")
Back to Journals » Journal of Experimental Pharmacology » Volume 12
Piperine Alters the Pharmacokinetics and Anticoagulation of Warfarin in Rats
Authors Zayed A , Babaresh WM, Darweesh RS, El-Elimat T , Hawamdeh SS
Received 12 April 2020
Accepted for publication 6 June 2020
Published 19 June 2020 Volume 2020:12 Pages 169—179
DOI https://doi.org/10.2147/JEP.S257919
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bal Lokeshwar
Aref Zayed,1 Wahby M Babaresh,1 Ruba S Darweesh,2 Tamam El-Elimat,1 Sahar S Hawamdeh1
1Department of Medicinal Chemistry and Pharmacognosy, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid 22110, Jordan; 2Department of Pharmaceutical Technology, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid 22110, Jordan
Correspondence: Aref Zayed Tel +962 2 7201000, Ext. 23240
Fax +962 2 7201075
Email [email protected]
Introduction: Piperine, the bioactive compound of black pepper, and warfarin are metabolized by cytochrome P450 enzymes and are both highly plasma protein-bound compounds. In this study, we evaluated the effect of co-administered piperine on the pharmacokinetics and anticoagulation of warfarin in rats.
Methods: We studied four Sprague-Dawley rat groups: a negative control group receiving only oral warfarin, a test group receiving warfarin plus piperine, a positive control group receiving warfarin plus sulfaphenazole (CYP2C inhibitor), and another positive control group receiving warfarin plus ketoconazole (CYP3A inhibitor). We also analyzed plasma concentrations of warfarin and its major metabolite, 7-hydoxywarfarin. Blood clotting time, calculated as international normalized ratio (INR), was also measured.
Results: Our results showed that although co-administration of piperine produced a non-significant decrease in warfarin concentrations, it resulted in significantly lower 7-hydroxywarfarin metabolite concentrations. Piperine significantly decreased, by sixfold, AUC0–∞, by eightfold, Cmax, but significantly increased, by fivefold, CL/F and, by sixfold, Vd/F of 7-hydroxywarfarin. The INR values were consistent with the decrease in warfarin concentration in the presence of piperine and showed a significant decrease at 24 h after warfarin dose.
Conclusion: We conclude that piperine could be a potent inhibitor of cytochrome P450 metabolism of warfarin in vivo and, contrary to the expectation, may reduce the plasma concentration and anticoagulation of warfarin. This interaction could have a clinical significance and should be investigated in patients.
Keywords: warfarin, 7-hydroxywarfarin, piperine, pharmacokinetics, herb–drug interaction, black pepper
Plain Language Summary
Warfarin is among the most prescribed anticoagulants around the world and has a famous drug-drug and drug–food interaction profile. Many patients nowadays self-medicate with natural remedies that may interact with their chronic medications. Piperine, the active constituent of pepper, has been historically used for a wide array of ailments. Black pepper is also commonly used in many cuisines. Due to the shared metabolism of warfarin and piperine by Cytochrome P450 (CYP) enzymes, and since both are highly bound to plasma proteins, we hypothesized that an interaction may be present between them on the pharmacokinetic and pharmacodynamic levels. Based on a rat study, we find the following:
- Co-administered piperine causes a significant decrease in the concentrations of the main warfarin metabolite, 7-hydroxywarfarin.
- Co-administration of piperine causes a decrease, although statistically non-significant, in the total concentration of warfarin.
- INR values show a significant decrease at 24 hours after warfarin dose when co-administered with piperine.
Introduction
Despite its various drug-drug and drug–food interactions, warfarin continues to be the most commonly prescribed anti-coagulant around the world.1 Warfarin has been the subject of extensive study due to its unique interaction profile resulting from its metabolism by many enzymes of the Cytochrome P450 (CYP system), such as CYP2C9 and CYP3A4.2 Warfarin is a racemic mixture of two enantiomers and its metabolism was found to be stereoselective. The more potent enantiomer, S-warfarin, is mainly metabolized by CYP2C9 to 7-hydroxywarfarin, while R-warfarin is metabolized by CYP1A1, 1A2 and 3A4 to yield 6-, 8-, and 10-hydroxywarfarin.3–6 Overall, 7-hydroxywarfarin is the most predominant metabolite of warfarin in humans.7–9 It is not surprising, therefore, that the majority of the drugs causing interactions with warfarin at the level of hepatic metabolism inhibit the CYP2C9 pathway (eg, amiodarone, co-trimoxazole, metronidazole, and fluvoxamine), consequently enhancing the effect of warfarin and causing the need to lower its dose for most patients.10 Drugs that inhibit CYP1A1, 1A2, and 3A4 generally have less effect on anticoagulation control. About 99% of warfarin in blood is bound to plasma proteins such as albumin which results in alteration in its bioavailability when displaced by other protein-bound drugs. Determining the suitable dosage regimen for warfarin is therefore challenging considering its narrow therapeutic index and potential drug-drug/diet interactions mediated by CYPs and plasma protein binding.
Warfarin doses must be determined individually to achieve a therapeutic window of 2–3 INR. Once the dose is determined, patients are often advised to avoid drastic changes in diet where foods interacting with warfarin are concerned. Moreover, all concurrently used medications, including over-the-counter (OTC) drugs, supplements, and herbal medicines must be disclosed.11,12 The latter is of particular interest, as the use of herbal medicine has been gaining an increasing popularity over the years as a form of alternative medicine.13,14 It is estimated that at least 1 in 4 patients use natural products in combination with prescribed drugs,15 and more than two-thirds of patients do not reveal their use of these products to healthcare providers.16,17 Often described as “natural”, such products may be perceived by patients as safe and void from interactions.18
Among the commonly used alternative medicines is piperine, the alkaloid active ingredient of black pepper (Piper nigrum). Piperine has been shown to possess a wide array of therapeutic effects that stem from its anti-inflammatory, antioxidant, antiulcer, anticancer, anti-dyslipidemic, and antidepressant properties.19,20 This favorable activity combined with the safety of piperine allows it to be a strong candidate for novel therapeutics or supplementary (eg, black pepper extract) medicines.21–24 The use of piperine in medicinal supplements in addition to the widespread use of black pepper in different cuisines call for conducting studies on possible piperine interactions with commonly used drugs.
