data sources: Correlation of porin redox activity and expression of autism is based on extensive literature, especially studies of antibodies, identification of cytosolic nicotinamide adenine dinucleotide reduced (NADH) dehydrogenase activity in the VDAC, and evidence for extreme sensitivity of the dehydrogenase to a mercurial. Evidence for a coenzyme Q requirement came from extraction and analog inhibition of NADH ferricyanide reductase in the erythrocyte plasma membrane, done in 1994, and reinterpreted when it was identified in VDAC in 2004. The effects of ubiquinol (the QH2 – reduced form of coenzyme Q) in children with autism were studied.
Results: A new role for coenzyme Q in the porin channels has implications on autism. Ubiquinol, the more active form of coenzyme Q, produces favorable response in children with autism. Agents which affected electron transport in porin show parallel effects in autism.
Conclusion: We propose a hypothesis that autism is controlled by a coenzyme Q-dependent redox system in the porin channels; this conclusion is based on the effects of agents that positively or negatively affect electron transport and the symptoms of autism. The full understanding of the mechanism of their control needs to be established.
Keywords: porin, channel, oxidation, reduction, autism
Introduction
Plasma membrane redox control
This investigation started when a paper by Gonzales-Gronow et al1 appeared reporting that antibodies in the voltage dependent anion channel (VDAC) were increased in autistic children. The recent discovery by Baker et al2 that VDAC is a transplasma membrane ferricyanide reductase introduced the possibility that plasma membrane redox is involved in autism through inhibition of the reductase.
Most cells have two systems for transferring electrons from cytosolic nicotinamide adenine dinucleotide (NADH) through the plasma membrane, to the external surface. Each of these systems involves interaction with coenzyme Q. The first system to be discovered was an NADH-oxidase which was shown to be subject to hormonal and growth factor control, and which is involved in proliferation of transformed cells.3,4 The second was an NADH-ferricyanide reductase which transfers electrons from cytosolic NADH to reduce ferricyanide outside the cell. This later system has been identified as the protein polymer VDAC.2,5 Both of these systems oxidize cytosolic NADH to NAD+ and have been shown to increase cytosolic NAD+ concentrations.6 VDAC has been shown to selectively reduce ferricyanide, and does not react with oxygen so it is not an NADH-oxidase.7 VDAC also reduces coenzyme Q, but this reaction may be based on a functional requirement for coenzyme Q in the electron transport chain inside VDAC. In other words, coenzyme Q acts as part of the electron chain – as it does in the mitochondria – or as part of the VDAC holoenzyme, rather than as a substrate for VDAC. Therefore the natural and final electron acceptor for VDAC remains elusive, thus, it is tempting to speculate that a natural redox function for VDAC is involved in keeping coenzyme Q in a reduced state in the plasma membrane (Figure 1).
