")
Back to Archived Journals » Core Evidence » Volume 11
Pirfenidone in the treatment of idiopathic pulmonary fibrosis: an evidence-based review of its place in therapy
Authors Margaritopoulos G, Vasarmidi E, Antoniou K
Received 24 August 2015
Accepted for publication 21 March 2016
Published 1 July 2016 Volume 2016:11 Pages 11—22
DOI https://doi.org/10.2147/CE.S76549
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Garry Walsh
George A Margaritopoulos,1 Eirini Vasarmidi,2 Katerina M Antoniou2
1Interstitial Lung Disease Unit, Royal Brompton Hospital, London, UK; 2Department of Thoracic Medicine and Laboratory of Molecular and Cellular Pneumonology, Interstitial Lung Disease Unit, University Hospital of Heraklion, Heraklion, Greece
Abstract: The landscape of idiopathic pulmonary fibrosis (IPF) has changed. The significant progress regarding our knowledge on the pathogenesis of the disease together with the experience achieved after a series of negative trials has led to the development of two drugs for the treatment of IPF. Both pirfenidone and nintedanib can slow significantly the rate of disease progression. They are safe with side effects that can be either prevented by close collaboration between health care professionals and patients or treated successfully when they occur, rarely leading to treatment discontinuation. However, there are still few unanswered questions regarding the application of the beneficial results of pharmaceutical trials in the general population of IPF patients. Long-term “real-life” studies are being undertaken to answer these questions. In this article, we focus on the advances that have led to the development of the antifibrotic agents with particular focus on pirfenidone.
Keywords: antifibrotic, pirfenidone, photosensitivity, nausea
Core evidence clinical impact summary for pirfenidone
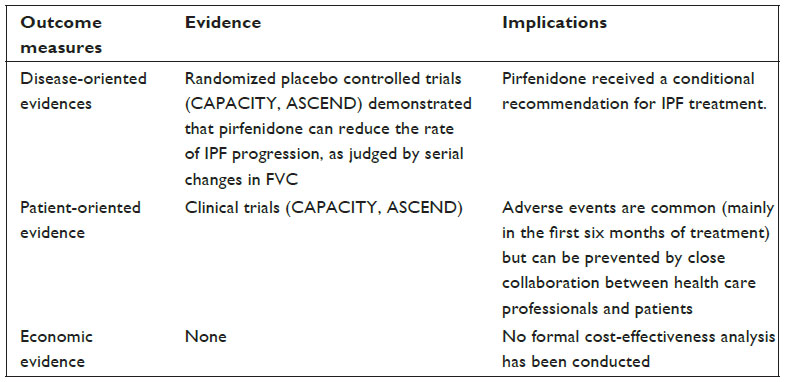
Introduction
There is no doubt that a new era has risen for idiopathic pulmonary fibrosis (IPF) treatment. The statement “a disease with no known effective treatment” will not be used anymore when we have to inform our patients about the management of their disease. The disappointment created by a series of negative trials has been followed by the completion of three successful randomized controlled trials and the licensing of two novel drugs, namely, pirfenidone and nintedanib, for this dreadful disease.1–3 Definitely, that was a significant accomplishment, which has led to the reform of the guidelines published only 4 years ago.4 In the most recent statement, both drugs have received the label of “conditional recommendation for IPF treatment”.5 It should be stressed that both drugs are not miraculous as they only manage to slow the rate of IPF progression, and issues regarding long-term efficacy and safety should be further studied and clarified. However, there is a light at the end of the tunnel, and future trials based on the knowledge and experience previously accumulated may lead to the development of more efficient drugs. In this article, we briefly review the pathogenesis of IPF, and the completed negative and positive trials in IPF, with particular focus on efficacy and safety of pirfenidone.
Pathogenesis of IPF
IPF is the most frequent and devastating form of idiopathic interstitial pneumonias.4 The median survival from the time of diagnosis is 3 years, explaining why IPF is considered more lethal than many cancers. It commonly affects males over 60 years old with a history of cigarette smoking. The main presenting symptoms are shortness of breath and chronic cough. In the US, 40,000 patients die of IPF every year, and both incidence and prevalence have been found to be higher in patients older than 65 years than previously reported.6
According to the recent official ATS/ERS/JRS/ALAT statement, the diagnosis of IPF requires the following: 1) exclusion of other known causes of interstitial lung disease (ILD) (eg, domestic and occupational environmental exposures, connective tissue disease, and drug toxicity); 2) in patients not subjected to surgical lung biopsy, the presence of a usual interstitial pneumonia (UIP) pattern on high-resolution computed tomography, which is characterized by subpleural, basal-predominant reticular changes, traction bronchiectasis, and honeycombing; and 3) specific combinations of high-resolution computed tomography and surgical lung biopsy pattern in patients subjected to surgical lung biopsy.4 Review of the cases by a multidisciplinary team meeting with pulmonologists, radiologists, and pathologists with special interest in the field of ILDs represents the “gold standard” diagnostic approach.