Previous studies reported that piperine is a potent inhibitor of CYP450 enzymes, particularly CYP3A4, CYP2C9, and CYP1A2, thus increasing the bioavailability of several drugs.25–28 The bioavailability of different antibiotics, eg, amoxicillin has been reported to be enhanced by piperine in animal models.29 Studies have also shown area under the curve (AUC) and maximum concentration (Cmax) of resveratrol, a natural polyphenolic antioxidant, to significantly increase in mice when administered with piperine.30 Similarly, clinical studies showed that a single daily dose of piperine (20 mg/day) for 7 days increased the oral bioavailability of propranolol and theophylline in healthy volunteers.31
Other drugs, however, have been associated with reduced bioavailability such as isoniazid and magnolol.32,33 Piperine can exert both inhibitory and stimulatory effects on drug metabolism, depending on the dosage regimen.34 Drug uptake via intestinal transporters has also been suggested to be affected by piperine.35 Piperine is a modulator of the efflux transporter P-glycoprotein (P-gp), suggesting its implication in interactions with P-gp substrate drugs.36–39
As described, warfarin is among the drugs commonly metabolized by CYP450 (mainly through hydroxylation) which could be affected by piperine. In addition to inhibiting CYP450, piperine has been reported to displace warfarin from plasma proteins in vitro.40 We therefore hypothesize that warfarin coadministration with piperine would affect warfarin metabolism and alter its PK. In this study, we aimed to assess the effect of piperine on the pharmacokinetics (PK) and anticoagulation of warfarin using rat as an animal model. Since piperine was reported to affect CYP3A4 and 2C9 hepatic metabolism, ketoconazole (CYP3A inhibitor) and sulfaphenazole (CYP2C inhibitor) were employed as positive controls to compare the effect piperine to their inhibitory effect.
Materials and Methods
Chemicals and Equipment
Racemic warfarin, 7-hydroxywarfarin, ketoconazole, sulfaphenazole, and piperine were purchased from Sigma Aldrich, St. Louis MO, USA. Naproxen was supplied from Hikma pharmaceuticals, Amman, Jordan. All organic solvents (acetonitrile, ethylacetate and methanol) were purchased from Fisher Scientific, Pittsburgh, PA, USA. Acetic acid was obtained from Scharlau, Spain. Phosphate buffer and potassium monobasic and dibasic were obtained from Sigma-Aldrich (St. Louis MO, USA) provided.
Pharmacokinetic and Anticoagulation Studies
We carried out the PK studies using four Sprague-Dawley (SD) rat groups (n=6 rats per group). All experimental procedures were carried out under the approval of the Animal Care and Use Committee (ACUC) at Jordan University of Science and Technology (Irbid, Jordan) and in compliance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the United States National Institutes of Health. Rats were maintained in a clean room at a temperature of 25°C, with a 12-hour (h) light-dark cycle and 50% relative humidity. Similar studies have used the rat model to assess drug–drug interactions.11–14 The groups used were: a negative control group receiving only oral warfarin (2 mg/kg), a test group receiving warfarin at 2 mg/kg and piperine at 20 mg/kg orally, a positive control group receiving warfarin at 2 mg/kg plus sulfaphenazole (SPZ) (human CYP2C9 inhibitor) at 120 mg/kg, and another positive control group receiving warfarin at 2 mg/kg plus ketoconazole (KCZ) (human CYP3A4 inhibitor) at 30 mg/kg orally. We administered warfarin orally as a single dose of an aqueous solution of warfarin (containing 4 mg/mL in Dimethylsulfoxide (DMSO)) to each animal at a dose of 2 mg/kg after 30 min of administration of piperine, KCZ, SPZ, or an equal volume of DMSO (no warfarin). The dose of piperine, sulfaphenazole (SPZ), a known 2C9 inhibitor, and ketoconazole (KCZ), a known 3A4 inhibitor, have been selected based on previous pharmacokinetic drug interaction studies.39,41,42 Blood samples were collected from tail vein at 0, 0.5, 1, 2, 3, 4, 6, 8, 24, 30, 48, 54, 72, 96, and 120 h after warfarin treatment. Blood samples were immediately separated by centrifugation at 13,000 rpm for 5 min, and plasma was transferred to sample tubes and kept frozen at −80°C until subsequent analysis. Then, we measured plasma concentrations of warfarin and 7-hydroxywarfarin to estimate pharmacokinetic parameters. Blood clotting time was calculated as international normalized ratio (INR). INR values were measured using the collected fresh blood samples before warfarin administration and immediately at 6, 24, and 72 h after warfarin dose.
HPLC Analysis of Warfarin
We carried out quantification of warfarin and 7-hydroxywarfarin by a high-performance liquid chromatography with fluorescence detection (HPLC-FLD) method that was previously validated.43 In brief, plasma samples were spiked with internal standard solution and then acidified by acetic acid before extraction with ethylacetate. The upper organic phase was isolated and evaporated and the residue was then reconstituted with mobile phase and injected into the HPLC system. Chromatographic separation was achieved using Fortis® reversed-phase diphenyl column 150 × 4.6 mm, particle size 3μm. The mobile phase consisted of a mixture of phosphate buffer (pH 7), methanol, and acetonitrile (70:20:10, v/v/v), and was delivered at a flow rate of 0.8 mL/min. Naproxen was used as an internal standard. The retention time of warfarin, 7-hydroxywarfarin, and naproxen were found to be approximately 14, 9.5, and 12 min, respectively. To conduct the analysis, Shimadzu HPLC system (Tokyo, Japan), equipped with a fluorescence detector was used and adjusted at excitation and emission of 310 nm and 390 nm, respectively. Data acquisition and integration were performed using the Shimadzu CLASS-VP v. 6.12 software.