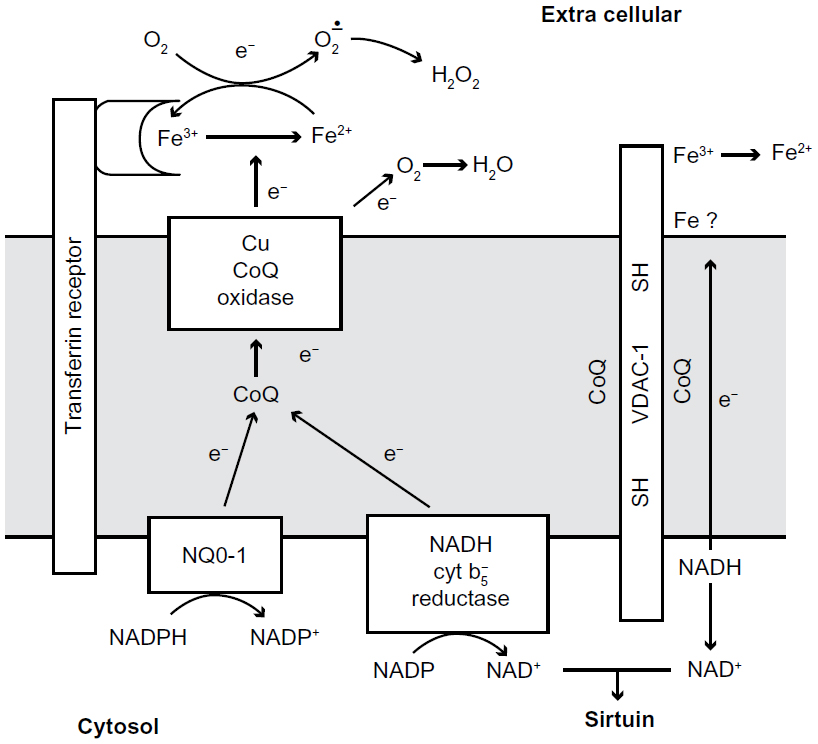 | Figure 1 Diagram of proposed transplasma membrane NADH oxidizing enzymes. |
A third system for transplasma membrane electron transport, which involves ascorbate recycling, has been proposed.8,9 Although no ascorbate is used with isolated plasma membrane in this study, ascorbate recycling is a biologically relevant process that should be noted in transplasma membrane electron transport. In particular, an ascorbic oxidase could provide a good supply of ascorbate free radicals (AFR).10–12 AFR for the VDAC system are the reason for the high concentrations of naturally occurring ascorbate in the brain.10–14
Following its original recognition in mitochondria, coenzyme Q has been found in all membranes (Table 1) in amounts sufficient to have significant function in membrane structure or redox activity. It has also been found bound to individual proteins, as in the mitochondrial uncoupling protein (UCP).15–17
 | Table 1 Coenzyme Q in membranes |
The NADH oxidase in the plasma membrane involves two possible dehydrogenases, NADH cytochrome b5 reductase18 and NO-01 (DT diaphorase) located on the cytosolic side of the membrane. Each dehydrogenase acts as a NADH coenzyme Q reductase to reduce the coenzyme Q within the plane of the lipid bilayer. The reduced QH2 is then oxidized by a reduced coenzyme Q oxidase on the extracellular side of the plasma membrane. The reduction by NQO1 has been confirmed by increased reduction with genetic duplication of NQO1. Experimentally, the requirement for coenzyme Q is often demonstrated by depletion or inhibition of coenzyme Q followed by a rescue effect of coenzyme Q reintroduction. For example, extraction of coenzyme Q from membrane preparations decreases NADH-oxidase activity, which is restored by re-addition of coenzyme Q (Table 2). This experimental concept will be further discussed in the context of VDAC (Table 3).19
 | Table 2 Heptane extraction of erythrocyte membrane |
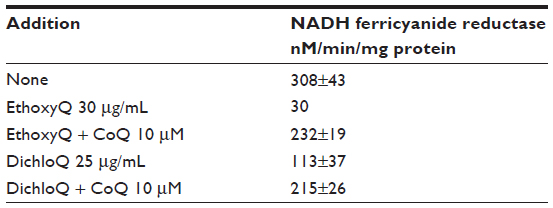 | Table 3 Analog inhibition of erythrocyte NADH ferricyanide reductase |
The second transplasma membrane NADH dehydrogenase was discovered when it separated from the oxidase during purification.4 It was a known protein, VDAC-1,2 previously purified from mitochondria, and a focus of this report. The VDAC has no oxidase activity but it transfers electrons from inside the cell to the external oxidant ferricyanide.2,5 In the VDAC the only known groups for electron transport across the membrane are two cysteine residues (-SH) on the inside of the channel.20 At this time, ferricyanide is the only oxidant that works as an external electron acceptor, but it is not normally available to cells; a likely natural electron acceptor would be semi-dehydroascorbate,21 especially in neural tissue where ascorbate concentration is found to be high,14 and ascorbate oxidase can produce the semi dehydroascorbate. Coenzyme Q could be the receptor but it is more likely to be a part of the electron transport chain and attached to a binding site.