Our understanding regarding the pathogenesis of IPF has shifted significantly in the recent years.7 Initially, it was believed that fibrosis was the result of chronic inflammation. However, the current belief is that the disease is the result of an epithelial-driven and fibroblast-activated process in which inflammation may have only an ancillary role.8 An interplay between host and environmental factors leads to repetitive microinjuries of alveolar epithelial cells, simultaneous activation of multiple pathogenetic pathways, activation of myofibroblasts, exaggerated deposition of extracellular matrix, development of fibrosis with architectural distortion, and consequently irreversible loss of lung function.9
Recent studies have suggested that the loss-of-function mutations of telomerase, an enzyme that compensates telomere shortening,10 a common polymorphism in the promoter region of MUC5B gene which confers an increased susceptibility to develop IPF but surprisingly is associated with a survival benefit11 and a single-nucleotide polymorphism in the gene for TOLLIP and SPPL2C that also promote fibrogenesis,12 are involved in the pathogenesis of IPF. The most well-known environmental factors are smoking, viral and bacterial infections, and gastroesophageal reflux.13–17
Myofibroblasts are the key cells in the process of fibrogenesis. They are a specialized contractile type of cells that predominate in areas of active fibrosis, namely, fibroblastic foci. There are three possible sources of myofibroblasts. Local fibroblasts can migrate in the site of injury attracted by mediators such as platelet-derived growth factor (PDGF), transforming growth factor-β (TGF-β), and tumor necrosis factor-α (TNF-α) produced by injured alveolar epithelial cells, and differentiate into myofibroblasts. Myofibroblasts may also derive from resident epithelial cells through a process called epithelial-to-mesenchymal transition as well as from circulating fibrocytes through a CXCR4-CXCL12-dependent mechanism.18,19
Dysregulations of developmental pathways, dysfunction of the immune system and coagulation cascade, loss of the balance between oxidants and antioxidants, and dysregulation of microRNAs are all mechanisms involved in the pathogenesis of IPF.9 Approximately 10% of the microRNAs are significantly changed in IPF lungs. Significantly downregulated microRNAs are members of let-7, mir-29, and mir-30 families and the miR-17-92 cluster whereas mir-155 and mir-21 are significantly upregulated.20 Therefore, it becomes conceivable that targeting a single pathway in order to slow the progression of the disease is not realistic and may have been one of the reasons of the failure of previous trials. The balance of abnormalities in each of these key pathways may vary between affected individuals. This would explain the range of clinical, radiological, and pathological phenotypes observed in IPF. Both pirfenidone and nintedanib target more than one pathway, and this may explain their effect on the progression of the disease. However, the combination of these compounds with others may be more beneficial. A growing body of evidence suggests that IPF has many clinical and biological similarities to cancer.21 The current treatment recommendation for cancer includes a combination of agents with different mechanisms of action, and keeping this in mind, future studies of combination therapies in IPF could be designed.
Negative randomized controlled trials in IPF
Different compounds have been studied in IPF (Table 1). Most of these studies have failed to show a significant effect in the progression of IPF. It can be speculated that a number of reasons have contributed to the failure of previous trials, but on the other hand, the knowledge gained has definitely helped in the design of trials that have led to the approval of two drugs as an IPF-specific therapy. Until few years ago, it was believed that IPF was an inflammatory-driven disease, whereas in the last decade, it has been viewed as an epithelial-driven disease. Therefore, it is only recently that clinical research in IPF has shifted focus from immunomodulatory to antifibrotic and antiproliferative compounds. Moreover, the knowledge that multiple pathways are involved in the pathogenesis of IPF has changed the therapeutic target. There has been a shift from using compounds that inhibit individual mediators or signaling pathways to compounds that are pleiotropic in their antifibrotic properties. Inclusion criteria have been more precise in the recently completed trials. Central review of the diagnosis of IPF performed by radiologists and pathologists experienced in ILD allowed the inclusion of well-defined populations of patients. Another important issue is the choice of an end point. Different end points have been used in the past, and that has made comparing clinical efficacy quite difficult.
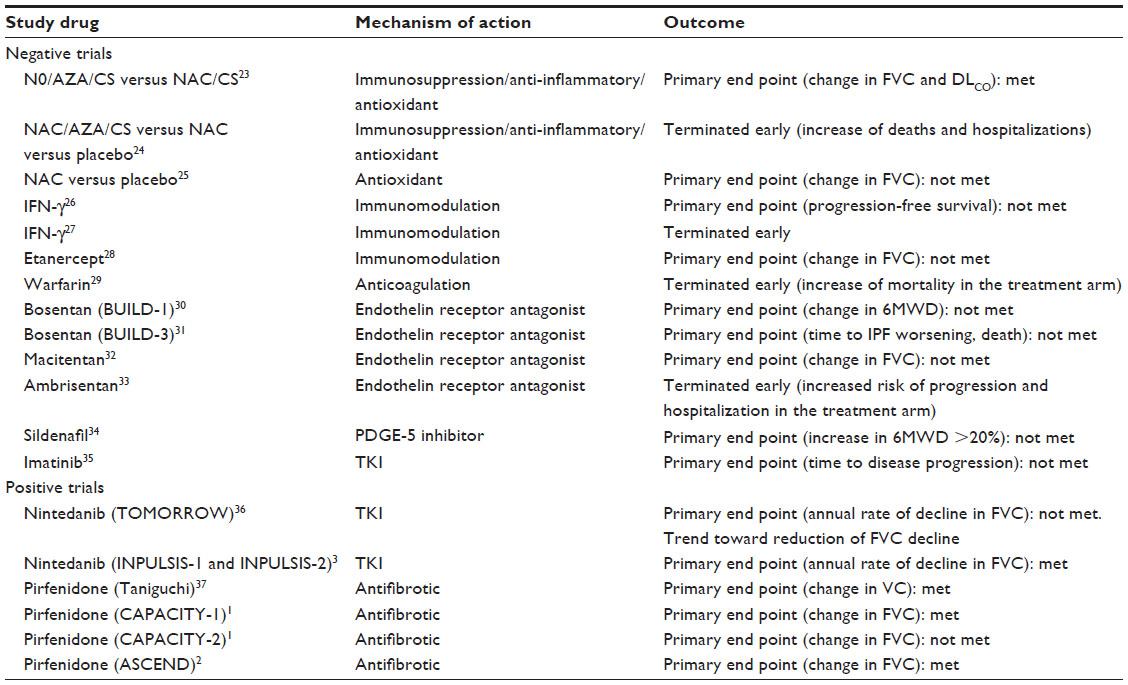 | Table 1 Completed clinical trials in IPF |
In the 2000 ATS/ERS guidelines, the use of anti- inflammatory agents, namely, prednisolone and azathioprine, was recommended as the standard of care.22 Subsequently, a much criticized study, in which the combination of prednisolone, azathioprine, and N-acetyl-cysteine (NAC) was compared with prednisolone and azathioprine, showed a significant reduction in the rate of decline of the forced vital capacity (FVC) and of the diffusing capacity for carbon monoxide (DLCO).23 Recent advances in the understanding of the pathogenesis of IPF together with the disappointing results of the recent PANTHER-IPF trial, in which the triple-therapy arm (combination of prednisolone, azathioprine, and NAC) was terminated early because of an increase of the number of deaths and hospitalizations as compared to the placebo arm, have led to a strong negative recommendation against the use of this regime in IPF.5,24 Similarly, in the subsequent modified PANTHER-IPF trial, the use of NAC versus placebo failed to meet the primary end point of change in FVC after 60 weeks of treatment.25
Interferon-γ (IFN-γ) is a cytokine with immunomodulatory and antifibrotic properties. A large randomized control trial that followed other smaller ones did not show any benefit in overall survival, and the trial was terminated early.26,27,38
Etanercept, a TNF-α antagonist, failed to show a significant effect on FVC from baseline to 48 weeks, although the drug was generally well tolerated.28
The knowledge that coagulation factors are overexpressed in the fibrotic lung has led to the belief that anticoagulation could have a positive effect in IPF. A study characterized by several limitations has shown a 1-year survival benefit of anticoagulation therapy (either warfarin or heparin).39 However, even in this case, a better designed trial not only failed to meet the primary end point but was also terminated early due to increased number of deaths in the treated arm.29 The discrepancy between the two studies could be attributed to differences in the genetic background of the study population and in the methodology of the two studies.40 Currently, anticoagulants are not recommended in IPF.5
Vasodilators, such as bosentan, macitentan, and ambrisentan, have been demonstrated ineffective in IPF trials in which they have been used in the hope of a combined vascular and interstitial effect.30–33 Moreover, ambrisentan has been related with disease progression. Only sildenafil has shown an encouraging effect in patients with advanced IPF who were more likely to have an element of pulmonary vasculopathy.34 Although in this study the primary end point has not been achieved (ie, improvement in 6-minute walking distance), there have been some encouraging results with regard to the secondary end points. An improvement in DLCO, gas transfer coefficient, and arterial oxygenation, in the absence of a simultaneous effect on FVC, indicated a significant effect on the pulmonary vessels.
