")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Plasma disturbance of phospholipid metabolism in major depressive disorder by integration of proteomics and metabolomics
Authors Gui SW, Liu YY, Zhong XG, Liu XY, Zheng P, Pu JC, Zhou J, Chen JJ , Zhao LB, Liu LX, Xu GW, Xie P
Received 30 January 2018
Accepted for publication 6 April 2018
Published 6 June 2018 Volume 2018:14 Pages 1451—1461
DOI https://doi.org/10.2147/NDT.S164134
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Si-Wen Gui,1,2,* Yi-Yun Liu,1–3,* Xiao-Gang Zhong,1,2,4,* Xinyu Liu,5,* Peng Zheng,1–3 Jun-Cai Pu,1–3 Jian Zhou,1,2 Jian-Jun Chen,1,2 Li-Bo Zhao,6 Lan-Xiang Liu,1–3 Guowang Xu,5 Peng Xie1–3
1Chongqing Key Laboratory of Neurobiology, Chongqing, China; 2Institute of Neuroscience and the Collaborative Innovation Center for Brain Science, Chongqing Medical University, Chongqing, China; 3Department of Neurology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China; 4School of Public Health and Management, Chongqing Medical University, Chongqing, China; 5CAS Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China; 6Department of Neurology, Yongchuan Hospital of Chongqing Medical University, Chongqing, China
*These authors contributed equally to this work
Introduction: Major depressive disorder (MDD) is a highly prevalent mental disorder affecting millions of people worldwide. However, a clear causative etiology of MDD remains unknown. In this study, we aimed to identify critical protein alterations in plasma from patients with MDD and integrate our proteomics and previous metabolomics data to reveal significantly perturbed pathways in MDD. An isobaric tag for relative and absolute quantification (iTRAQ)-based quantitative proteomics approach was conducted to compare plasma protein expression between patients with depression and healthy controls (CON).
Methods: For integrative analysis, Ingenuity Pathway Analysis software was used to analyze proteomics and metabolomics data and identify potential relationships among the differential proteins and metabolites.
Results: A total of 74 proteins were significantly changed in patients with depression compared with those in healthy CON. Bioinformatics analysis of differential proteins revealed significant alterations in lipid transport and metabolic function, including apolipoproteins (APOE, APOC4 and APOA5), and the serine protease inhibitor. According to canonical pathway analysis, the top five statistically significant pathways were related to lipid transport, inflammation and immunity.
Conclusion: Causal network analysis by integrating differential proteins and metabolites suggested that the disturbance of phospholipid metabolism might promote the inflammation in the central nervous system.
Keywords: major depressive disorder, plasma proteomics, iTRAQ, metabolomics, integrative analysis
Introduction
Major depressive disorder (MDD) is a severe and highly prevalent psychiatric disorder affecting 3% of the global population.1 It is a complex disease characterized by pervasive and persistent low mood and loss of interest or pleasure.2 A survey on the global burden of 220 diseases using disability-adjusted life years suggested that MDD was a key contributor to disability among the nonfatal consequences of disease and injury.3 Furthermore, consequences of depressive episodes include serious impairments in social functioning4 and even suicidal ideation and attempts.5 Indeed, approximately 2%–7% of MDD patients commit suicide.6 The pathophysiology of depression is not yet understood, but current theories center around monoaminergic systems,7 immunological dysfunction8 and hypothalamic–pituitary–adrenocortical (HPA) axis dysfunction.9 However, a clear causative etiology of MDD remains largely unknown. Therefore, improved understanding of the molecular mechanisms of disease progression and discovery of novel therapeutic targets are urgently needed for patients with MDD.
The central nervous system (CNS) and peripheral tissues have interactions. In recent years, multi-omics techniques including genomics,10 proteomics11 and metabolomics12 have been used to discover peripheral biomarkers of depression, such as neurotrophic factors, neurotransmitters, amino acids, lipids and carbohydrates. In contrast to the single-omics approach, researchers have used multiple combinatorial approaches to explore the disease state.13 Combined proteomics and metabolomics analysis is particularly attractive. Metabolomics data enable functional interpretation of proteomics data and aid in understanding their regulatory relationships. Accordingly, combined proteomics and metabolomics analysis has been used to determine the molecular mechanisms of various diseases such as cancer,14 cardiovascular disease15 and mental disorder.16 Moreover, in our previous study, we performed combined analysis of proteomics and metabolomics data from cerebellar tissue of chronic mild stress (CMS)-treated depressed rats and CON.17 We found abnormal cerebellum energy metabolism in depression, including disturbance of amino acid, glycolytic and tricarboxylic acid (TCA) cycle enzymes and mitochondrial respiratory chain dysfunction. Although this provides clues to the pathophysiology of depression, rat models do not adequately explain changes in the human body during disease conditions. Consequently, combined proteomics and metabolomics analysis in human plasma samples is needed to further explore molecular changes in depressed patients and how peripheral tissues affect CNS changes.
Here, we aimed to identify critical protein alterations in the plasma of patients with MDD. Furthermore, we integrated our proteomics and previous metabolomics data of depressed patients18 to explore significantly perturbed pathways in MDD.
Participants and methods
Ethics approval and informed consent
The ethics committee of Chongqing Medical University reviewed and approved the protocol of this study and the procedures used for sample collection and analysis. All subjects provided written informed consent after detailed introduction of the study. All procedures were performed according to the Declaration of Helsinki.