Coenzyme Q is required for VDAC
The evidence for coenzyme Q involvement with VDAC is more tenuous because we used the activity in the red cell membrane and it should be subjected to direct testing with isolated recombinant (VDAC) enzyme. The methods referenced in this critical review are a reinterpretation of original research.19 Brief experimental overviews and important details regarding concentrations of drugs are provided as part of the experimental results; however, the original research papers should be referred to for further experimental details.22
NADH dehydrogenase in red cell membranes is based only on VDAC, as is indicated by inhibition with thiol reagents.23 This electron transport by ferricyanide reductase of erythrocytes is 100% inhibited by micromolar levels of (1.0 μM) p-Chloromercuribenzoate (PCMB), whereas rat liver plasma membrane ferricyanide reduction is only partially inhibited (15%) by a PCMB concentration 100 times greater (0.1 mM). These larger 0.1 mM PCMB concentrations were also needed to inhibit ferricyanide reduction of Human Epitheloid Carcinoma (HeLa) cell plasma membrane; a 55% inhibition was noted, highlighting the unique sensitivity of the erythrocyte plasma membrane system used in this study. The contrasting sensitivities for mercurial compounds between the cell lines tested indicates that erythrocyte membranes have key differences, likely attributed to VDAC exclusively when compared with other plasma membranes, which have an ancillary ferricyanide reductase capacity based on the oxidase and less sensitive to mercurial inhibition (Table 4).
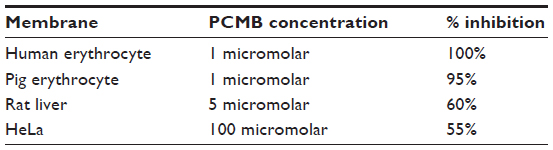 | Table 4 Inhibition of NADH ferricyanide reductase in isolated plasma membrane by PCMB |
Further evidence for coenzyme Q function in VDAC is shown by two additional lines of investigation. The first experiment in this series used direct extraction of erythrocyte plasma membrane with heptane; this resulted in a large decrease in the NADH ferricyanide reductase activity (Table 2), an activity that was largely restored by rescue with coenzyme Q addition. Thus it appeared that coenzyme Q was required for the VDAC ferricyanide reductase activity as proposed.24 The second experiment featured coenzyme Q analogs, such as Dichloroquinone or diethoxycoenzyme Q, which were shown to inhibit NADH-ferricyanide reduction in erythrocyte membranes (Table 3); this effect was partially reversed by rescue experiments with addition of coenzyme Q, indicating a direct contribution of coenzyme Q to NADH-ferricyanide reductase activity.22
The requirement for coenzyme Q for the function of a pore protein, such as VDAC, is not without precedence; for example, binding of coenzyme Q to the UCP in the mitochondria is required for optimal function.16,17 Binding coenzyme Q in VDAC may lower the effective redox potential of coenzyme Q from 100 mV to a value closer to the redox potential of thiol groups (approximately –225 mV). It is noted that there is also evidence for residual, chelator-sensitive iron in the plasma membrane, which may bridge any remaining redox potential gap.25,26
Activation of plasma membrane redox results in cellular growth
Activation of plasma membrane NADH oxidase or increasing transplasma membrane electron flow by supply of impermeable external oxidants stimulates the growth of transformed cells in serum deficient media.27,28 The transformed cells can grow without added growth factors. Untransformed cells can respond to oxidants if additional growth factors are added (Table 5). The plasma membrane oxidase can also prevent apoptosis.29,30 Which plasma membrane redox systems contribute to growth or apoptosis has not been established. There are indications that both the oxidase and the reductase are involved in growth control. For example the best growth stimulation is with ferricyanide, which can be reduced by both systems as compared to lesser growth with indophenol, which does not accept electrons from VDAC (Table 6). Furthermore, growth factor stimulation of the oxidase, for example, by addition of insulin or diferric transferrin, would not involve VDAC. If growth stimulation is based on increasing NAD in the cytosol to activate sirtuin-1, then additive effects should be expected. The increase in NAD in cells by oxidation of NADH in response to diferric transferrin as an activator of the oxidase, or by reduction of ferricyanide, has also been shown.6 More recent studies have shown that ascorbate has a role in electron transfer across the plasma membrane.10–13
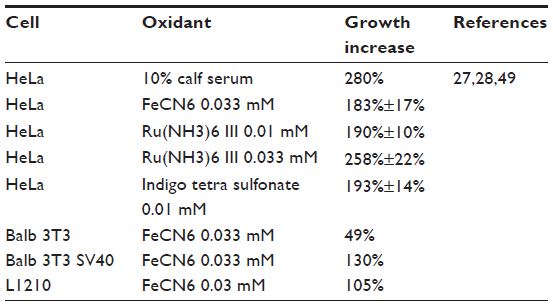 | Table 5 Oxidant stimulation of cell proliferation |
 | Table 6 Relative redox activity with ferricyanide, indophenol and oxygen nanomoles/min/mg protein NADH acceptor reductase |
Transmembrane electron transport from NADH or ascorbate can reduce AFRs outside the cell; conversely, AFRs can be reduced inside the cell. Since AFRs can act as electron acceptors for electrons from internal NADH, they may be the natural acceptors for ferricyanide reductase. The redox system may control opening of the channel.31
Antioxidant effects
The role of reduced coenzyme Q in antioxidant protection has been extensively discussed.32 The terminal oxidase for NADH oxidation is a copper protein, external NADH oxidase (ECTO-NOX), which carries electrons through to oxygen to produce H2O. Production of H2O decreases the production of oxygen radicals.