The tyrosine kinase inhibitor, imatinib mesylate, has also failed to demonstrate effectiveness in IPF with no significant effect on time to disease progression or time to death.35
Positive randomized controlled trials in IPF
Nintedanib
Nintedanib is an intracellular inhibitor that targets multiple tyrosine kinases, including the PDGF receptor, vascular endothelial growth factor receptors 1 and 2, and fibroblast growth factor receptors 1–3.41 Initially developed for cancer treatment, it has been shown to additionally possess antifibrotic activities. A double-blind, randomized, placebo-controlled Phase II trial of the efficacy and safety of nintedanib showed that the annual rate of decline of FVC in the group of patients who received 150 mg daily was 0.06 L compared with 0.19 L in the placebo arm. Moreover, fewer acute exacerbations and preserved quality of life were observed in the treatment arm. Gastrointestinal side effects and liver toxicity were moderate in the high-dose arm compared to placebo.36 The INPULSIS trials were two parallel 52-week, randomized, double-blind, Phase III studies (INPULSIS-1 and INPULSIS-2) that further evaluated the efficacy and safety of nintedanib (at the dose of 150 mg twice daily) compared with placebo in patients with IPF.3 One thousand and sixty-six patients were randomly assigned in a 3:2 ratio to receive nintedanib or placebo. In INPULSIS-1, the adjusted annual rate of decline of FVC was –114. mL in the nintedanib arm and –239.9 mL in the placebo arm (difference: 125.3 mL; P<0.001). In INPULSIS-2, the respective rates were –113.6 and –207.3 mL (difference: 93.7 mL; P<0.001). Difference in time to the first acute exacerbation of IPF was statistically significant in INPULSIS-2 (P=0.005) but not in INPULSIS-1 (P=0.67). In a prespecified pooled analysis, no significant difference in death from any cause or death from a respiratory cause was observed (hazard ratio [HR] 0.70; P=0.14). However, neither trial was powered to detect statistically significant differences in mortality. The most frequent side effect in the treatment arms was diarrhea, but most patients tolerated the full dose of nintedanib during the study.
Pirfenidone
Preclinical studies
Pirfenidone (5-methyl-1-phenyl-2-[1H]-pyridone) is an orally administered drug with antifibrotic, anti-inflammatory, and antioxidant effects. These properties have been initially evaluated in preclinical studies.
In vitro studies of normal human lung fibroblasts have shown that pirfenidone abrogates TGF-β1-stimulated collagen synthesis by inhibiting the upregulation of HSP47 and Col1 RNA, in a concentration-dependent manner.42 In vitro experiments with rat hepatic stellate cells showed that pirfenidone blocks the proliferative effects of PDGF in a concentration-dependent manner.43 In a more recent study in primary human lung fibroblasts, pirfenidone has reduced fibroblast proliferation, production of alpha smooth muscle cells actin induced by TGF-β, and the level of procollagen 1.44
In the in vivo model of murine lipopolysaccharide-induced acute lung injury and endotoxic shock, pirfenidone downregulated proinflammatory cytokines, such as TNF-α, IFN-γ, and interleukin (IL)-6, and improved survival.45–47 In both in vitro human peripheral blood lymphocytes and in vivo mice model, pirfenidone exerts a similar anti-inflammatory effect inhibiting staphylococcal enterotoxin B-induced proliferation and synthesis of TNF-α, IFN-γ, IL-1β, and IL-6.48
With regard to the antioxidant properties, it has been observed that pirfenidone can act as a scavenger of hydroxyl (OH–) and superoxide anion (O–) free radicals.49 In sheep liver microsomes, pirfenidone blocks NADPH-dependent lipid peroxidation in a concentration-dependent manner.50
The effects of pirfenidone in animal models of bleomycin-induced pulmonary fibrosis suggest an attenuation of the development of the disease. However, it should be stressed that despite the bleomycin model being at the forefront of basic research into the regulation of pulmonary fibrogenesis, it does not reproduce the phenotype exhibited by IPF. In the bleomycin model, the initial insult results in widespread epithelial cell apoptosis and necrosis, which subsequently triggers a marked neutrophilic inflammatory infiltrate. The extent of epithelial cell death and the subsequent inflammatory response determine the severity of the fibrosis. In hamsters, pirfenidone has been administered 2 days prior to a single dose of bleomycin (defined as prophylactic administration) or after the second of three weekly consecutive administrations. It has significantly attenuated the development of fibrosis and reduced the markers of oxidative stress and neutrophilic inflammation.51,52 Importantly, it has been shown that pirfenidone works even when administered at a dose of 400 mg/kg/day, 14 days after the first of five administrations of bleomycin for five consecutive days, suggesting that the drug can also act therapeutically.53
Clinical trials in IPF
The first clinical trial of pirfenidone in IPF was a Phase II open-label trial performed by Raghu et al.54 They enrolled 54 patients with a mean FVC of 58.8% predicted and a mean DLCO of 34.3% predicted, who had been previously treated unsuccessfully with corticosteroids with or without the addition of an immunosuppressant. Pirfenidone was administered at a dose of 40 mg/kg up to a maximum of 3,600 mg/day, in divided doses for an average of 25 months. One- and 2-year survival was 78% and 63%, respectively. However, the major limitation of the study has been the lack of a placebo arm.