Data Analysis
Non-compartmental pharmacokinetic analysis was performed using the WinNonlin 5.3. software. The pharmacokinetic parameters estimated included the peak plasma concentration (Cmax), the time to reach peak plasma concentration (Tmax), the terminal-phase half-life (t1/2), the total plasma clearance (CL), the apparent volume of distribution (Vd), the total area under the plasma drug concentration-time curve (AUC), and the mean residence time (MRT). We determined Cmax and Tmax by a visual inspection of data. AUC0–∞ was determined by the trapezoidal rule using data of plasma drug concentration versus time comprising data points from time zero to the last experimental time plus the excess area (from the last experimental time to infinity). We also calculated t1/2 as t1/2 = 0.693/k, where (k) is the terminal-phase rate constant which we calculated from the slope of the terminal phase of plasma concentration versus time profile using linear regression analysis. CL/F, the ratio of the plasma clearance (CL) to the fraction of the dose absorbed (F), was determined as Dose/AUC0–∞. Vd/F was determined as Dose/(AUC0–∞ x k). MRT was determined as AUMC0–∞/AUC0–∞, where AUMC0–∞ is the total area under the first moment of the plasma concentration-time curve, from time zero to time infinity.
Statistical Analysis
Data were expressed in the form of “mean ± Standard error of mean (SEM)”. We analyzed differences between groups for continuous variables by one-way ANOVA with post hoc Dunnett t-student using SPSS17.0 software. A p-value of less than 0.05 was considered statistically significant.
Ethical Approval
We conducted the study and carried out all experimental procedures under the approval of the Institutional Animal Care and Use Committee (IACUC) of the Jordan University of Science and Technology (Approval No. 16/3/3/749) and in compliance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the United States National Institutes of Health.
Results
Effect of Piperine on the Pharmacokinetics of Warfarin
To assess the effect of piperine on the pharmacokinetics of warfarin, we administered a single oral dose of piperine to rats followed by a single oral dose of warfarin. The plasma concentrations of warfarin and 7-hydoxywarfarin were then analyzed and the pharmacokinetics parameters were estimated. The mean plasma concentration-time profiles of warfarin and 7-hydroxywarfarin for the four studied groups, ie, warfarin alone, warfarin plus piperine, warfarin plus SPZ, and warfarin plus KCZ are shown in Figures 1 and 2, respectively. The pharmacokinetic parameters of warfarin and 7-hydroxywarfarin for the four rat groups are shown in Tables 1 and 2, respectively.
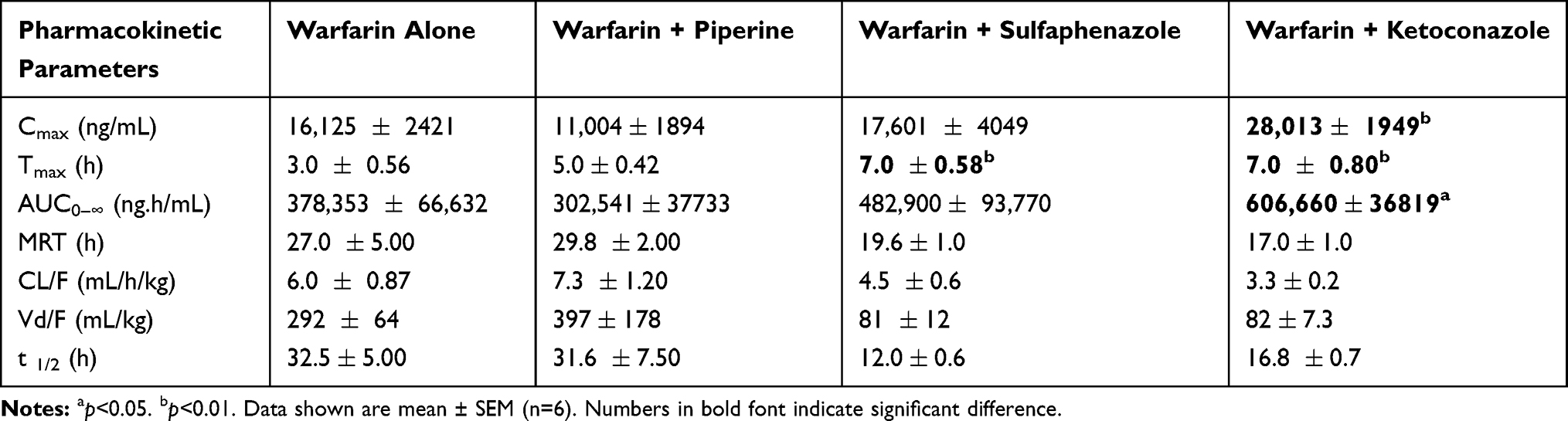 |
Table 1 Pharmacokinetics of Warfarin (2 mg/kg, p.o.) After Co-Administration of Piperine (20 mg/kg), Sulfaphenazole (120 mg/kg) and Ketoconazole (30 mg/kg) |
 |
Table 2 Pharmacokinetics of 7-Hydroxywarfarin After Co-Administration of Piperine (20 mg/kg), Sulfaphenazole (120 mg/kg) and Ketoconazole (30 mg/kg) |
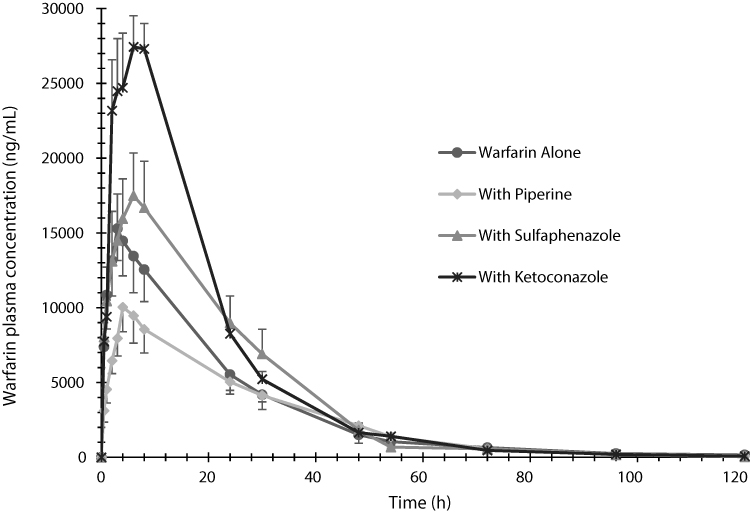 |
Figure 1 The plasma mean concentration versus time profiles of warfarin determined after administration of a single oral dose of warfarin alone (2 mg/kg), warfarin (2 mg/kg) with piperine (20 mg/kg, p.o.), warfarin (2 mg/kg) with sulfaphenazole (120 mg/kg, p.o.), and warfarin (2 mg/kg) with ketoconazole (30 mg/kg, p.o.) in Sprague-Dawley rats. Data are shown in mean ± SEM, n=6. |
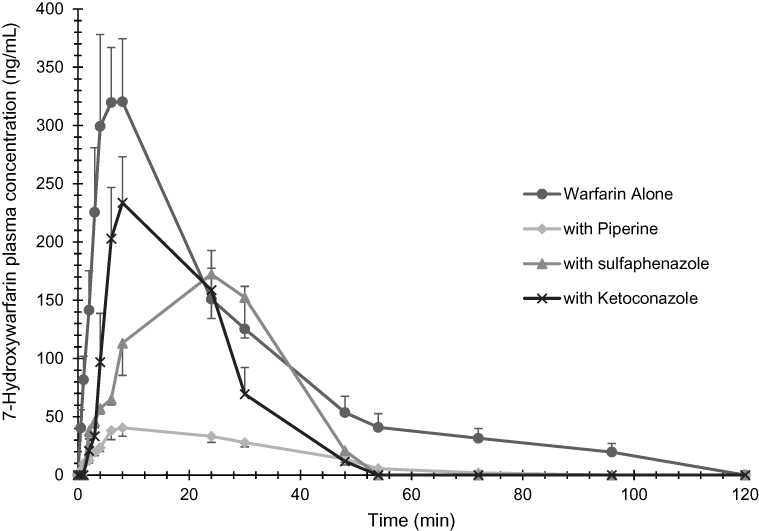 |