Participants
Patients with MDD were recruited from the Psychiatric Department of the First Affiliated Hospital of Chongqing Medical University, Chongqing, China. The inclusion criteria were diagnosis of MDD according to Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV) criteria for MDD,19 first-episode and treatment-naïve patients, patients aged 18–60 years, patients with Hamilton depression score (on the 17-item scale) >18 and patients with no comorbidity. Healthy CON matched for age, sex and body mass index (BMI) were enrolled from the Medical Examination Center at Chongqing Medical University. In total, 20 first-episode, drug-naïve, depressed patients and 20 demographically matched CON were included.
Samples for proteomics and metabolomics analyses were all screened from our clinical sample database (contains ~2,000 samples) with the same inclusion criteria. Table S1 gives information regarding patients used for metabolomics analysis.
Sample collection and preparation
Peripheral blood samples (5 mL) were collected in EDTA tubes (BD Vacutainer catalog no 367863; BD, Franklin Lakes, NJ, USA) by venipuncture between 8:00 and 10:00 am, and then immediately placed on ice and centrifuged at 3,000 rpm at 4°C for 15 min. The aliquoted plasma samples were stored at −80°C within 1 h of collection. Pooled plasma samples were generated by combining equal volumes of all 20 individual plasma samples from both groups. The protein concentration of each pooled sample was determined by using a commercial Bradford Protein Assay Kit (Beyotime, Shanghai, China). Protein normalization was confirmed by 12% sodium docecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Isobaric tags for relative and absolute quantification (iTRAQ) labeling and strong cation exchanger (SCX)-based fractionation
Before labeling, protein digestion for resultant peptide mixtures was performed according to the pooled sample preparation procedure described previously.20 Briefly, the proteins (100 μg) in each sample were reduced, blocked on cysteine, alkylated and subsequently digested with trypsin overnight at 37°C. Prior to iTRAQ labeling, digested samples were desalted with a C18 cartridge (Sigma-Aldrich Co., St Louis, MO, USA; EMD Millipore, Billerica, MA, USA) to remove urea, salt and other reagents. Then, the purified sample was evaporated to dryness with a SpeedVac (RVT4104; Thermo Fisher Scientific, Waltham, MA, USA). After resuspension with 30 μL 0.5 M triethylammonium bicarbonate (pH 8.5), samples were labeled with iTRAQ reagents following the protocol provided by the manufacturer. Proteins obtained from control samples were labeled with iTRAQ Reagent-8plex Multiplex Kit (AB Sciex, Framingham, MA, USA), which contained 113, 114 and 115 reporter tags. Proteins obtained from patients with MDD were labeled with 116, 117 and 118 reporter tags. iTRAQ-labeled peptides were mixed and fractionated by SCX chromatography using the AKTA Purifier 100 system (GE Healthcare UK Ltd, Little Chalfont, UK). For each test, 33 SCX fractions were collected followed by a C18 cartridge (66872-U; Sigma-Aldrich Co.; EMD Millipore) for desalination.21
Liquid chromatography–mass spectrometry (LC–MS)/MS-based quantitative protein analysis
The labeled peptides were analyzed on a Q Exactive mass spectrometer (Thermo Fisher Scientific) coupled with an EASY-nanoLC system. A nanoViper C18 trap column (100 μm × 2 cm, 5 μm particle size; Thermo Fisher Scientific) and a C18 analytical column (10 cm × 75 μm, 3 μm particle size; Thermo Fisher Scientific) were used. The peptide mixtures were first injected into buffer A (0.1% formic acid) at a flow rate of 300 nL/min. The separation gradient was 0–55 min, buffer B (0.1% formic acid and 84% acetonitrile) from 0% to 55%; 55–57 min, buffer B from 55% to 100% and 57–60 min, buffer B remained 100%. Then, the eluted peptides were analyzed by a Q Exactive mass spectrometer. The mass range of positive ion mode was from 300 to 1,800 m/z, the number of scan ranges was 20, the primary mass spectrometry resolution was 70,000 (m/z 200), the resolution for high-energy collisional dissociation (HCD) spectra was 17,500 (m/z 200) and the maximum inject times were set at 10 and 60 ms. Dynamic exclusion time was 40.0 s. The top 10 abundant precursor ions were used for MS/MS analysis with normalized collision energy at 30 eV and an underfill ratio of 0.1%.