A small Phase II open-label Japanese study of ten patients with advanced IPF and scleroderma-associated pulmonary fibrosis has shown that after 1 year of treatment, there was no significant deterioration of the disease; the drug was well tolerated, but the survival in 2 years has not been significantly changed.55
Subsequently, a Phase II multicenter, randomized, placebo-controlled trial was carried out. One hundred and seven patients were randomized 2:1 and received 600 mg of pirfenidone three times daily or placebo for 12 months.56 The mean vital capacity was 81.6%±20.3%, and the DLCO was 57.6%±17.2%. The primary end point was change in the lowest arterial oxygen saturation during the 6-minute walk test (6MWT). The trial was terminated prematurely at 9 months because of excess acute exacerbations in the placebo group (5/35) compared with the pirfenidone arm (0/72). Although the primary end point was not met, a significantly slower decline of FVC was observed in the treatment arm than in placebo (–0.03 vs –0.13 L; P=0.0366).
Later, a double-blind, randomized, placebo-controlled Phase III trial was designed in Japan to test the efficacy and safety of the drug over 52 weeks.37 The patients had mild-to-moderate IPF with a mean vital capacity of 77.3%±16.8% predicted and a mean DLCO of 52.1%±16.8% predicted, and were assigned to receive a high dose (1,800 mg/day) or a low dose of pirfenidone (1,200 mg/day) or placebo in the ratio of 2:1:2. The primary end point was change in FVC from baseline to 52 weeks, and secondary end points included progression-free survival, defined as the time until the first progression event (decline in FVC >10%, or death), and change in the lowest arterial oxygen saturation measured by pulse oximetry during the 6-minute steady-state exercise test. The primary end point was met with a significantly slower rate of FVC decline in the high-dose group than in the placebo arm (–0.09 vs –0.16 L; P=0.04). The progression-free survival was significantly longer in the high-dose arm compared with the placebo arm (P=0.02). It should be stressed that the study has received a lot of criticism because there has been a change in the initial end point (ie, change in the lowest arterial oxygen saturation measured by pulse oximetry during the 6-minute steady-state exercise test) regardless of whether this was decided before unblinding the study. There was also concern regarding the handling of missing values in the statistical analysis. The major side effect was photosensitivity, which presented almost equally in the two treatment arms. Following this study, the drug was licensed in Japan, in 2008.
The two CAPACITY trials (studies 004 and 006) were conducted in North America, Australia, and Europe.1 Patients with mild-to-moderate IPF were recruited, and the inclusion criteria included FVC ≥50% predicted, DLCO ≥35% predicted, and 6MWT distance ≥150 m. In study 004, 435 patients were assigned to receive either high-dose pirfenidone (2,403 mg/day) or low-dose pirfenidone (1,197 mg/day) or placebo in a 2:1:2 dosing ratio; in study 006, 344 patients were assigned to receive high-dose pirfenidone (2,403 mg/day) or placebo. In both studies, the primary end point was the change of FVC% predicted from baseline to week 72. Only the study 004 met this primary end point. Mean FVC change at week 72 was –8.0% in the high-dose pirfenidone arm compared to –12.4% in the placebo arm (P=0.001). Moreover, high-dose pirfenidone reduced the proportion of patients with FVC decline ≥10% compared to placebo (20% vs 35%, respectively; P<0.001). In study 006, mean changes in% predicted FVC were almost identical between the two groups (–9.0% and –9.6% in the treatment and placebo arms, respectively). However, it should be noted that the rate of FVC decline was identical in the treatment arms of both trials, whereas the placebo arm of study 006 presented an attenuated FVC decline. According to the baseline characteristics in studies 004 and 006, the patients enrolled in study 006 had, on average, a recent diagnosis of early and maybe milder IPF, and the placebo group had a greater proportion of patients with obstructive airway disease, a characteristic associated with reduced FVC decline. These baseline imbalances could partly explain the attenuated rate of FVC decline in the placebo group in study 006. In the study 006, a significant decline from baseline to week 72 in 6MWT distance was observed in patients assigned to pirfenidone (absolute difference 31.8 m, 95% confidence interval [CI] 3.2–60.4). In the pooled analysis of the data, pirfenidone reduced the decline of FVC% predicted compared to placebo (mean change: –8.5% in the treatment arm vs –11% in the placebo arm; P<0.005). Moreover, a smaller proportion of patients had a decline in FVC ≥10% in the pooled pirfenidone group. A 31% reduction in the mean decline in 6MWT distance (P<0.001) and a 26% reduction in the risk of death or disease progression (HR 0.74, 95% CI 0.57–0.96; P=0.025) were also observed. Pirfenidone was generally well tolerated, with the most common side effects being gastrointestinal discomfort and photosensitivity. A recent Cochrane review, including the four studies mentioned earlier, has shown that treatment with pirfenidone reduced the risk of disease progression by 30% (HR 0.70, 95% CI 0.56–0.88).57
Following these studies, pirfenidone has been licensed in Europe for IPF patients with mild-to-moderate disease. However in the US, doubts over the unsuccessful 006 study led to the decline of the application for licensing. The US Food and Drug Administration (FDA) requested a subsequent double-blind, randomized, placebo-controlled Phase III trial (ASCEND) to be carried out.2
In the ASCEND study, 555 IPF patients were randomized 1:1 to either pirfenidone 2,403 mg/day (n=278) or placebo (n=277). Compared to the CAPACITY trials, the inclusion criteria were altered to enroll patients at higher risk of disease progression. Patients with a ratio of the forced expiratory volume in 1 second/FVC <0.80 were excluded, and the minimum baseline DLCO (as an inclusion criterion) was reduced from 35% to 30% of the predicted value. As a result, ~22% of the patients had a DLCO <35% predicted. The primary end point was the change from baseline to week 52 in the percentage of predicted FVC. The treatment effect was achieved, and moreover, it was evident by week 13 and increased throughout the duration of the trial. The mean decline from baseline in FVC was 235 mL in the pirfenidone group and 428 mL in the placebo group (absolute difference 193 mL, relative difference 45.1%; P<0.001). Moreover, pirfenidone also significantly reduced the proportion of patients who had a decline of ≥10% in the percentage of the predicted FVC or who died, and increased the proportion of patients who had no decline in the percentage of the predicted FVC as compared with placebo (16.5% vs 31.8% [P<0.001] and 22.7% vs 9.7% [P<0.001], respectively). Pirfenidone reduced the decline of the distance walked during the 6MWT and improved progression-free survival (P=0.04 and P<0.001, respectively).