Figure 2 The plasma mean concentration versus time profiles of 7-hydroxywarfarin determined after administration of a single oral dose of warfarin alone (2 mg/kg), warfarin (2 mg/kg) with piperine (20 mg/kg, p.o.), warfarin (2 mg/kg) with sulfaphenazole (120 mg/kg, p.o.), and warfarin (2 mg/kg) with ketoconazole (30 mg/kg, p.o.) in Sprague-Dawley rats. Data are shown in mean ± SEM, n=6. |
The mean plasma concentration-time profiles of warfarin in the presence or absence of piperine can be seen in Figure 1. The mean peak concentration of warfarin in the control group was reached around 3 h, whereas it increased to 5 h with the coadministration of piperine. Total plasma concentrations of warfarin were decreased by piperine. In comparison, warfarin concentration increased with the coadministration of the CYP450 inhibitors KCZ and SPZ in the positive control groups.
Table 1 summarizes the pharmacokinetic parameters of warfarin in the presence and absence of piperine. According to our data, the co-administration of piperine (20 mg/kg) resulted in alteration in warfarin’s PK parameters, although differences were not statistically significant. Cmax and AUC0–∞ were decreased by ~30% and 20%, respectively, whereas CL/F and Vd/F were increased by ~20% and ~35%, respectively (Table 1). Values of t1/2 were almost the same (~32 h) with and without co-administration of piperine. In comparison, AUC0–∞ of warfarin was increased in the presence of SPZ (by ~28%) and KCZ (by ~60%), although it was only statistically significant for the latter. CL/F, Vd/F, and t1/2 values were lower for both SPZ and KCZ groups, but the differences were not statistically significant (Table 1).
The mean plasma concentration-time profiles of 7-hydroxywarfarin in the presence and absence of piperine can be seen in Figure 2. We noticed that piperine resulted in a substantial decrease in the plasma concentrations of 7-hydroxywarfarin. Both SPZ and KCZ also resulted in a decrease in 7-hydroxywarfarin concentrations, but not to the same extent as that caused by piperine. 7-hydroxywarfarin metabolite peak plasma concentration was delayed in the SPZ group compared to the other groups.
7-hydroxywarfarin metabolite data (Table 2) show that piperine significantly affected the pharmacokinetic parameters of warfarin. Piperine significantly decreased Cmax (~8-fold decrease) and AUC0–∞ (~6-fold decrease) and significantly increased CL/F (~5-fold increase) and Vd/F (~6-fold increase) of the metabolite (Table 2). t1/2 of 7-hydroxywarfarin was not changed whereas Tmax was doubled in the presence of piperine although it was not significantly different. Both SPZ and KCZ significantly decreased AUC0–∞ (~2-fold decrease). Although Cmax values were decreased in both groups, only SPZ resulted in statistical significance (Table 2). Both CL/F and t1/2 decreased whereas Vd/F increased in the presence of SPZ and KCZ, but without any statistical significance.
To evaluate the effect of piperine on warfarin metabolism, we determined and compared the percent of 7-hydroxywarfarin metabolite’s AUC0–∞ values relative to AUC0–∞ values of parent drug (AUC7-hydroxywarfarin/AUCwarfarin) as seen in Figure 3. Piperine resulted in the lowest 7-hydroxywarfarin formation (21%) and was significantly different (p < 0.01) compared to the control group. Both SPZ and KCZ also decreased 7-hydroxywarfarin formation significantly (41% and 33%, respectively) (p < 0.05), but not to the same levels produced by piperine (Figure 3).