Data analysis
Obtained MS/MS spectra were processed with Proteome Discoverer 1.4 (Thermo Fisher Scientific) and subsequently searched against the UniProt Human database, which contained 156,639 sequences, downloaded on January 5, 2017 (http://www.uniprot.org), using Mascot engine (version 2.2; Matrix Science, London, UK). Search parameters were as follows: trypsin as digestion enzyme, allowance of two missed cleavages, oxidized methionine and iTRAQ 8-plex (Y) as variable modifications with carbamidomethyl (C), iTRAQ modification at the peptide N-terminus and iTRAQ 8-plex (K) as fixed modifications. The peptide mass tolerance and fragment mass tolerance were set to 20 ppm and 0.1 Da, respectively. Furthermore, all peptide ratios were normalized by median protein ratio, which should be 1. A decoy database search strategy was adopted to estimate the false discovery rate (FDR) for peptide identification. For this study, a high peptide confidence (1% FDR) was selected based on the assumption that expression of most proteins does not change. This bias correction mitigates the systematic errors arising from samples in each experimental condition that were not combined in exactly equal amounts. The software identified a median average protein ratio, which was corrected to unity, and then, this factor was applied to all quantification results.22
Protein identifications were grouped depending on peptide matches from all samples. Protein ratios were calculated based on the median of unique peptides. Proteins with p-values ≤0.05 and fold changes ≥1.2 were considered as significantly regulated.23 Differential plasma proteins were profiled by functional classification through PANTHER (http://www.pantherdb.org/).42
Demographic data and clinical characteristics were analyzed using two-tailed Student’s t-test and Fisher’s exact test by SPSS 21.0 (IBM Corporation, Armonk, NY, USA).
Ingenuity Pathway Analysis (IPA)
IPA software (Qiagen NV, Venlo, the Netherlands) was used to integrative proteomics and metabolomics analyses (including biological function annotation, canonical pathways and relevant networks). The association between uploaded data and canonical pathways was measured by two means: 1) statistical significance – Fisher’s exact test was used to calculate p-values to determine the probability that the association between the uploaded molecules and the canonical pathways is explained by chance and 2) the ratio of the number of uploaded molecules that map to the given pathway divided by the total number of molecules in the canonical pathway.
Results
Demographic and clinical characteristics of participants
In total, 20 patients with MDD and 20 healthy CON were included. There were no significant differences in the age, sex or BMI. Demographic and clinical characteristics of the participants are summarized in Table 1.
 | Table 1 Demographic and clinical features of recruited subjectsa |
Quantitative protein analysis
After SCX fractionation and subsequent LC–MS/MS analysis, 370,351 MS/MS spectra were obtained, of which 21,213 peptide spectrum matches were assigned to 3,829 peptides (2,816 unique peptides) with an FDR of 1%. Moreover, these identified peptides corresponded to a set of 669 proteins. Ultimately, 74 differentially expressed proteins were identified (p < 0.05, unique peptide >1, 1.2-fold change; Figure 1A). Among these 74 significantly differentially expressed proteins, 11 showed upregulation and 63 downregulation (Table S2). The differential proteins in both sample groups were evaluated by hierarchical clustering analysis (Figure 1B) and classified by gene ontology (GO) categories of biological processes (Figure 1C). Functions of the differential proteins were mainly concentrated in three areas: metabolic process (GO: 0008152), cellular process (GO: 0009987) and biological regulation (GO: 0065007). Hence, significant changes in these biological functions may relate to disease development.
 | Figure 1 Differential proteins in patients with depression. |
Bioinformatics analysis
To further investigate the function of these 74 differentially expressed proteins, IPA software was used. According to canonical pathway analysis, the top five statistically significant pathways were related to lipid transport, inflammation and immunity (Figure 2A). As the top-ranking canonical pathway, the liver X receptor (LXR)/retinoid X receptor (RXR) activation pathway is involved in the regulation of lipid metabolism, inflammation and cholesterol catabolism. This finding is also supported by our previous study.24
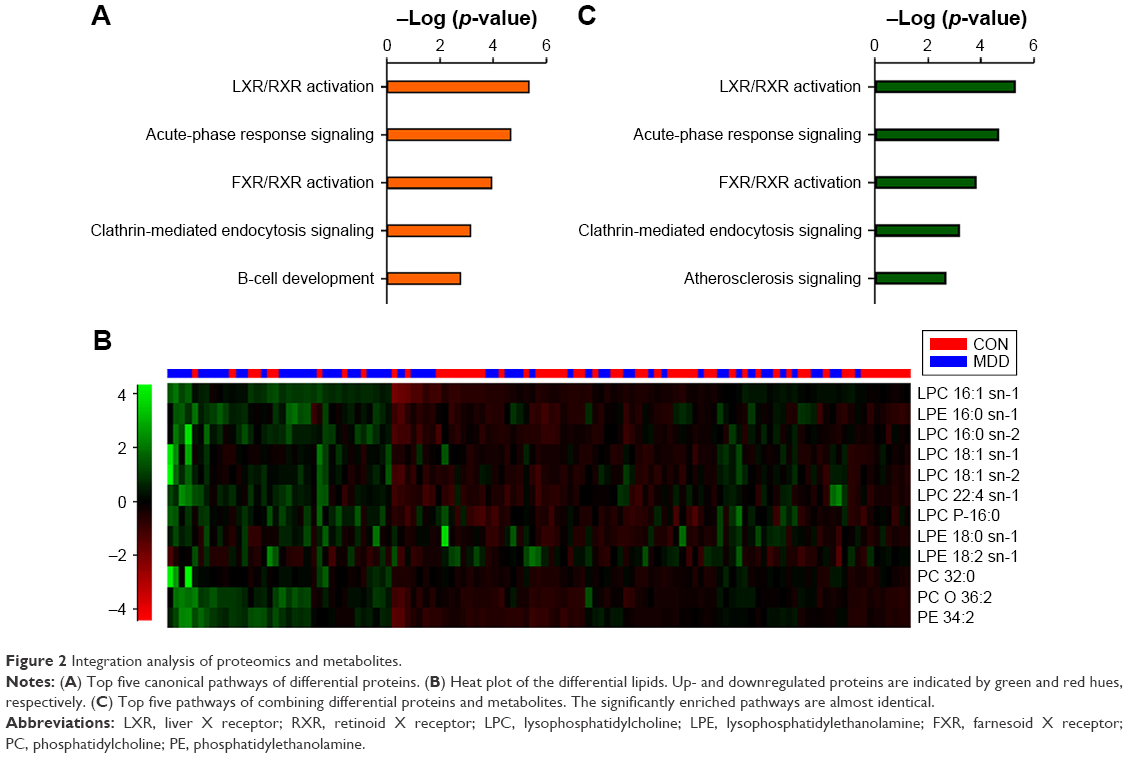 | Figure 2 Integration analysis of proteomics and metabolites. |
Thus, to explore the regulatory relationship between differential proteins and lipids, we uploaded significant lipid metabolites from LC–MS/MS analysis to the IPA database and performed an integrative analysis based on a causal network.18 The metabolites were classified into four categories: fatty acyls, amino acids, phospholipids and bile acids, alcohols and derivatives based on their chemical structure and properties (Table S3). Of these metabolites, phospholipids showed a general increase (Figure 2B).