In a prespecified pooled analysis, in 1,247 patients (555 from the ASCEND study and 692 from the CAPACITY studies), pirfenidone reduced the risk of death at 1 year by 48% compared to placebo (HR 0.52, 95% CI 0.31−0.87; P=0.01). In addition, in the pooled population, the risk of treatment-emergent death due to IPF at 1 year was reduced by 68% in the pirfenidone group compared to the placebo group (HR 0.32, 95% CI 0.14−0.76; P=0.006). The positive results from the ASCEND study together with the results of the INPULSIS trials have led to the licensing of both drugs by the FDA on October 2014 for treatment of IPF regardless of the severity of the disease.
Adverse events
The recommended daily dose of pirfenidone for adult patients with IPF is three 267 mg capsules three times a day, for a total of 2,403 mg/day.58 There is an uptitration of the dose: in the first week, one capsule (267 mg) three times a day; in the second week, two capsules (534 mg) three times a day; and from the third week onward, full dose of three capsules (801 mg) three times a day. It is recommended that pirfenidone is taken typically during food in order to prevent drug-related side effects.
The most commonly reported adverse drug reactions in the CAPACITY and ASCEND trials were gastrointestinal (nausea, dyspepsia, vomiting, anorexia) and skin related (rash and photosensitivity). All the adverse events (AEs) were dose dependent and were mild to moderate in severity. When IPF deterioration has been excluded as a cause of treatment withdrawal, ~1% of the patients had discontinued treatment because of AEs with rash, nausea, and increasing liver enzymes having been the most frequent. The incidence of these events is higher in the first 6 months of treatment and declines significantly thereafter. Therefore, appropriate education and adherence of the patients to the protective measures are very important especially in the first six months because it will prevent the development of side effects, which can lead to discontinuation of the drug and to disappointment and discouragement of the patients (Table 2).
 | Table 2 Prevention of side effects related to pirfenidone |
In order to prevent gastrointestinal AEs, it is recommended that the drug should be taken during a meal.55 In addition, it is better if the tablets are split through the meal or between the courses in case of a larger meal. If despite these measures, an AE develops, then the dose should be reduced starting from the dose that corresponds to the time when the event is more evident (for instance, morning nausea). If this proves ineffective, then the drug should be discontinued for 10–15 days and reintroduced gradually. Use of prokinetics, such as domperidone and metoclopramide, and protein pump inhibitors can be helpful.
Skin-related AEs are usually associated with sun exposure.59 These appear as erythematous or as a phototoxic burn-like skin rash occurring on sun-exposed body areas, which differentiate them from the allergic reactions which occur also in areas that are not exposed to sun. The patients have to avoid sun exposure mainly from midday until late afternoon and for a few hours after taking the drug to allow decrease of plasma concentration. In animal models, it has been observed that phototoxicity is linked to plasma concentration. It should be noted that, although dense cloud, thick clothing, and glasses may offer some protection against ultraviolet (UV) exposure, UV-A rays can penetrate such barriers, and therefore, even on a cloudy day or when the patient drives, protective measures are always necessary. The use of wide-brimmed hats, sunglasses, long-sleeved shirts, trousers, gloves if possible, and sunscreens with high sun protection factor (ie, >50) with UV-A/UV-B protection is recommended.60 If despite the protective measures the rash develops, the dose could be initially reduced. In case that the rash persists after 7 days, the drug should be discontinued for 15 days and reintroduced gradually when the rash resolves. Whenever the rash has urticarial characteristics or develops in the parts of the body that are not exposed to sun, then it should be considered allergic. In this case, the drug should be discontinued permanently, and additional treatment with antihistamines and oral prednisone should be considered.
Blood tests in order to monitor liver enzymes should be undertaken before the introduction of treatment and repeated monthly during the first 6 months. Subsequently, these can be repeated every 3 months. Elevations above three times of the upper limit of normal are significant, and the dose should be adjusted accordingly.
Long-term safety and tolerability trials
As shown in clinical trials, pirfenidone is efficacious because it can slow the rate of the progression of the disease. Treatment benefits seen in pharmaceutical trials are not always applicable to the general IPF population, because patients recruited may not be truly representative of those seen in daily clinical practice due to strict exclusion criteria. It is unknown whether the beneficial effect observed after a year in the recent trials can be maintained and whether long-term use is associated with a higher incidence of AEs. In order to provide answers to these questions, long-term safety and tolerability trials have been carried out.
Patients who completed the CAPACITY trials were eligible to participate in an open-label, long-term, follow-up extension study, namely, RECAP.61,62 Plainly, the inclusion criteria were the same as those in the CAPACITY trials. Patients who took part in the ASCEND trial were also eligible to participate in the RECAP trial. Six hundred and three patients have been recruited and received open-label pirfenidone until August 2013 when an interim analysis was performed. The median time on treatment (in addition to the 72 weeks during which 65% of subjects from the active treatment arms of CAPACITY received treatment) was 163.3 weeks, and the mean daily dose of pirfenidone was 2,332 mg/day. The most frequent treatment AEs were similar to those reported in CAPACITY and rarely led to treatment discontinuation. Interestingly, survival was 69% at week 228 (4.4 years), and after 5 years of follow-up, ~50% of the patients initially randomized to pirfenidone and subsequently included into the RECAP trial were still receiving therapy.