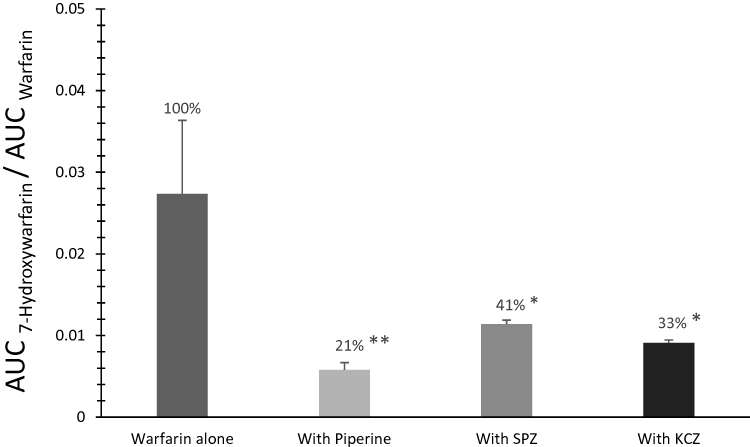 |
Figure 3 Relative formation of 7-hydroxywarfarin to warfarin, calculated as AUC7-hydroxywarfarin/AUCwarfarin, after administration of a single oral dose of warfarin alone (2 mg/kg), warfarin (2 mg/kg) with piperine (20 mg/kg, p.o.), warfarin (2 mg/kg) with sulfaphenazole (120 mg/kg, p.o.), and warfarin (2 mg/kg) with ketoconazole (30 mg/kg, p.o.) in Sprague-Dawley rats. Data are shown in mean ± SEM. *p<0.05, **p<0.01. |
Effects of Piperine on the Anticoagulation of Warfarin
Figure 4 shows the INR values for warfarin alone, warfarin with piperine, warfarin with SPZ, and warfarin with KCZ treatment groups. The INR values that we measured throughout the experiment were in the range of 1.2–8.0. The values peaked at 24 h for the four groups. Mean INR values prior to warfarin administration (0 h) were around 1.0 for all groups. There were no significant differences between the four groups at 6 h, and the mean INR values were around 1.4. The piperine group, however, was significantly lower at 24 h (6.5) compared to the control, SPZ, and KCZ treatment groups which all scored the maximum INR value (8). Moreover, at 72 h, the piperine treatment group showed again the lowest INR value (1.4), although it was not significantly different compared to the control group (2.8). Despite the fact that SPZ and KCZ groups showed a decrease in their INR values at 72 h, they were significantly higher (6.5 and 6.0, respectively), compared to the control treatment group.
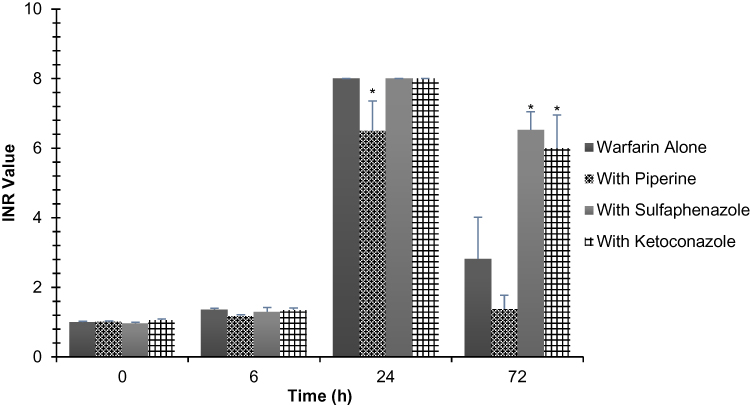 |
Figure 4 Comparison of INR values for warfarin between different rat groups following a single oral dose of warfarin alone (2 mg/kg), warfarin (2 mg/kg) with piperine (20 mg/kg, p.o.), warfarin (2 mg/kg) with sulfaphenazole (120 mg/kg, p.o.), and warfarin (2 mg/kg) with ketoconazole (30 mg/kg, p.o.). Data are shown in mean ± SEM, n=6. *p<0.05. |
Discussion
Warfarin is one of the most commonly used anticoagulants due to its efficacy and low cost. However, its infamous drug–drug and drug–food interaction profile, resulting from its significant protein binding and large interindividual variability, presents a challenge in dosing regimen and safety. In addition, piperine has been shown to interact with several drugs at different levels,27,33,39,44,45 but its effect on the pharmacokinetics and pharmacodynamics of warfarin has not been investigated yet.
There are several potential mechanisms where piperine and warfarin may interact, some of which may have opposite influences, and therefore investigating these effects in vivo is essential to determine the net effects of any pharmacokinetic and pharmacodynamic alterations. Piperine is known to inhibit the metabolism of many drugs in rats and humans by inhibition of CYP450 enzymes.26,31,46 Since warfarin is metabolized mainly by CYP450 2C and 3A, mainly through hydroxylation, it is expected that its co-administration with piperine would affect warfarin metabolism. Monitoring 7-hydroxywarfarin levels is particularly important, since it is the most predominant metabolite of racemic warfarin and it is the product of CYP 2C metabolic route of the pharmacologically active S-warfarin enantiomer. Altering plasma protein binding is another potential interaction mechanism since piperine was shown to displace warfarin from plasma proteins in vitro and increase its uptake into endothelial cells.40 Moreover, piperine is known to affect the bioavailability of several drugs through alteration of their absorption resulting in increasing plasma levels of some36,46,47 while decreasing these of others.32,33 The involvement of one or more of the previous interactions should be demonstrated by alteration of pharmacological action of warfarin or changes in levels of the drug and its metabolite. In this study, therefore, we aimed to examine the effect of piperine on the pharmacokinetics of warfarin and 7-hydroxywarfarin and anticoagulation of warfarin in vivo.