By combining differential proteins and lipids, the top five significant pathways were highly similar to the pathways of differential proteins (Figure 2C). Network analysis identified a causal network of lipid metabolism that showed the interaction between proteins and lipids (Figure 3). In this network, apolipoprotein E (APOE) was the central node and showed downregulation (Figure 3). Furthermore, hemoglobin subunit beta (HBB), haptoglobin (HP), serotransferrin (TF), apolipoprotein A-V (APOA5), complement factor H (CFH), and immunoglobulin gamma (IgG) formed a downregulated co-expression pattern with APOE.
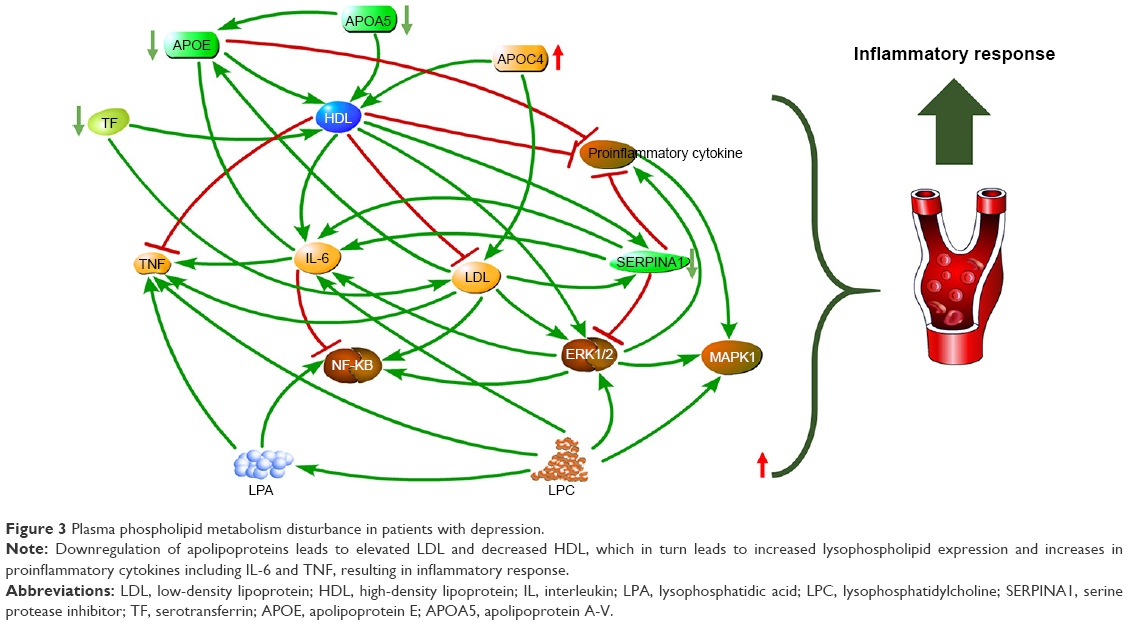 | Figure 3 Plasma phospholipid metabolism disturbance in patients with depression. |
Discussion
This study is the first to combine the proteomics and lipid metabolomics data to investigate the pathophysiological mechanisms underlying MDD in human plasma. Our IPA analysis results show that LXR/RXR activation is the top-ranking pathway for combined differentially expressed proteins and lipids. Previously, we obtained a similar result in proteomics study of the chronic unpredictable mild stress (CUMS) mouse model of depression,25 also finding a significant change in the LXR/RXR activation pathway. The RXR is a nuclear receptor that mediates the biological effects of retinoid. Moreover, RXRα is a dimeric partner of the type II nuclear receptor that contains LXR. Furthermore, LXR can be activated by oxysterol ligands and forms a heterodimer with RXR. Together, LXR and RXR are involved in lipid metabolism.26 Notably, depression is highly related to disturbance of lipid metabolism, as found in chronic restraint stress (CRS)-treated rats27 and also previously by us in blood from depressed patients.21 Furthermore, our previous research in CUMS-treated mice also showed that proteins involved in lipid metabolism may simultaneously participate in the immune regulation process.28 This suggests that disrupted lipid metabolism and neuroinflammation may coordinate together, leading to depression.