Reduced Q is effective by itself in reducing radical oxygen species (ROS) or by regenerating tocopherol and ascorbate after they have been oxidized by ROS. Protection against free radical damage requires a pool of reduced coenzyme Q, which is therefore needed for antioxidant protection. Coenzyme Q is unique among antioxidants in that enzymes in the membranes can keep it reduced.
Relationship with autism
Several lines of evidence indicate that VDAC and its associated coenzyme Q cofactor are involved in autism; most important is the finding that antibodies to VDAC have been reported in autism patients and that the antibodies inhibit the NADH ferricyanide reductase activity of VDAC.1
Disorders in mitochondrial respiratory chain complexes (Complex I–Complex V), decreased coenzyme Q10 concentration, as well as abnormalities of mtDNA, are associated with autism spectrum disorders;33–35 one case of coenzyme Q10 treatment (from 600 to 1200 mg/d) showed improvement in autistic behavior.34
In our previous pilot study, a beneficial effect of ubiquinol in children with autism was demonstrated for the first time.36,37 We included 24 children (17 boys, 7 girls; male to female ratio 2.4:1; aged 3–6 years) in the study according to the DSM IV criteria (Diagnostic and Statistic Manual of Mental Diseases) and using CARS (Childhood Autism Rating Scale, a test combining parent reports and direct observation by professionals).38 All parents signed an informed consent when including their children in the study. Children were treated at home. Before the beginning of the study, all children with neurological disturbances were examined by either psychologists or neurologists. Children with obesity, diabetes, or epilepsy were not included in the study. Parents provided data about their child’s behavior before and after 3 months of ubiquinol supplementation. The screening tool was designed by a psychologist specifically for the purposes of this study.
Children with autism were supplemented during the first week at a daily dose of 50 mg ubiquinol (liquid liposomal ubiquinol [Li-QH™ liposomal liquid reduced coenzyme Q10, produced by Tishcon Corp., Westbury, New York, USA] was diluted in milk, tea, or juice). The following week, the daily dose was increased to twice daily 50 mg doses of ubiquinol (morning and at lunch after meal). If children were hyperactive, ubiquinol was given in the evening before sleeping instead of at lunchtime. Children with autism were then treated with 100 mg/day ubiquinol for 3 months with the last dose of ubiquinol 24 hours before blood collection. Differences between baseline values of coenzyme Q10-TOTAL and after 3 months of treatment were considered statistically significant if corrected P<0.05, using paired Student’s t-test. A placebo group of children with autism was not included in the study for ethical reasons. Baseline values for mean coenzyme Q10-TOTAL was 0.512±0.164 μmol/L plasma; after 3 months supplementation, mean coenzyme Q10-TOTAL was 3.016±0.307 μmol/L plasma, meaning coenzyme Q10-TOTAL concentration increased 489%. A significant improvement in autistic symptomology was observed after 3 months of ubiquinol supplementary therapy in children maintained at over 2.5 μmol/L CoQ10-TOTAL plasma concentration. The improvement details were as follows: 12% of children improved communication with parents, 21% improved verbal communication, playing games improved in 42% of children, sleeping improved in 34%, and food rejection decreased in 17%. In some cases, aggressiveness increased in the early stages of the study but changing the dosage schedule changed the response to a positive one.34,36,37 Other authors have published a report detailing increased plasma coenzyme Q10 concentration (+153%) in children with autism after 3 months of coenzyme Q10 supplementation in daily doses of 50 mg, together with several other vitamins.39 We cannot compare our results as it is not clear if the children were treated with ubiquinol or ubiquinone, nor is it clear if plasma coenzyme Q10 concentrations were as ubiquinone, ubiquinol, or coenzyme Q10-TOTAL.