Our results show a decrease, although statistically non-significant, in the total concentration of warfarin, when co-administered with piperine. This can be observed by the ~30% and ~20% reduction in AUC0–∞ and Cmax values, respectively, of warfarin in piperine-treated group (Table 2). It should be noted that warfarin levels measured in this study are total concentration (Ctotal) of warfarin which represents the sum of the concentrations of free (Cfree) and plasma protein bound (Cbound) warfarin. Therefore, both or either concentration levels could have been altered as a result of piperine coadministration. The fact that we also observed an increase, although statistically non-significant, in the volume of distribution (~35%) and clearance (~20%) of warfarin among the piperine group (Table 2) may indicate that the free drug fraction (fu) which represents Cfree/Ctotal might have increased. Since warfarin is a highly protein bound drug (>99%), being displaced from albumin may cause such results. In fact, piperine has been shown to displace plasma bound warfarin from albumin and α-acid glycoprotein40 which may cause an increase in its clearance in vivo and thus decrease total plasma concentration of warfarin. It should be noted that an increase in free drug fraction (fu) does not necessarily result in an increase in, and should not be confused with, free drug concentration (Cfree). This is particularly important in the in vivo situation, since unlike the case in vitro, total drug concentration is not fixed and the increased drug-free fraction as a result of drug displacement from proteins may not change free drug concentration, but results in reduction in total concentration as it might be the case in our study. An excellent explanation of this pharmacokinetic relation was provided by Toutain et al.47
Perhaps the most notable observation in this study is the significant decrease in the concentrations of the main warfarin metabolite, 7-hydroxywarfarin, as a result of the coadministration of piperine. Both Cmax and AUC0–∞ of 7-hydroxywarfarin were significantly decreased, while CL/F and Vd/F were significantly increased. This can be explained by the inhibition of the CYP450 metabolic pathway by piperine, resulting in a decrease in the 2C-mediated formation of 7-hydroxywarfarin.48 We noticed that piperine has even resulted in lower 7-hydroxywarfarin Cmax compared to SPZ, the positive control used as 2C9 inhibitor. The extent of metabolite inhibition by piperine is clearly illustrated in Figure 3 which demonstrates the relative formation of 7-hydroxywarfarin to warfarin, calculated as AUC7-hydroxywarfarin/AUCwarfarin. Piperine has the lowest metabolite formation with a value of 21% whereas SPZ and KCZ produced 41% and 33%, respectively. SPZ and KCZ are commonly used in interaction studies, in vitro and in vivo, to compare the potential inhibitory effect of test compounds to their selective inhibition of CYP2C and 3A, respectively. In addition to the CYP450 metabolic inhibition, reports suggested that piperine inhibits a family of cell membrane transporters called the organic-anion-transporting polypeptides (OATPs) expressed in the liver and intestines.49 This potential inhibition could cause a reduction in warfarin uptake by hepatic cells when co-administered with piperine, consequently preventing its metabolism into 7-hydroxywarfarin.35 Conversely, inhibition of intestinal OATPs could lead to decreased warfarin absorption into the bloodstream, which may further explain the decrease in the AUC0–∞ of warfarin described previously.
Our study also finds an increase, although a non-significant one, in Tmax of warfarin in the piperine group. This has been suggested by a similar study to result from the delayed gastric emptying and inhibition of intestinal uptake transporters such as p-gp associated with piperine.39 The study assessed the interactions between piperine and the antihistamine, fexofenadine, and found an increase in Tmax of fexofenadine with piperine co-administration. However, since warfarin is not reported to be a substrate of intestinal p-gp, we suggest that the delay in Tmax seen in our study is attributed to the increased gastric emptying time alone.50
From a pharmacodynamic point of view, INR is the main tool for assessing warfarin efficacy and adjusting dosing regimen among patients. A too high INR value may indicate warfarin toxicity, while a too low value is considered subtherapeutic.1 Careful consideration of concomitant drugs, herbs, and diet is usually warranted to ensure warfarin safety and efficacy. In our study, we find a significant decrease in INR values in the piperine-treated group at 24 h compared to the positive and negative controls. This decrease is consistent with the decline in warfarin total concentration seen in the piperine group, which suggests that piperine could change pharmacodynamics profile of warfarin in rats. The significantly decreased INR values, upon piperine co-administration at 24 h, suggest a decrease in the anticoagulant action of warfarin. Peak INR values were achieved at 24 h for all groups, a result consistent with previous reports.4,11
Piperine is available both as a dietary supplement and from its natural dietary source, black pepper.46 Dietary intake of black pepper varies considerably from one population group to another. The individual in United States consumes an average of 359 mg black pepper each day, equivalent to 18 to 32 mg of piperine depending on the content of piperine in black pepper (5–15%).46 Thus, interactions may occur even at the dietary level, as a piperine plasma concentration of 20 mg/day is sufficient to cause 3A and 2C inhibition.36
Our study has a few limitations. First, we took measurements for the total concentrations of warfarin rather than measuring both free and total concentrations. Free warfarin fraction may be more clinically relevant and thus offers a more meaningful look into the clinical consequences of piperine co-administration. Furthermore, we administered and measured warfarin as a racemic mixture. The S and R isoforms undergo different metabolic pathways within the P450 system, and thus the effects of piperine on their respective kinetics may differ. However, both metabolizing enzymes for S and R (2C and 3A, respectively) are inhibited by piperine, so results may not vary broadly if separation is achieved. Lastly, this is an animal study, and its results may not accurately reflect the kinetics in humans. Differences in CYP enzyme specificity are present between humans and rats, and the enzymes responsible for warfarin metabolism in each are not 100% homologous. However, the warfarin rats model offers considerable resemblance between the two species.49 Future studies will involve intravenous pharmacokinetic experiments together with oral administration to give more clear interpretations about piperine effect on absorption, in addition to in vitro mechanistic studies involving recombinant CYP450 or liver microsomes.
Conclusions
Piperine is a modulator of multiple intestinal and hepatic metabolism pathways. Our results confirm that piperine may inhibit the CYP2C enzyme system in rats, which is mainly responsible for 7-hydroxylation of warfarin. However, The PK interaction study between warfarin and piperine showed unexpectedly lower plasma levels of warfarin, although it was not statistically significant, as indicated by Cmax and AUC0–∞ when compared to the control group. These effects may be attributed to reduction in warfarin oral bioavailability as a result of inhibition of gastrointestinal motility by piperine, thus preventing any increase in warfarin concentration due to potential inhibition of its metabolism. In addition, piperine may have displaced warfarin from plasma protein thus increasing its clearance. Furthermore, we found piperine to change the anticoagulation of warfarin in rats. The significantly decreased INR values at 24 h, upon piperine co-administration, suggested a decreased anticoagulant action of warfarin and is consistent with the slight decrease in the total warfarin concentration. The overall findings of our study suggest that alteration of anticoagulant effect of warfarin when co-administered with piperine is the net effect of various changes in pharmacodynamic and pharmacokinetic factors. The current study represents an alert of potential risk for food–drug interactions. It is therefore important to investigate the relevance of this study to human by performing further studies in clinical settings to evaluate risk of any potential interaction in patients.
Acknowledgments
The authors would like to extend their appreciation to German Academic Exchange Service (DAAD) for financially supporting W. B. during his MSc studies.