Additionally, due to their structural function, lipids may contribute to neuronal processes by influencing various signaling pathways directing neuronal survival or death.29 Lipids play an important role in CNS as modulators of the redox state and inflammation. Certain lipids are downregulated in an experimental model of Parkinson’s disease (PD) and, in particular, lysophosphatidylcholine (LPC; 16:0) and LPC (18:1), which are both important to neuroinflammatory signaling, appear to be upregulated.30
APOE is a key regulator of lipid metabolism. As the ligand for low-density lipoprotein (LDL) receptors, APOE is closely related to lipoprotein metabolism and immunomodulation. A growing body of research indicates that APOE is involved in many immunological processes, including inhibition of T-cell proliferation, regulation of macrophage function, promotion of lipid antigen presentation (via CD1) to natural killer T cells as well as modulation of inflammation and oxidation.31,32 APOE is produced by macrophages, and its secretion has been shown to be confined to typical monocytes in peripheral blood mononuclear cells (PBMCs). Secretion of APOE by monocytes is downregulated by inflammatory cytokines and upregulated by transforming growth factor (TGF)-beta.33 In the nervous system, non-neuronal cell types, most notably astroglia and microglia, are the primary producers of APOE, while neurons preferentially express the receptors for APOE.34 Here, APOE was found to be downregulated in an integrative network (Figure 3). We could hypothesize that this is the result of astroglia and microglia dysfunction. As a part of the blood–brain barrier (BBB), astroglia dysfunction may lead to enhanced permeability of BBB. Consequently, more peripheral inflammatory factors may enter the brain causing neuroinflammation. On the other hand, the reduction of apolipoprotein-affected lipid transport in plasma may result in decreased high-density lipoprotein (HDL) and increased LDL.35 HDL helps inhibit oxidation36 and inflammation,37 and promotes the platelet aggregation.38 Therefore, the activation of oxidation and inflammation in MDD may be caused by the downregulation of HDL. Our results suggest that upregulation of the pro-inflammatory cytokine interleukin (IL)-6 may be caused by a general increase in lysophospholipids released by LDL. Lysophosphatides result from hydrolysis of phospholipids by enzymatic action of phospholipase A2. Indeed, LPC, lysophosphatidylethanolamine (LPE) and other hemolytic molecules can produce lysophosphatidic acid (LPA). The role of LPA in astrocytes suggests that LPA may alter the permeability of the BBB under physiological or pathological conditions.39,40 This may involve increased mRNA levels in the entire proinflammatory network, as found in autopsied brain from patients with depression.41 Additionally, LPA increases neurotoxicity of excitatory amino acids and inhibits glutamate uptake by glial cells. The downregulation of apolipoproteins leads to decreased HDL, which can accept and promote excretion of lysophospholipids. This process may promote these neurotoxic substances, which are more detrimental to the CNS.
There are several limitations to our study. First, the cohorts used in the proteomics and metabolomics analyses are different. Consequently, we used a method based on biological function of pathways for integration analysis instead of investigating the relationship between differential proteins and metabolites at the mathematical level. Second, iTRAQ and label free, as two very popular quantitative proteomic analysis methods, each has its own advantages and can complement each other. We only used an iTRAQ approach here, and the combination of these two methods should be considered in future studies. Third, our proteomics findings were not validated by a secondary method such as Western blot. It is worth noting that this study primarily combined proteomics and metabolomics analyses of plasma samples from patients with depression and healthy CON. Following bioinformatics analysis, phospholipid metabolism dysregulation was identified as significantly altered in patients with depression. Thus, the results showed links between differential proteins and metabolite biomarkers, and may provide novel insight into further research on antidepressants, such as potential treatment targets. Further study in this filed should include more investigation to validate regulations among molecules.
Conclusion
In this study, we used an iTRAQ-based quantitative proteomics approach to compare plasma protein expression between patients with depression and healthy CON. Altogether, we identified 74 differentially expressed proteins, mainly associated with dysregulation of lipid metabolism pathway. In particular, aberrant expression of apolipoproteins may play a role in the pathogenesis of depression. Moreover, combined analysis of differential proteins and metabolites suggests that disturbance of phospholipid metabolism coupled with abnormal expression of apolipoproteins may promote the inflammation in the CNS and ultimately lead to depression.
Acknowledgments
This work was supported by Major Scientific Instrument and Equipment Development Project of China (Grant No 2012YQ120044) and National Key R&D Program of China (Grant No 2017YFA0505700).
Disclosure
The authors report no conflicts of interest in this work.
