")
Back to Journals » OncoTargets and Therapy » Volume 12
Pim-1 inhibitor SMI-4a suppresses tumor growth in non-small cell lung cancer via PI3K/AKT/mTOR pathway
Authors Jiang W, Chen Y, Song X, Shao Y, Ning Z, Gu W
Received 28 January 2019
Accepted for publication 8 April 2019
Published 23 April 2019 Volume 2019:12 Pages 3043—3050
DOI https://doi.org/10.2147/OTT.S203142
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Wenjie Jiang,1 Yuan Chen,1 Xing Song,1 Yingjie Shao,1 Zhonghua Ning,1 Wendong Gu1
1Department of Radiation Oncology, The Third Affiliated Hospital of Soochow University, Jiangsu, Changzhou 213003, People’s Republic of China
Background: In the present study, we aimed to investigate the effect of proviral integration site for moloney murine leukemia virus-1 (Pim-1) inhibitor (SMI-4a) on the progression of non-small cell lung cancer (NSCLC).
Materials and methods: The effects of SMI-4a on proliferation, apoptosis, and cell cycle of NSCLC cells were examined by in vitro experiments using human NSCLC cell lines (A549 and Ltep-a-2). The pathway regulated by SMI-4a was detected using Western blot. Furthermore, we performed in vivo experiments to assess the effects of SMI-4a on tumor growth using mouse models with NSCLC.
Results: Our data demonstrated that SMI-4a could inhibit the proliferation of A549 and Ltep-a-2 cells markedly in a dose-dependent manner (P<0.05). Treatment with 80 μmol/L of SMI-4a for 48 h significantly induced the apoptosis rate of NSCLC cells (P<0.05), and blocked the cell cycle of NSCLC cells in G2/M phase (P<0.05). The phosphorylation levels of PI3K, AKT, and mTOR in NSCLC cells were significantly downregulated by SMI-4a (P<0.05). Result from in vivo experiments demonstrated that SMI-4a could suppress the tumor growth in mouse models with NSCLC (P<0.05).
Conclusions: SMI-4a suppresses the progression of NSCLC by blocking the PI3K/AKT/mTOR pathway.
Keywords: Pim-1, SMI-4a, NSCLC, tumor progression
Introduction
In recent years, lung cancer has become the leading cause of cancer-related death in the world.1 Non-small cell lung cancer (NSCLC) comprises 80% of all lung cancer cases.2 Surgical resection is the most effective treatment of NSCLC. However, because of the lack of early and effective diagnostic methods, most NSCLC patients are diagnosed at an advanced stage and loss the opportunities of radical surgery for the early stage.3 In addition, the development and clinical application of platinum-based chemotherapy regimens have gradually entered a plateau.4
With the discovery of multiple oncogenes, more and more molecular targeted drugs showed satisfactory effects in NSCLC.5 The treatment of NSCLC has entered a new era of precision medicine. Among molecular targeted drugs, epithelial growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) have been widely used in NSCLC patients.6 Studies have shown that EGFR-TKIs are more effective than conventional chemotherapy drugs in NSCLC patients with EGFR sensitive mutation.7–10 However, with the widespread use of EGFR-TKIs, resistance has emerged inevitably. Therefore, it is necessary to develop more molecular drugs targeting different oncogenes of NSCLC.
Proviral integration site for moloney murine leukemia virus-1 (Pim-1), as a kind of serine threonine kinase, functions in signal transduction.11 Pim-1 regulates the activities and subcellular localizations of certain proteins by phosphorylating their serine and threonine sites.12 For example, downregulated expression level of Pim-1 induced by specific monoclonal antibody (mAb) led to decreased phosphorylation of AKT at Ser473 and increased cleavage of caspase-9, that activated the mitochondrial cell death pathway.13 Previous studies showed that Pim-1, acting as an oncogene, is highly expressed in various tumors and promotes their progression, and that overexpression of Pim-1 in NSCLC tissues is associated with advanced clinical parameters.14–16 However, the therapeutic effect of Pim-1 inhibitor in NSCLC remains unclear.
The aim of the present study is to investigate the effects of Pim-1 inhibitor (SMI-4a) on the progression of NSCLC in vitro and in vivo. We found that SMI-4a significantly suppressed the proliferation and cell cycle, and induced the apoptosis of NSCLC cells in vitro. Besides, SMI-4a could also suppress the tumor growth in mouse models with NSCLC. PI3K/AKT/mTOR pathway was involved in the anti-tumor process induced by SMI-4a. Pim-1 inhibitor may serve as a new molecular targeted drug for NSCLC patients.
Materials and methods
Cell culture
Human NSCLC cell lines (A549 and Ltep-a-2) obtained from the Shanghai Institutes of Biological Sciences Cell Bank were cultured in DMEM (Thermo Fisher Scientific, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA) and 1% penicillin/streptomycin (Gibco) at 37 °C in a humidified incubator of 5% CO2.