Disclosure
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
1. Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med. 2005;165(10):1095–1106. doi:10.1001/archinte.165.10.1095
2. Chu Y, Zhang L, Wang XY, Guo JH, Guo ZX, Ma XH. The effect of compound danshen dripping pills, a Chinese herb medicine, on the pharmacokinetics and pharmacodynamics of warfarin in rats. J Ethnopharmacol. 2011;137(3):1457–1461. doi:10.1016/j.jep.2011.08.035
3. Lehmann DF. Enzymatic shunting: resolving the acetaminophen-warfarin controversy. Pharmacotherapy. 2000;20(12):1464–1468. doi:10.1592/phco.20.19.1464.34860
4. Guo C, Xue S, Zheng X, et al. The effect of fenofibric acid on the pharmacokinetics and pharmacodynamics of warfarin in rats. Xenobiotica. 2018;48(4):400–406. doi:10.1080/00498254.2017.1306760
5. Kaminsky LS, Zhang ZY. Human P450 metabolism of warfarin. Pharmacol Ther. 1997;73(1):67–74. doi:10.1016/s0163-7258(96)00140-4
6. Ufer M. Comparative pharmacokinetics of vitamin K antagonists. Clin Pharmacokinet. 2005;44(12):1227–1246. doi:10.2165/00003088-200544120-00003
7. Ufer M, Kammerer B, Kirchheiner J, Rane A, Svensson JO. Determination of phenprocoumon, warfarin and their monohydroxylated metabolites in human plasma and urine by liquid chromatography-mass spectrometry after solid-phase extraction. J Chromatogr B Anal Technol Biomed Life Sci. 2004;809(2):217–226. doi:10.1016/j.jchromb.2004.06.023
8. Zhou Q, Yau WP, Chan E. Enantioseparation of warfarin and its metabolites by capillary zone electrophoresis. Electrophoresis. 2003;24(15):2617–2626. doi:10.1002/elps.200305441
9. Zuo Z, Wo SK, Lo CM, Zhou L, Cheng G, You J. Simultaneous measurement of S-warfarin, R-warfarin, S-7-hydroxywarfarin and R-7-hydroxywarfarin in human plasma by liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal. 2010;52(2):305–310. doi:10.1016/j.jpba.2010.01.005
10. Juurlink DN. Drug interactions with warfarin: what clinicians need to know. CMAJ. 2007;177(4):369–371. doi:10.1503/cmaj.070946
11. John J, John M, Wu L, Hsiao C, Abobo CV, Liang D. Effects of etravirine on the pharmacokinetics and pharmacodynamics of warfarin in rats. Br J Pharmacol. 2013;168(8):1851–1858. doi:10.1111/bph.12082
12. Eriksson N, Wadelius M. Prediction of warfarin dose: why, when and how? Pharmacogenomics. 2012;13(4):429–440. doi:10.2217/pgs.11.184
13. Mazzari ALDA, Prieto JM. Herbal medicines in Brazil: pharmacokinetic profile and potential herb-drug interactions. Front Pharmacol. 2014;9(5):162. doi:10.3389/fphar.2014.00162
14. Eisenberg DM, Davis RB, Ettner SL, et al. Trends in alternative medicine use in the United States, 1990–1997: results of a follow-up national survey. JAMA. 1998;280(18):1569–1575. doi:10.1001/jama.280.18.1569
15. Gardiner P, Phillips R, Shaughnessy AF. Herbal and dietary supplement-drug interactions in patients with chronic illnesses. Am Fam Physician. 2008;77(1):73–78.
16. Aggarwal A, Ades PA. Interactions of herbal remedies with prescription cardiovascular medications. Coron Artery Dis. 2001;12(7):581–584. doi:10.1097/00019501-200111000-00009
17. Miller LG. Herbal medicinals: selected clinical considerations focusing on known or potential drug-herb interactions. Arch Intern Med. 1998;158(20):2200–2211. doi:10.1001/archinte.158.20.2200
18. Srinivasan K. Black pepper and its pungent principle-piperine: a review of diverse physiological effects. Crit Rev Food Sci Nutr. 2007;47(8):735–748. doi:10.1080/10408390601062054
19. Moses G. What’s in complementary medicines? Aust Prescr. 2019;42(3):82–83. doi:10.18773/austprescr.2019.024
20. Borre YE, Panagaki T, Koelink PJ, et al. Neuroprotective and cognitive enhancing effects of a multi-targeted food intervention in an animal model of neurodegeneration and depression. Neuropharmacology. 2014;79:738–749. doi:10.1016/j.neuropharm.2013.11.009
21. Thiel A, Buskens C, Woehrle T, et al. Black pepper constituent piperine: genotoxicity studies in vitro and in vivo. Food Chem Toxicol. 2014;66:350–357. doi:10.1016/j.fct.2014.01.056
22. Bhat BG, Chandrasekhara N. Lack of adverse influence of black pepper, its oleoresin and piperine in the weanling rat. J Food Saf. 1986;7(4):215–223. doi:10.1111/j.1745-4565.1986.tb00543.x
23. Ganesh Bhat B, Chandrasekhara N. Studies on the metabolism of piperine: absorption, tissue distribution and excretion of urinary conjugates in rats. Toxicology. 1986;40(1):83–92. doi:10.1016/0300-483X(86)90048-X
24. Piyachaturawat P, Glinsukon T, Toskulkao C. Acute and subacute toxicity of piperine in mice, rats and hamsters. Toxicol Lett. 1983;16(3–4):351–359. doi:10.1016/0378-4274(83)90198-4
25. Bhardwaj RK, Glaeser H, Becquemont L, Klotz U, Gupta SK, Fromm MF. Piperine, a major constituent of black pepper, inhibits human P-glycoprotein and CYP3A4. J Pharmacol Exp Ther. 2002;302(2):645–50.
26. Wadhwa S, Singhal S, Rawat S. Bioavailability enhancement by piperine: a review. AJBPS. 2014;4:36.
27. Volak LP, Ghirmai S, Cashman JR, Court MH. Curcuminoids inhibit multiple human cytochromes P450 (CYP), UDP-glucuronosyltransferase (UGT), and sulfotransferase (SULT) enzymes, while piperine is a relatively selective CYP3A4 inhibitor. Drug Metab Dispos. 2008;36(8):1594–1605. doi:10.1124/dmd.108.020552
28. Shamsi S, Tran H, Tan RS, Tan ZJ, Lim LY. Curcumin, piperine, and capsaicin: a comparative study of spice-mediated inhibition of human cytochrome P450 isozyme activities. Drug Metab Dispos. 2017;45(1):49–55. doi:10.1124/dmd.116.073213
29. Hiwale AR, Dhuley JN, Naik SR. Effect of co-administration of piperine on pharmacokinetics of β-lactam antibiotics in rats. Indian J Exp Biol. 2002;40(3):277–281.