References
Collaborators GDaIIaP. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2016;388(10053):1545. | ||
Health TNIoM [webpage on the Internet]. Depression. 2016. Available from: https://www.nimh.nih.gov/health/topics/depression/index.shtml. Accessed December 28, 2017. | ||
Salomon JA, Vos T, Hogan DR, et al. Common values in assessing health outcomes from disease and injury: disability weights measurement study for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2129. | ||
Kline ER, Seidman LJ, Cornblatt BA, et al. Depression and clinical high-risk states: baseline presentation of depressed vs. non-depressed participants in the NAPLS-2 cohort. Schizophr Res. 2017;192:357–363. | ||
Subramaniam M, Abdin E, Seow EL, Picco L, Vaingankar JA, Chong SA. Suicidal ideation, suicidal plan and suicidal attempts among those with major depressive disorder. Ann Acad Med Singapore. 2014;43(8): 412–421. | ||
Ohara MW. Oxford Handbook of Depression and Comorbidity. Oxford, England: Oxford University Press; 2014. | ||
Ruhé HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: a meta-analysis of monoamine depletion studies. Mol Psychiatry. 2007;12(4):331. | ||
Köhler O, Benros ME, Nordentoft M, et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry. 2014;71(12):1381–1391. | ||
Phillips LJ, Mcgorry PD, Garner B, et al. Stress, the hippocampus and the hypothalamic-pituitary-adrenal axis: implications for the development of psychotic disorders. Aust N Z J Psychiatry. 2006;40(9):725–741. | ||
Leniculescu H, Kurian SM, Yehyawi N, et al. Identifying blood biomarkers for mood disorders using convergent functional genomics. Mol Psychiatry. 2009;14(2):156–174. | ||
Yang Y, Yang D, Tang G, et al. Proteomics reveals energy and glutathione metabolic dysregulation in the prefrontal cortex of a rat model of depression. Neuroscience. 2013;247(13):191–200. | ||
Peng Z, Ying W, Liang C, et al. Identification and validation of urinary metabolite biomarkers for major depressive disorder. Mol Cell Proteomics. 2013;12(1):207. | ||
Snyder M, Mias G, Stanberry L, Kolker E. Metadata checklist for the integrated personal OMICS study: proteomics and metabolomics experiments. OMICS. 2014;18(1):81–85. | ||
Ma Y, Zhang P, Wang F, Liu W, Yang J, Qin H. An integrated proteomics and metabolomics approach for defining oncofetal biomarkers in the colorectal cancer. Ann Surg. 2012;255(4):720. | ||
Mayr M, Yusuf S, Weir G, et al. Combined metabolomic and proteomic analysis of human atrial fibrillation. J Am Coll Cardiol. 2008;51(5):585. | ||
Filiou MD, Zhang Y, Teplytska L, et al. Proteomics and metabolomics analysis of a trait anxiety mouse model reveals divergent mitochondrial pathways. Biol Psychiatry. 2011;70(11):1074–1082. | ||