Proliferation assays
A549 and Ltep-a-2 cells (2×103 in 100 μL/well) were seeded in 96-well plates and treated with Pim-1 inhibitor (SMI-4a; Sigma, USA) at different concentrations (0, 5, 10, 20, 40, and 80 μmol/L) for 48 h. Then 10 μL of CCK8 solution was added to each well for 4 h incubation. The absorbance at 450 nm was measured using Multiscan Plate Reader (Thermo Fisher Scientific).
Apoptosis assay
Cells were washed with phosphate buffered saline (PBS) twice, centrifuged at 1,000 r/min for 5 min, and then resuspended in 500 μL of binding buffer. Then 5 μL of Annexin V-FITC and 5 μL of PI were added. After incubation in dark at 37 °C for 15 min, the apoptosis of cells was analyzed by flow cytometry (BD Biosciences, San Diego, CA, USA) according to the manufacturer’s instructions.
Cell cycle assay
The synchronized cells were washed with PBS twice, fixed with cold 75% ethanol, and then cultured at 4 °C overnight. The cells were again washed with PBS, followed by staining with propidium iodide (PI; 50 µg/ml, Sigma-Aldrich®, St. Louis, MO, USA) in the presence of RNase A (100 µg/ml; Fermentas®, Shanghai, China). After incubation in dark at 37 °C for 30 min, the cell cycle was measured by flow cytometry according to the manufacturer’s instructions.
Western blotting analysis
Mammalian protein extraction agent (Thermo Fisher Scientific) mixed with alt protease inhibitor cocktail (Thermo Fisher Scientific) (100:1) was used to lyse cells. Equal amounts of proteins were loaded onto the 10% sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE). Then interest proteins were transferred from the gels onto the polyvinylidene fluoride (PVDF) membranes (Millipore, USA). Membranes were blocked with 5% bovine serum albumin (BSA) for 1 h at 37 °C. Then membranes were incubated in primary antibodies overnight at 4 °C and subsequently in horseradish peroxidase-conjugated secondary antibodies for 2 h at 37 °C. Protein bands were visualized with electrochemiluminescence assay (Millipore). ImageJ (National Institutes of Health, USA) was used to quantify the protein bands.17
In vivo study
Approved by Animal Ethics Committee of Soochow University, animal experiments were performed following the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication). Male C57BL/6 mice (6 weeks old) were purchased from Shanghai Experimental Animal Center (Shanghai, China). A549 or Ltep-a-2 cells (1×107) resuspended in PBS were subcutaneously inoculated into the armpit. After one week, mice were randomly divided into five groups treated with 5/10/20/40 mg/kg of SMI-4a or vehicle control (0 mg/kg) (5 mice per group). Every three days, the length and width of the tumors were measured. Tumor volume was calculated using the following formula: (width2 × height)/2. After the last measurement of the volume, the tumors of mice were immediately removed, weighed, and then lysed for Western blotting analysis.
Statistical analysis
Statistical analysis was performed using SPSS v21.0 (SPSS Inc., USA). Difference between two groups was assessed using Independent sample t-test. When comparing more than two groups, one-way analysis of variance (ANOVA) followed by Dunnett’s test was used. Each experiment was repeated three times. Data were presented as the means ± standard deviation (SD). A P<0.05 was considered as statistically significant.
Results
SMI-4a inhibits the proliferation of NSCLC cells
CCK-8 assay was used to examine the cell proliferation. The results showed that SMI-4a inhibited the proliferation of A549 and Ltep-a-2 cells in a dose-dependent manner (P<0.05; Figure 1). Because 80 μmol/L of SMI-4a showed a strongest inhibitory effect on the proliferation of both cell lines, 80 μmol/L was chosen as the SMI-4a concentration in the following in vitro experiments.
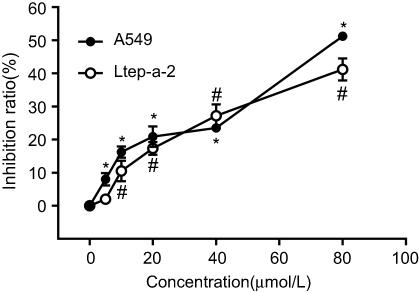 | Figure 1 SMI-4a inhibits the proliferation of NSCLC cells. The inhibition ratios of A549 and Ltep-a-2 cells were detected at 48h after treatment with various concentrations of SMI-4a (0, 5, 10, 20, 40, or 80 μmol/L). Each value represents the mean of triplicate experiments; bars, SD. ANOVA; *P<0.05, vs 0 μmol/L, A549 group; #P<0.05, vs 0 μmol/L, Ltep-a-2 group. |
SMI-4a induces the apoptosis of NSCLC cells
Flow cytometry assay was used to examine the cell apoptosis. The results showed that treatment with 80 μmol/L of SMI-4a for 48 h significantly induced the apoptosis rate of NSCLC cells (Figure 2). The apoptosis rate of A549 cells increased from 1.1±0.8 to 17.0±0.9 (P<0.001). The apoptosis rate of Ltep-a-2 cells increased from 1.3±0.7 to 10.0±0.8 (P=0.001).