PASSPORT is an ongoing, post-authorization safety registry with a follow-up period of 2 years, following the approval of pirfenidone in Europe in 2011.63 The aim of the registry is to provide information about the long-term safety profile of pirfenidone. Until September 2014, 1,009 patients have been recruited from over 100 active sites in ten European countries. Routine clinical care visits take place approximately every 3 months. In December 2013, an interim analysis was performed. Five hundred and thirty patients have received at least one dose of pirfenidone with a median exposure of 5.48 months. In accordance with the results from the Phase III trials already completed, the most common adverse drug reactions observed have been gastrointestinal symptoms (nausea, decreased appetite, weight loss) and skin-related symptoms (rash, photosensitivity reactions). Sixteen percent of patients have discontinued treatment because of an adverse drug reaction. However, it should be stressed that at the time of the interim analysis, the median average exposure was half of that in the completed Phase III trials. The authors have observed that modification of the dose, either reduction or temporary discontinuation, has been the most efficient approach in order to treat the various side effects and carry on with the long-term treatment. In fact, 69% of the patients have continued the treatment after having experienced an adverse reaction, and the dose has been modified and only 20% have discontinued the drug. The PASSPORT study is anticipated to be completed in September 2016, and final results will be published shortly thereafter.
Multicenter observational studies have evaluated the effect of pirfenidone in a general population of patients who received the drug through the European named patient program. In the first study, which has been carried out in 12 Italian ILD centers, pulmonary function parameters (%FVC, %DLCO) and the distance walked during the 6MWT have been evaluated in the year before and after treatment with pirfenidone was commenced.64 In the time period before the introduction of pirfenidone, patients may have been treated with combination of steroids, azathioprine, and NAC. In this study, there was an almost significant difference between the mean reduction of FVC in the year prior to and after pirfenidone introduction (6.3% vs 1.3%; P=0.06), whereas no differences have been observed with regard to DLCO and 6MWT. Moreover, the authors have shown that the rate of FVC decline did not differ significantly before and after pirfenidone has been introduced in patients with an FVC >75%, whereas there was a significant reduction of the rate of decline in patients with more severe disease and FVC <75%. The same result has been observed when the patients were stratified according to the GAP system which derives from gender (G), age (A) and two physiologic parameters (P).65 The authors concluded that the drug may also have a beneficial effect in more severe cases. Interestingly, in a subgroup of patients, the treatment has resulted in a >10% increase of FVC, something that has not been observed in the pharmaceutical trials.
Another “real-life” study has been conducted in Belgium and the Netherlands, and included patients with comorbidities such as COPD, pulmonary embolism, and pulmonary hypertension who are usually excluded in clinical trials.66 The authors have observed that the rate of FVC decline 6 months before and 6 months after the introduction of pirfenidone has been significantly different (4.8% vs 0.8%). More patients have experienced a decline <5% and less patients >10% after the introduction of pirfenidone. Loss of appetite and nausea were the most frequent gastrointestinal side effects, and nausea was the most common reason for the discontinuation of the drug. Effectiveness and safety were similar to those reported in the ASCEND study.
Two other studies have confirmed that the most frequent side effects are gastrointestinal and skin related, which occur mainly during the first 6 months of treatment and may lead to the discontinuation of the drug.67,68 Few important issues have been highlighted: firstly, the importance of a frequent review of the patients by a specialist nurse and secondly, the importance of a contact number given to the patients so that they can communicate whenever they have questions about the drug or when they experience side effects. In the case of side effects, adjustment of the dose according to the opinion of an expert in IPF may be the best way to overcome the difficulties mainly during the first 6 months of treatment.68 Communication is vital to ensure compliance and adherence of the patients to the treatment as they can benefit as much as possible from pirfenidone.
Open questions
It is now well recognized that there are delays in the referral of patients with IPF to expert centers and consequently in the diagnosis of the disease.69 Therefore, when the diagnosis has been confirmed, the disease may already be advanced with an FVC <50%. Moreover, some patients present with an acute exacerbation or can have an inexorably progressive disease. Are these patients eligible to be treated with pirfenidone? Plainly, the FDA has approved both pirfenidone and nintedanib regardless of disease severity, but the fact remains that patients with an FVC <50% have been excluded from the recent trials and it is unknown, at least for the time being, if the drugs are effective and safe in this group of patients.
Another open question is whether patients who do not fulfill the criteria for the diagnosis of definite IPF, should be treated with pirfenidone.4 According to the guidelines, a definite diagnosis of IPF could be reached by integrating clinical, radiological and histological data in the multidisciplinary meeting. However, the performance of surgical lung biopsy is usually not possible because of advanced age and stage of the disease and because of the presence of comorbidities. Therefore, the diagnosis is based on the clinical behavior and on radiological features. In that case only 50% of the cases can be diagnosed with definite IPF and receive pirfenidone. It is not certain whether the drug works in the rest of IPF patients with a possible UIP pattern in HRCT, as they were excluded from the clinical trials for pirfenidone. It should be noted though, that in INPULSIS trial a number of this type of patients was included. It was recently observed that patients aged over 65 with a possible UIP pattern in HRCT are more likely to have a UIP pattern on biopsy, but this was a single study and definitely needs to be replicated before being widely applied.70 Another group of patients who were excluded from the clinical trials are patients with an unclassifiable ILD after a MDT evaluation. In this case, empirical treatment should be based on the most probable diagnosis, disease behavior and response to pragmatic therapy.71
In most IPF studies, patients aged over 80 years have been excluded. These patients usually also have other comorbidities for which they may already receive treatment at the time of IPF diagnosis. The risk of interaction between these drugs and the antifibrotic agents is possible. Administration of the new antifibrotic agents should be based on the balance between risk and benefit. A subgroup analysis of patients older than 65 years in the CAPACITY studies did not show an increased incidence of adverse side effects or reduced efficacy, and a similar benefit of pirfenidone over placebo on FVC decline was observed for age subgroups 65, 65–74, and >74 years when pooled data from the ASCEND and CAPACITY trials were recently analyzed.2
Both pirfenidone and nintedanib have managed to reduce the rate of progression by 50%. Whereas it is highly unlikely that a trial of direct comparison between the two compounds will ever be carried out, studies in which the combination of the drugs will be evaluated have already been designed. The results of a randomized, double-blind, Phase II, dose escalation trial that assessed the safety, tolerability, and pharmacokinetics of nintedanib, alone and when added to ongoing pirfenidone therapy, in Japanese patients with IPF have been recently published.72 Patients receiving pirfenidone at the beginning of the trial were stratified to every nintedanib dose group and placebo. AEs were reported in nine out of 17 patients receiving nintedanib alone and ten out of 21 patients receiving nintedanib added to pirfenidone. All AEs were mild or moderate in intensity with gastrointestinal disorders such as nausea and vomiting having been the most common ones. Nintedanib had no effect on the pharmacokinetics of pirfenidone, but a trend toward lower exposure of nintedanib when it was added to pirfenidone has been observed. Plainly, the short duration of the study is an important limitation, and in the future, longer studies are needed to confirm these results, and moreover, to provide evidence regarding the efficacy of the combination.