Shao WH, Chen JJ, Fan SH, et al. Combined metabolomics and proteomics analysis of major depression in an animal model: perturbed energy metabolism in the chronic mild stressed rat cerebellum. OMICS. 2015;19(7):383. | ||
Liu X, Zheng P, Zhao X, et al. Discovery and validation of plasma biomarkers for major depressive disorder classification based on liquid chromatography- mass spectrometry. J Proteome Res. 2015;14(5):2322–2330. | ||
Uher R, Payne JL, Pavlova B, Perlis RH. Major depressive disorder in DSM-5: implications for clinical practice and research of changes from DSM-IV. Depress Anxiety. 2014;31(6):459–471. | ||
Wisniewski J, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6(5): 359–362. | ||
Xu HB, Zhang RF, Luo D, et al. Comparative proteomic analysis of plasma from major depressive patients: identification of proteins associated with lipid metabolism and immunoregulation. Int J Neuropsychopharmacol. 2012;15(10):1413. | ||
Han X, Shao W, Liu Z, et al. iTRAQ-based quantitative analysis of hippocampal postsynaptic density-associated proteins in a rat chronic mild stress model of depression. Neuroscience. 2015;298:220. | ||
Ren Y, Hao P, Dutta B, et al. Hypoxia modulates A431 cellular pathways association to tumor radioresistance and enhanced migration revealed by comprehensive proteomic and functional studies. Mol Cell Proteomics. 2013;12(2):485. | ||
Liu YY, Zhou XY, Yang LN, et al. Social defeat stress causes depression- like behavior with metabolite changes in the prefrontal cortex of rats. PLoS One. 2017;12(4):e0176725. | ||
Yang C, Zhou C, Li J, et al. Quantitative proteomic study of the plasma reveals acute phase response and LXR/RXR and FXR/RXR activation in the chronic unpredictable mild stress mouse model of depression. Mol Med Rep. 2017;17(1):93–102. | ||
Edwards PA, Kennedy MA, Mak PA. LXRs; oxysterol-activated nuclear receptors that regulate genes controlling lipid homeostasis. Vascul Pharmacol. 2002;38(4):249–256. | ||
Liu L, Zhou X, Zhang Y, et al. The identification of metabolic disturbances in the prefrontal cortex of the chronic restraint stress rat model of depression. Behav Brain Res. 2016;305:148. | ||
Wu Y, Tang J, Zhou C, et al. Quantitative proteomics analysis of the liver reveals immune regulation and lipid metabolism dysregulation in a mouse model of depression. Behav Brain Res. 2016;311:330–339. | ||
Catani MV, Gasperi V, Bisogno T, Maccarrone M. Essential dietary bioactive lipids in neuroinflammatory diseases. Antioxid Redox Signal. Epub 2017 Jul 24. | ||