VDAC NADH dehydrogenase is extremely sensitive to mercurial inhibition, which is consistent with the low-level effects of mercury in autism.40 A sensitivity to lead23 at VDAC SH groups could also point to lead-poisoning-related initiation of autism.
Compounds with a positive effect on autism show a stimulation of plasma membrane redox activity. Ascorbate had favorable results in autism, and semi-dehydroascorbate can be an acceptor for VDAC41 and can increase the rate of plasma membrane oxidase.12 Glutathione is low in some children with autism, which may decrease electron transport through VDAC by removing an electron donor. The positive effect of hyperbaric oxygen on autism42 can be based on stimulation of the plasma membrane oxidase. On the other hand deficiencies in other compounds which may be involved in control of autism have been observed; triiodothyronine (T3) and other thyroid hormones, which are necessary for neuronal migration and fetal brain development, have been found to be low in some children with autism.43,44 It has also been reported that T3 stimulates the plasma membrane oxidase; this could represent an important link between thyroid hormone deficiencies and reduced plasma membrane redox function.27 Differic transferrin, which also stimulates the oxidase, is low in some children with autism so the oxidase may not be fully active in autism.45,46
The cytokines tumor necrosis factor alpha (TNFα) and interleukin 2 (IL2) that inhibit plasma membrane ferricyanide reduction47 are increased in autism45,48 and the consequent inhibition could be a basis for autism. Coenzyme Q at 0.03 mM doubles the growth of HeLa cells in serum-free media; ferricyanide results in an additional 50% increase (Table 7). Redox is similarly stimulated so that coenzyme Q may provide an approach to increasing autism-related redox status in order to decrease symptoms.36 One can project specific treatments of functional elements in the electron transport, rather than a general condition such as lipid peroxidation. This may lead to remarkable therapy wherein both CoQH2 and ascorbate and vitamins are used in combination to get maximum redox activity.
 | Table 7 Coenzyme Q stimulation of growth and redox activity |
Acknowledgments
This paper was supported by the Grant Ministry of Education of Slovakia, VEGA 1/0614/12, FLC 1992 support. The authors would like to acknowledge the Folkers Foundation. The authors would also like to acknowledge Professor Anthony W Linnane (Centre of Molecular Biology and Medicine, Melbourne, VIC, Australia) for reviewing the manuscript.
Disclosure
The authors report no conflicts of interest in this work. Work was done between 1990–1992 and 2012–2013 and has not been previously published, with one exception – in 1992 Sun et al published extraction studies.
References
Gonzales-Gronow M, Cuchacovich M, Francos R, et al. Antibodies against the voltage-dependent anion channels (VDAC) and its protective ligand hexokinase-I in children with autism. J Neuroimmunol. 2010;227(1–2):153–161. | |
Baker MA, Lane DJ, Ly DJ, De Pinto V, Lawen A. VDAC1 is a transplasma membrane NADH-ferricyanide reductase. J Biol Chem. 2004;279(6):4811–4819. | |
Löw H, Crane FL. Hormone regulated redox function in plasma membranes. FEBS Lett. 1976;68(2):157–159. | |
Brightman AO, Wang J, Miu RK, et al. A growth factor- and hormone-stimulated NADH oxidase from rat liver plasma membrane. Biochim Biophys Acta. 1992;1105(1):109–117. | |
Baker MA, Lawen A. Plasma membrane NADH-oxidoreductase system: a critical review of the structural and functional data. Antioxid Redox Signal. 2000;2(2):197–212. | |