30. Johnson JJ, Nihal M, Siddiqui IA, et al. Enhancing the bioavailability of resveratrol by combining it with piperine. Mol Nutr Food Res. 2011;55(8):1169–1176. doi:10.1002/mnfr.201100117
31. Bano G, Raina R, Zutshi U, Bedi K, Johri R, Sharma S. Effect of piperine on bioavailability and pharmacokinetics of propranolol and theophylline in healthy volunteers. Eur J Clin Pharmacol. 1991;41(6):615–617. doi:10.1007/BF00314996
32. Singh A, Verma S, Al Jarari NMH, et al. Effect of piperine on pharmacokinetics of rifampicin and isoniazid: development and validation of high performance liquid chromatography method. J Appl Pharm Sci. 2018;8(3):72–81. doi:10.7324/JAPS.2018.8311
33. Chen XY, Yang GH, Li CL, et al. Pharmacokinetic interaction between magnolol and piperine in rats. Trop J Pharm Res. 2016;15(3):631–638. doi:10.4314/tjpr.v15i3.27
34. Chopra B, Dhingra AK, Kapoor RP, Prasad DN. Piperine and its various physicochemical and biological aspects: a review. Open Chem J. 2017;3(1):75–96. doi:10.2174/1874842201603010075
35. Frymoyer A, Shugarts S, Browne M, Wu AH, Frassetto L, Benet LZ. Effect of single-dose rifampin on the pharmacokinetics of warfarin in healthy volunteers. Clin Pharmacol Ther. 2010;88(4):540–547. doi:10.1038/clpt.2010.142
36. Bedada SK, Boga PK. The influence of piperine on the pharmacokinetics of fexofenadine, a P-glycoprotein substrate, in healthy volunteers. Eur J Clin Pharmacol. 2017;73(3):343–349. doi:10.1007/s00228-016-2173-3
37. Feng X, Liu Y, Wang X, Di X. Effects of piperine on the intestinal permeability and pharmacokinetics of linarin in rats. Molecules. 2014;19(5):5624–5633. doi:10.3390/molecules19055624
38. Athukuri BL, Neerati P. Enhanced oral bioavailability of domperidone with piperine in male wistar rats: involvement of CYP3A1 and P-gp inhibition. J Pharm Pharm Sci. 2017;20:28–37. doi:10.18433/j3mk72
39. Jin MJ, Han HK. Effect of piperine, a major component of black pepper, on the intestinal absorption of fexofenadine and its implication on food-drug interaction. J Food Sci. 2010;75(3):H93–6. doi:10.1111/j.1750-3841.2010.01542.x
40. Dubey RK, Leeners B, Imthurn B, Merki-Feld GS, Rosselli M. Piperine decreases binding of drugs to human plasma and increases uptake by brain microvascular endothelial cells. Phytother Res. 2017;31(12):1868–1874. doi:10.1002/ptr.5929
41. Ogiso T, Iwaki M, Tanaka H, et al. Pharmacokinetic drug interactions between ampiroxicam and sulfaphenazole in rats. Biol Pharm Bull. 1999;22(2):191–196. doi:10.1248/bpb.22.191
42. Wang L, Wang S, Chen M, et al. Inhibitory effect of ketoconazole and voriconazole on the pharmacokinetics of carvedilol in rats. Drug Dev Ind Pharm. 2015;41(10):1661–1666. doi:10.3109/03639045.2014.983930
43. Zayed A, Babaresh W, Darweesh R, El-Elimat T. Simultaneous determination of warfarin and 7-hydroxywarfarin in rat plasma by HPLC-FLD. Acta Pharm. 2019;70(2020):343–357. doi:10.2478/acph-2020-0025
44. De Oliveira RG, Vasconcellos MLAA, Alencar-Filho EB. [The influence of piperine on the bioavailability of drugs: a molecular approach]. Quim Nova. 2014;37(1):69–73. Portuguese. doi:10.1590/S0100-40422014000100013
45. Volak LP, Hanley MJ, Masse G, et al. Effect of a herbal extract containing curcumin and piperine on midazolam, flurbiprofen and paracetamol (acetaminophen) pharmacokinetics in healthy volunteers. Br J Clin Pharmacol. 2013;75(2):450–462. doi:10.1111/j.1365-2125.2012.04364.x
46. Bhardwaj RK, Glaeser H, Becquemont L, et al. Piperine, a major constituent of black pepper, inhibits human P-glycoprotein and CYP3A4. J Pharmacol Exp Ther. 2002;302(2):645–650. doi:10.1124/jpet.102.034728
47. Toutain PL, Bousquet-Melou A. Free drug fraction vs. free drug concentration: a matter of frequent confusion. J Vet Pharmacol Ther. 2002;25(6):460–463. doi:10.1046/j.1365-2885.2002.00442.x
48. Ding T, Zhang Y, Chen A, Tang Y, Liu M, Wang X. Effects of cucurbitacin E, a tetracyclic triterpene compound from cucurbitaceae, on the pharmacokinetics and pharmacodynamics of warfarin in rats. Basic Clin Pharmacol Toxicol. 2015;116(5):385–389. doi:10.1111/bcpt.12329
49. Cheong J, Halladay JS, Plise E, Sodhi JK, Salphati L. The effects of drug metabolizing enzyme inhibitors on hepatic efflux and uptake transporters. Drug Metab Lett. 2017;11(2):111–118. doi:10.2174/1872312811666171010101248
50. Gschwind L, Rollason V, Daali Y, Bonnabry P, Dayer P, Desmeules JA. Role of P-glycoprotein in the uptake/efflux transport of oral vitamin K antagonists and rivaroxaban through the caco-2 cell model. Basic Clin Pharmacol Toxicol. 2013;113(4):259–265. doi:10.1111/bcpt.12084.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.