Farmer K, Smith CA, Hayley S, Smith J. Major alterations of phosphatidylcholine and lysophosphotidylcholine lipids in the substantia nigra using an early stage model of Parkinson’s disease. Int J Mol Sci. 2015; 16(8):18865–18877. | ||
van den EP, Garg S, León L, et al. Apolipoprotein-mediated pathways of lipid antigen presentation. Nature. 2005;437(7060):906. | ||
Zhang H, Wu J, Zhu J. The role of apolipoprotein E in Guillain-Barré syndrome and experimental autoimmune neuritis. J Biomed Biotechnol. 2010;2010(1110–7243):357412. | ||
Braesch-Andersen S, Paulie S, Smedman C, Mia S, Kumagai-Braesch M. ApoE production in human monocytes and its regulation by inflammatory cytokines. PLoS One. 2013;8(11):e79908. | ||
Zhang Z, Mu J, Li J, Li W, Song J. Aberrant apolipoprotein E expression and cognitive dysfunction in patients with poststroke depression. Genet Test Mol Biomarkers. 2013;17(1):47–51. | ||
Shimano H, Yamada N, Katsuki M, et al. Overexpression of apolipoprotein E in transgenic mice: marked reduction in plasma lipoproteins except high density lipoprotein and resistance against diet-induced hypercholesterolemia. Proc Natl Acad Sci U S A. 1992;89(5):1750. | ||
Ribas V, Sánchez-Quesada JL, Antón R, et al. Human apolipoprotein A-II enrichment displaces paraoxonase from HDL and impairs its antioxidant properties: a new mechanism linking HDL protein composition and antiatherogenic potential. Circ Res. 2004;95(8):789–797. | ||
Vaisar T, Pennathur S, Green PS, et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. 2007;117(3):746. | ||
Takahashi Y, Chiba H, Matsuno K, et al. Native lipoproteins inhibit platelet activation induced by oxidized lipoproteins. Biochem Biophys Res Commun. 1996;222(2):453–459. | ||
Keller JN, Steiner MR, Holtsberg FW, Mattson MP, Steiner SM. Lysophosphatidic acid-induced proliferation-related signals in astrocytes. J Neurochem. 1997;69(3):1073–1084. | ||
Xaio H, Banks WA, Niehoff ML, Morley JE. Effect of LPS on the permeability of the blood-brain barrier to insulin. Brain Res. 2001; 896(1–2):36. | ||
Shelton RC, Claiborne J, Sidorykwegrzynowicz M, et al. Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Mol Psychiatry. 2011;16(7):751–762. | ||
Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8(8):1551–1566. |
Supplementary materials
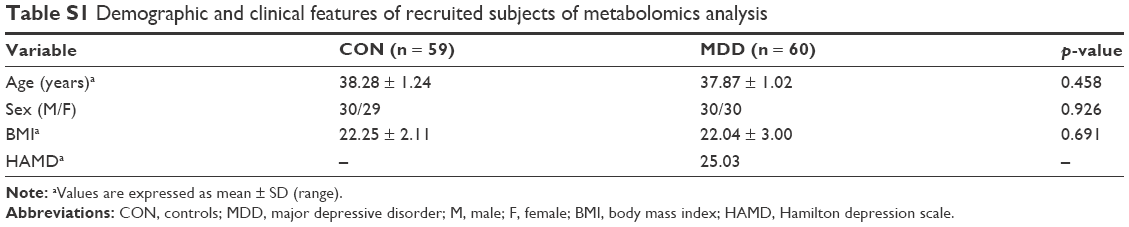 | Table S1 Demographic and clinical features of recruited subjects of metabolomics analysis |
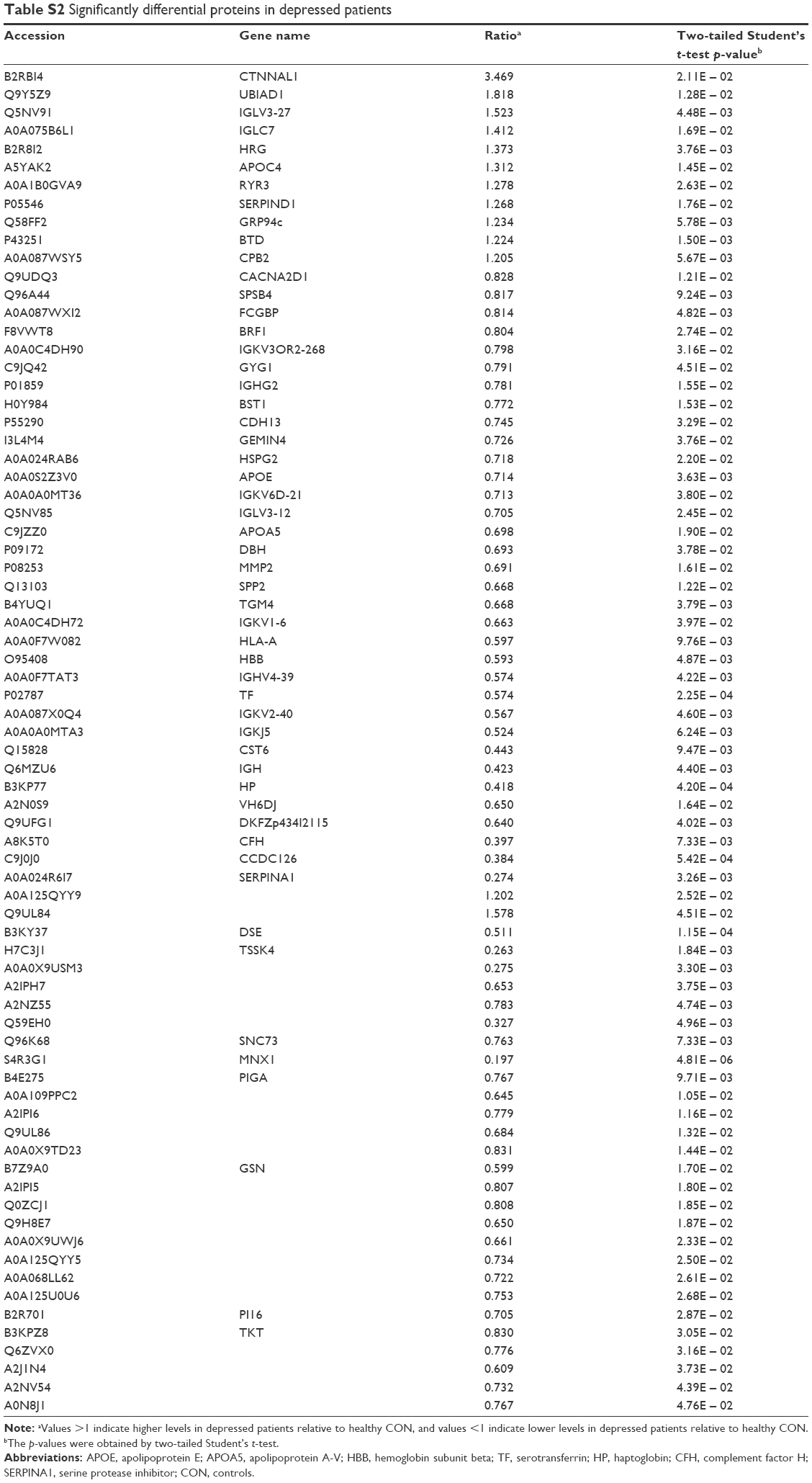 | Table S2 Significantly differential proteins in depressed patients |
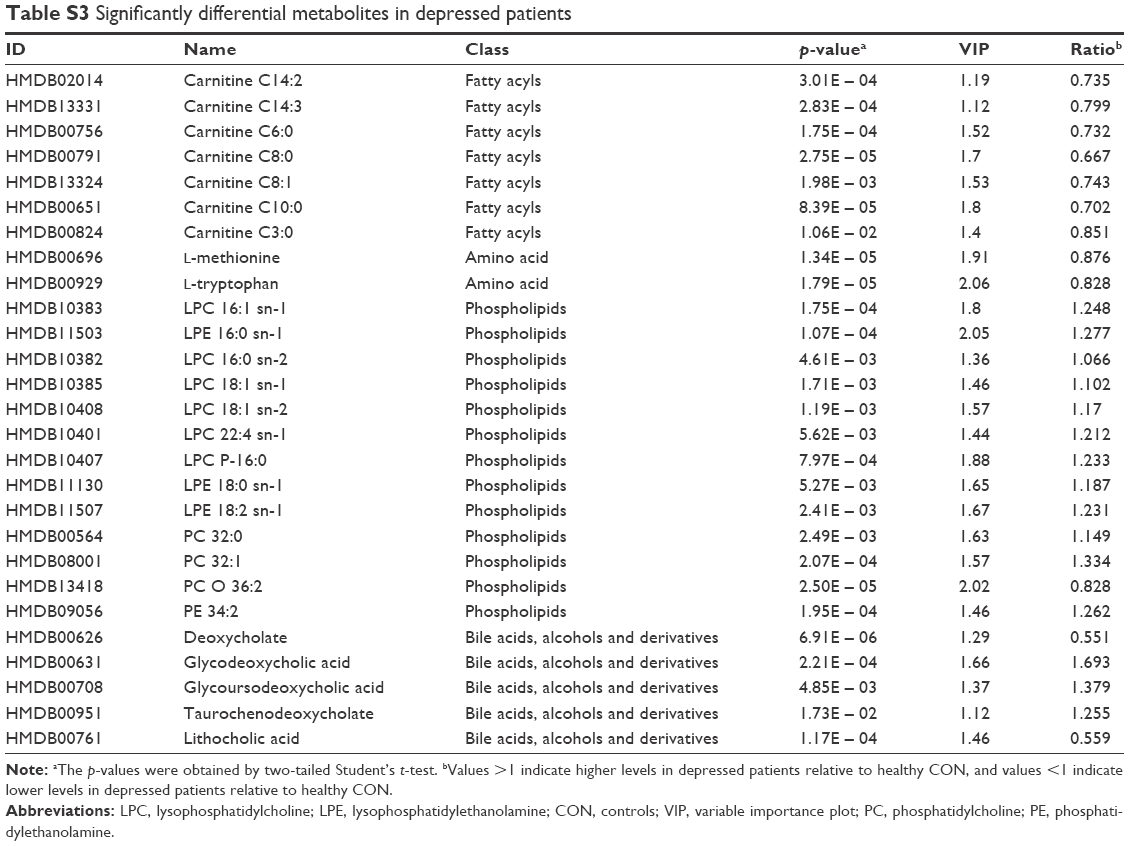 | Table S3 Significantly differential metabolites in depressed patients |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.