Navas P, Sun IL, Morré DJ, Crane FL. Decrease of NADH in HeLa cells in the presence of transferrin or ferricyanide. Biochem Biophys Res Commun. 1986;135(1):110–115. | |
Goldenberg H, Crane FL, Morre DJ. NADH oxidoreductase of mouse liver plasma membranes. J Biol Chem. 1979;254(7):2491–2498. | |
Lane DJ, Lawen A. Transplasma membrane electron transport comes in two flavors. BioFactors. 2008;34(3):191–200. | |
Lane DJ, Lawen A. Ascorbate and plasma membrane electron transport-enzymes vs efflux. Free Radic Biol Med. 2009;47(5):485–495. | |
May JM. Is ascorbic acid an antioxidant for the plasma membrane? FASEB J. 1999;13(9):995–1006. | |
May JM, Qu ZC. Ascorbate-dependent electron transfer across the human erythrocyte membrane. Biochim Biophys Acta. 1999;142(1):19–31. | |
May JM, Qu ZC, Whitesell RR. Ascorbic acid recycling enhances the antioxidant reserve of human erythrocytes. Biochemistry. 1995;34(39):12721–12728. | |
Van Dijin MM, Van de Zee J, Van Stevenick J, Van den Broek PJ. Ascorbate stimulates ferricyanide reduction in HL-60 cells through a mechanism distinct from the NADH-dependent plasma membrane reductase. J Biol Chem. 1998;273(22):13415–13420. | |
Smythies JR. The role of ascorbate in brain: therapeutic implications. J R Soc Med. 1996;89(5):241. | |
Klingenberg M, Winkler E, Echtay K. Uncoupling protein, H+ transport and regulation. Biochem Soc Trans. 2001;29(Pt6):806–811. | |
Echtay KS, Winkler E, Frischmuth K, Klingenberg M. Uncoupling proteins 2 and 3 are highly active H(+) transporters and highly nucleotide sensitive when activates by coenzyme Q (ubiquinone). Proc Natl Acad Sci U S A. 2001;98(4):1416–1421. | |
Echtay KS, Winkler E, Klingenberg M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature. 2000;408(6812):609–613. | |
Navarro F, Villalba JM, Crane FL, Mackellar WC, Navas P. A phospholipid-dependent NADH-coenzyme Q reductase from liver plasma membrane. Biochem Biophys Res Commun. 1995;212(1):138–143. | |
Sun IL, Sun EE, Crane FL, Morré DJ, Lindgren A, Löw H. Requirement for coenzyme Q in plasma membrane electron transport. Proc Natl Acad Sci USA. 1992;89(23):11126–11130. | |
De Pinto V, Messina A, Lane DJ, Lawen A. Voltage-dependent anion-selective channel (VDAC) in the plasma membrane. FEBS Lett. 2010;584(9):1793–1799. | |
Gómez-Díaz C, Rodríguez-Aguilera JC, Barroso MP, et al. Antioxidant ascorbate is stabilized by NADH-coenzyme Q10 reductase in the plasma membrane. J Bioenerg Biomembr. 1997;29(3):251–257. | |
Grebing C, Crane FL, Löw H, Hall K. A transmembranous NADH-dehydrogenase in human erythrocyte membranes. J Bioenerg Biomembr. 1984;16(5–6):517–533. | |
Prins JM, Brooks DM, Thompson CM, Lurie DI. Chronic low-level Pb exposure during development decreases the expression of the voltage-dependent anion channel in auditory neurons of the brainstem. Neurotoxicology. 2010;32(6):662–673. | |
Matteucci E, Giampietro O. Electron pathways through erythrocyte plasma membrane in human physiology and pathology. Potential redox biomarker? Biomark Insights. 2007;2:321–329. | |
Alcain FJ, Löw H, Crane FL. Iron at the cell surface controls DNA synthesis in CCI 39 cells. Biochem Biophys Res Commun. 1994;203(1):16–21. | |
Alcain FJ, Löw H, Crane FL. Iron reverses impermeable chelator inhibition of DNA synthesis in CCI 39 cells. Proc Natl Acad Sci U S A. 1994;91(17):7903–7906. | |
Crane FL, Sun IL, Clark MG, Grebing C, Löw H. Transplasma-membrane redox systems in growth and development. Biochim Biophys Acta. 1985;811(3):233–264. | |
Sun IL, Crane FL, Grebing C, Löw H. Transmembrane redox in control of cell growth. Stimulation of HeLa cell growth by ferricyanide and insulin. Exp Cell Res. 1985;156(2):528–536. | |