As already mentioned, patients with comorbidities have been excluded from the trials. Pulmonary hypertension, infections, and gastroesophageal reflux are very common and have been associated with disease progression. A combination of an antifibrotic drug with antireflux agents, antibiotics, and drugs that target the vascular component has been recently suggested.73 However, prospective studies are definitely needed to evaluate this suggestion. In the recently revised guidelines, it has been suggested that clinicians use regular antiacid treatment for patients with IPF. On the other hand, there is no recommendation regarding the treatment of pulmonary hypertension in IPF, and specifically, there is a conditional recommendation against the use of sildenafil.
Conclusion
The year 2014 was an amazing year for IPF. There are two drugs, pirfenidone and nintedanib, which are currently recommended for the treatment of the disease, and since our knowledge on the pathogenesis of IPF is in continuous progress, we may expect the development of other efficacious agents in the future. Combination of drugs that target different pathogenetic pathways, a treatment approach widely used in other respiratory diseases, such as asthma, COPD, tuberculosis, and lung cancer, seems to be the basis for the design of future clinical trials. Until then and after years of waiting, we are more than happy to be able to offer our patients two weapons to combat this dismal disease. Both drugs are efficacious and safe with a close collaboration between doctors, nurses, and patients being important in order to overcome side effects that can develop, mainly at the beginning of treatment. This approach will allow patients to gain as much benefits as possible from the antifibrotic therapy.
Acknowledgment
This review was supported by the National Institute of Health Research Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London.
Disclosure
The authors report no conflicts of interest in this work.
References
Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377:1760–1769. | |
King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. | |
Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. | |
Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. | |
Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192:e3–e19. | |
Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2:566–572. | |
Margaritopoulos GA, Giannarakis I, Siafakas NM, et al. An update on idiopathic pulmonary fibrosis. Panminerva Med. 2013;55:109–120. | |
Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. | |
Maher TM, Wells AU, Laurent GJ. Idiopathic pulmonary fibrosis: multiple causes and multiple mechanisms? Eur Respir J. 2007;30:835–839. | |
Armanios M. Syndromes of telomere shortening. Ann Rev Genomics Hum Genet. 2009;10:45–61. | |
Peljto AL, Zhang Y, Fingerlin TE, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309:2232–2239. | |
Noth I, Zhang Y, Ma SF, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet. 2013;1:309–317. | |
Margaritopoulos GA, Harari S, Caminati A, Antoniou KM. Smoking-related idiopathic interstitial pneumonia: a review. Respirology. 2016;21:57–64. | |
Lasithiotaki I, Antoniou KM, Vlahava VM, et al. Detection of herpes simplex virus type-1 in patients with fibrotic lung diseases. PLoS One. 2011;6:e27800. | |
Molyneaux PL, Cox MJ, Willis-Owen SA, et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190:906–913. | |
Tcherakian C, Cottin V, Brillet PY, et al. Progression of idiopathic pulmonary fibrosis: lessons from asymmetrical disease. Thorax. 2011;66:226–231. | |
Lee JS, Collard HR, Anstrom KJ, et al. Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: an analysis of data from three randomised controlled trials. Lancet Respir Med. 2013;1(5):369–376. | |
Andersson-Sjöland A, de Alba CG, Nihlberg K, et al. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. 2008;40:2129–2140. | |
Antoniou KM, Papadaki HA, Soufla G, et al. Investigation of bone marrow mesenchymal stem cells (BM MSCs) involvement in Idiopathic Pulmonary Fibrosis (IPF). Respir Med. 2010;104:1535–1542. | |
Pandit KV, Milosevic J, Kaminski N. MicroRNAs in idiopathic pulmonary fibrosis. Transl Res. 2011;157:191–199. | |
Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. 2010;35:496–504. | |
American Thoracic Society; European Respiratory Society. Idiopathic pulmonary fibrosis: diagnosis and treatment: international consensus statement. Am J Respir Crit Care Med. 2000;161:646–664. | |
Demedts M, Behr J, Buhl R, et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005;353:2229–2242. | |
Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, et al. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968–1977. | |
Idiopathic Pulmonary Fibrosis Clinical Research Network; Martinez FJ, de Andrade JA, Anstrom KJ, King TE Jr, Raghu G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2093–3101. | |
Raghu G, Brown KK, Bradford WZ, et al. A placebo-controlled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2004;350:125–133. | |
King TE Jr, Albera C, Bradford WZ, et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet. 2009;374:222–228. | |
Raghu G, Brown KK, Costabel U, et al. Treatment of idiopathic pulmonary fibrosis with etanercept: an exploratory, placebo-controlled trial. Am J Respir Crit Care Med. 2008;178:948–955. | |
Noth I, Anstrom KJ, Calvert SB, et al. The Idiopathic Pulmonary Fibrosis Clinical Research Network (IPFnet). A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:88–95. | |
King TE, Behr J, Brown KK, et al. BUILD-1: a randomized placebocontrolled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177:75–81. | |
King TE, Brown KK, Raghu G, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184:92–99. | |
Raghu G, Million-Rousseau R, Morganti A, et al. Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomized controlled MUSIC trial. Eur Respir J. 2013;42:1622–1632. | |
Raghu G, Behr J, Brown KK, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158:641–649. | |
Zisman DA, Schwarz M, Anstrom KJ, et al. Idiopathic Pulmonary Fibrosis Clinical Research Network. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010;363:620–628. | |
Daniels CE, Lasky JA, Limper AH, et al. Imatinib treatment for idiopathic pulmonary fibrosis: randomized placebo-controlled trial results. Am J Respir Crit Care Med. 2010;181:604–610. | |
Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–1087. | |
Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35:821–829. | |
Ziesche R, Hofbauer E, Wittmann K, Petkov V, Block LH. A preliminary study of long-term treatment with interferon gamma-1b and low-dose prednisolone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 1999;341:1264–1269. | |
Kubo H, Nakayama K, Yanai M, et al. Anticoagulant therapy for idiopathic pulmonary fibrosis. Chest. 2005;128:1475–1482. | |
Tzouvelekis A, Margaritopoulos G, Loukides S, Bouros D. Warfarin in idiopathic pulmonary fibrosis: friend or foe, is it a matter of genes and heparin? Am J Respir Crit Care Med. 2013;187:213–214. | |
Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68:4774–4782. | |
Nakayama S, Mukae H, Sakamoto N, et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 2008;82:210–217. | |
Di Sario A, Bendia E, Svegliati Baroni G, et al. Effect of pirfenidone on rat hepatic stellate cell proliferation and collagen production. J Hepatol. 2002;37:584–591. | |
Conte E, Gili E, Fagone E, Fruciano M, Iemmolo M, Vancheri C. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur J Pharm Sci. 2014;58:13–19. | |
Nakazato H, Oku H, Yamane S, Tsuruta Y, Suzuki R. A novel anti-fibrotic agent pirfenidone suppresses tumor necrosis factor-alpha at the translational level. Eur J Pharmacol. 2002;446:177–185. | |
Oku H, Nakazato H, Horikawa T, Tsuruta Y, Suzuki R. Pirfenidone suppresses tumor necrosis factor-alpha, enhances interleukin-10 and protects mice from endotoxic shock. Eur J Pharmacol. 2002;446:167–176. | |
Cain WC, Stuart RW, Lefkowitz DL, Starnes JD, Margolin S, Lefkowitz SS. Inhibition of tumor necrosis factor and subsequent endotoxin shock by pirfenidone. Int J Immunopharmacol. 1998;20:685–695. | |
Hale ML, Margolin SB, Krakauer T, Roy CJ, Stiles BG. Pirfenidone blocks the in vitro and in vivo effects of staphylococcal enterotoxin B. Infect Immun. 2002;70:2989–2994. | |
Giri SN, Leonard S, Shi X, Margolin SB, Vallyathan V. Effects of pirfenidone on the generation of reactive oxygen species in vitro. J Environ Pathol Toxicol Oncol. 1999;18:169–177. | |
Misra HP, Rabideau C. Pirfenidone inhibits NADPHdependent microsomal lipid peroxidation and scavenges hydroxyl radicals. Mol Cell Biochem. 2000;204:119–126. | |
Iyer SN, Wild JS, Schiedt MJ, Hyde DM, Margolin SB, Giri SN. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med. 1995;125:779–785. | |
Iyer SN, Margolin SB, Hyde DM, Giri SN. Lung fibrosis is ameliorated by pirfenidone fed in diet after the second dose in a three-dose bleomycin-hamster model. Exp Lung Res. 1998;24:119–132. | |
Kakugawa T, Mukae H, Hayashi T, et al. Pirfenidone attenuates expression of HSP47 in murine bleomycin-induced pulmonary fibrosis. Eur Respir J. 2004;24:57–65. | |
Raghu G, Johnson WC, Lockhart D, Mageto Y. Treatment of idiopathic pulmonary fibrosis with a new antifibrotic agent, pirfenidone: results of a prospective, open-label phase II study. Am J Respir Crit Care Med. 1999;159(4 Pt 1):1061–1069. | |
Nagai S, Hamada K, Shigematsu M, Taniyama M, Yamauchi S, Izumi T. Open-label compassionate use one year-treatment with pirfenidone to patients with chronic pulmonary fibrosis. Intern Med. 2002;41:1118–1123. | |
Azuma A, Nukiwa T, Tsuboi E, et al. Double-blind, placebo controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171:1040–1047. | |
Spagnolo P, Del Giovane C, Luppi F, et al. Non-steroid agents for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev. 2010;9:CD003134. | |
European Medicines Agency. EPAR – product information. London: European Medicines Agency; 2011. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/includes/document/document_detail.jsp?webContentId=WC500103049&mid=WC0b01ac058009a3dc. Accessed May 17, 2016. | |
Margaritopoulos G. Challenges in IPF diagnosis, current management and future perspectives: Patient case 2. Sarcoidosis Vasc Diffuse Lung Dis. 2015;32 (Suppl 1):38–39. | |
Costabel U, Bendstrup E, Cottin V, et al. Pirfenidone in idiopathic pulmonary fibrosis: expert panel discussion on the management of drug-related adverse events. Adv Ther. 2014;31:375–391. | |
Costabel U, Albera C, Cohen A, et al. The long-term safety of pirfenidone in patients with idiopathic pulmonary fibrosis (IPF): interim data from the RECAP extension study. Eur Respir J. 2011; 38 (Suppl 55):174s. | |
Costabel U, Albera C, Bradford WZ, et al. Analysis of lung function and survival in RECAP: an open-label extension study of pirfenidone in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2014;31:198–205. | |
Koschel D, Cottin V, Skold M, et al. Pirfenidone post-authorisation safety registry (PASSPORT) – interim analysis of IPF treatment. Eur Respir J. 2014;44 (Suppl 58):1904. | |
Harari S, Caminati A, Albera C, et al. Efficacy of pirfenidone for idiopathic pulmonary fibrosis: an Italian real life study. Respir Med. 2015;109:904–913. | |
Ley B, Ryerson CJ, Wittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156:684–691. | |
Wijsenbeek MS, Grutters JC, Wuyts WA. Early experience of pirfenidone in daily clinical practice in Belgium and the Netherlands: a retrospective cohort analysis. Adv Ther. 2015;32:691–704. | |
Valeyre D, Albera C, Bradford WZ, et al. Comprehensive assessment of the long-term safety of pirfenidone in patients with idiopathic pulmonary fibrosis. Respirology. 2014;19:740–748. | |
Chaudhuri N, Duck A, Frank R, et al. Real world experiences: pirfenidone is well tolerated in patients with idiopathic pulmonary fibrosis. Respir Med. 2014;108:224–226. | |
Antoniou KM, Symvoulakis EK, Margaritopoulos GA, et al. Early diagnosis of IPF: time for a primary-care case-finding initiative? Lancet Respir Med. 2014;2:e1. | |
Fell CD, Martinez FJ, Liu LX, et al. Clinical predictors of a diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181:832–837. | |
Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–748. | |
Ogura T, Taniguchi H, Azuma A, et al. Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1382–1392. | |
Wuyts WA, Antoniou KM, Borensztajn K, et al. Combination therapy: the future of management for idiopathic pulmonary fibrosis? Lancet Respir Med. 2014;2:933–942. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.