Elinder F, Akanda N, Tofighi R, et al. Opening of plasma membrane voltage-dependent anion channels (VDAC) precedes caspase activation in neuronal apoptosis induced by toxic stimuli. Cell Death Differ. 2005;12(8):1134–1140. | |
Larm JA, Vaillant F, Linnane AW, Lawen A. Up-regulation of the plasma membrane oxidoreductase as a prerequisite for the viability of human Namalwa rho 0 cells. J Biol Chem. 1994;269(48):30097–300100. | |
Sridharan M, Bowles EA, Richards JP, et al. Prostacyclin receptor-mediated ATP release from erythrocytes requires the voltage-dependent anion channel. Am J Physiol Heart Circ Physiol. 2012;302(3):H553–H559. | |
Navas P, Villalba JM, de Cabo R. The importance of plasma membrane coenzyme Q in ageing and stress responses. Mitochondrion. 2007; 7 Suppl:S34–S40. | |
Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry. 2012;17(3):290–314. | |
Tsao CY, Mendell JR. Autistic disorder in 2 children with mitochondrial disorders. J Child Neurol. 2007;22(9):1121–1123. | |
Poling JS, Frye RE, Shoffner J, Zimmerman AW. Developmental regression andmitochondrial dysfunction in a child with autism. J Child Neurol. 2006;21(2):170–172. | |
Gvozdjakova A, Kucharska J, Babinska K, et al. Effect of ubiquinol on oxidative stress, antioxidants and psychological manifestations in children with autism. Presented at: Seventh Conference of the International Coenzyme Q10 Association; November 8–11, 2012; Seville, Spain. Abstract book:92–93. | |
Gvozdjáková A, Kucharská J, Ostatníková D, Babinská K, Nakládal D, Crane FL. Ubiquinol supportive therapy in children with autism. 2nd International Conference and Exhibition on Nutritional Science and Therapy, Philadelphia, USA, July 15–17, 2013. Nutritional Science. 2013;3(4):115. | |
Schopler E, Reichler RJ, DeVellis RF, Daly K. Toward objective classification of childhood autism: Childhood Autism Scale (CARS). J Autism Dev Disord. 1980;10(1):91–103. | |
Adams JB, Audhya T, McDonough-Means S, et al. Efect of a vitamin/mineralsupplement on children and adults with autism. BMC Pediatrics. 2011;11:111. | |
Mutter J, Naumann J, Schneider R, Walach H, Haley B. Mercury and autism: accelerating evidence. Neuro Endocrinol Lett. 2005; 26(5):439–446. | |
Dolske MC, Spollen J, McKay S, Lancashire E, Tolbertz L. A preliminary trial of ascorbic acid as supplemental therapy for autism. Prog Neuropsychopharmacol Biol Psychiatry. 1993;17(5):765–774. | |
Rossignol DA, Bradstreet JJ, Van Dyke K, et al. Hyperbaric oxygen treatment in autism spectrum disorders. Med Gas Res. 2012;2(1):16. | |
Román GC. Autism: transient in utero hypothyroxinemia related to maternal flavonoid ingestion during pregnancy and to other environmental antithyroid agents. J Neurol Sci. 2007;262(1–2):15–26. | |
Stein MA, Weiss RE. Thyroid function tests and neurocognitive functioning in children referred for attention deficit/hyperactivity disorder. Psychoneuroendocrinology. 2003;28(3):304–316. | |
Tostes MH, Teixeira HC, Gattaz WF, Brandão MA, Raposo NR. Altered neurotrophin, neuropeptide, cytokines and nitric oxide levels in autism. Pharmacopsychiatry. 2012;45(6):241–243. | |
Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13(3):171–181. | |
Sun IL, Sun LE, Crane FL. Cytokine inhibition of transplasma membrane election transport. Biochem Mol Biol Int. 1996;38(1):175–180. | |
Cohly HH, Panja A. Immunological findings in autism. Int Rev Neurobiol. 2005;71:317–341. | |
Crane FL, Morre DJ, Löw H, editors. Plasma Membrane Oxidoreductases in Control of Animal and Plant Growth. New York: Plenum Press; 1988:443. | |
Crane FL, Löw H. NADH oxidation in liver and fat cell plasma membranes. FEBS Lett. 1976;68(2):153–156. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.