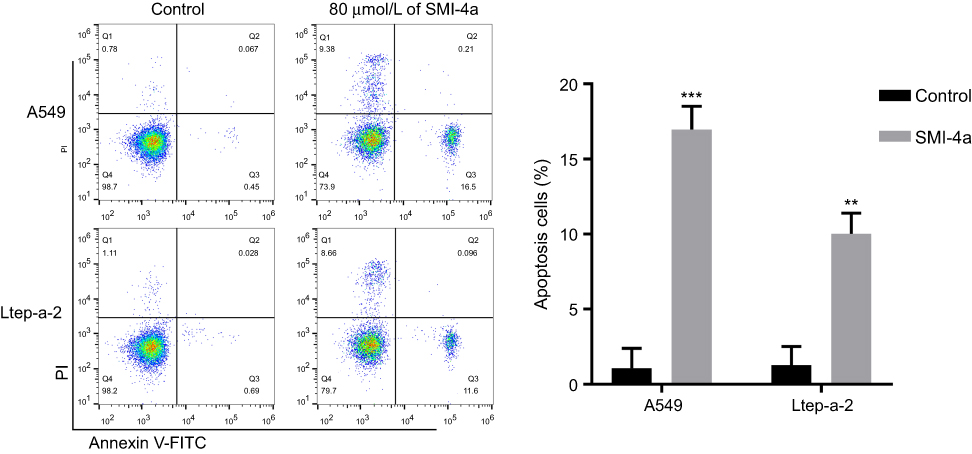 | Figure 2 SMI-4a induces the apoptosis of NSCLC cells. The apoptosis ratios of A549 and Ltep-a-2 cells were detected at 48h after treatment with 80 μmol/L of SMI-4a by flow cytometry. Representative images and quantitative data are shown. Each column is shown as the mean of triplicate experiments; bars, SD. Student’s t-test; **P<0.01, ***P<0.001. |
SMI-4a blocks the cell cycle of NSCLC cells
The effect of SMI-4a on the cycle distribution of NSCLC cells was also detected by flow cytometry assay. The results showed that SMI-4a blocked the cell cycle of NSCLC cells in G2/M phase (Figure 3). After 48h treatment of 80 μmol/L of SMI-4a, the portion of A549 cells in G2/M phase increased from 19.2±2.3 to 28.3±3.7 (P=0.021); the portion of Ltep-a-2 cells in G2/M phase increased from 19.6±3.2 to 32.0±5.5 (P=0.037).
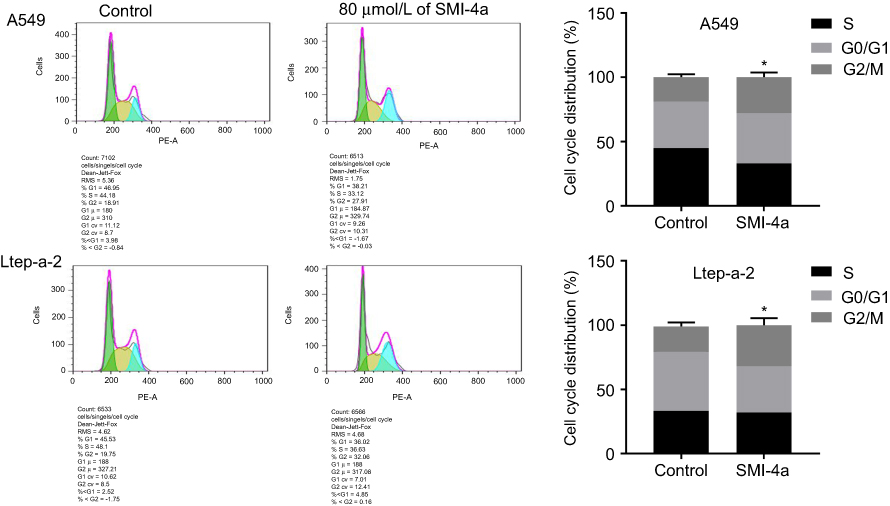 | Figure 3 SMI-4a blocks the cell cycle of NSCLC cells. The cell cycles of A549 and Ltep-a-2 cells were detected at 48h after treatment with 80 μmol/L of SMI-4a by flow cytometry. Representative images and quantitative data are shown. Each column is shown as the mean of triplicate experiments; bars, SD. Student’s t-test; *P<0.05. |
SMI-4a blocks the phosphorylation of PI3K/AKT/mTOR pathway
Given the complex relationship between Pim-1 and PI3K/AKT/mTOR pathway, we further investigated the role of Pim-1 inhibitor in the activation of this pathway. By Western blotting analysis, we found that the total expression of PI3K, AKT and mTOR was not altered by SMI-4a (P>0.05; Figure 4). However, the phosphorylation of PI3K, AKT and mTOR was significantly downregulated by SMI-4a (P<0.05; Figure 4).
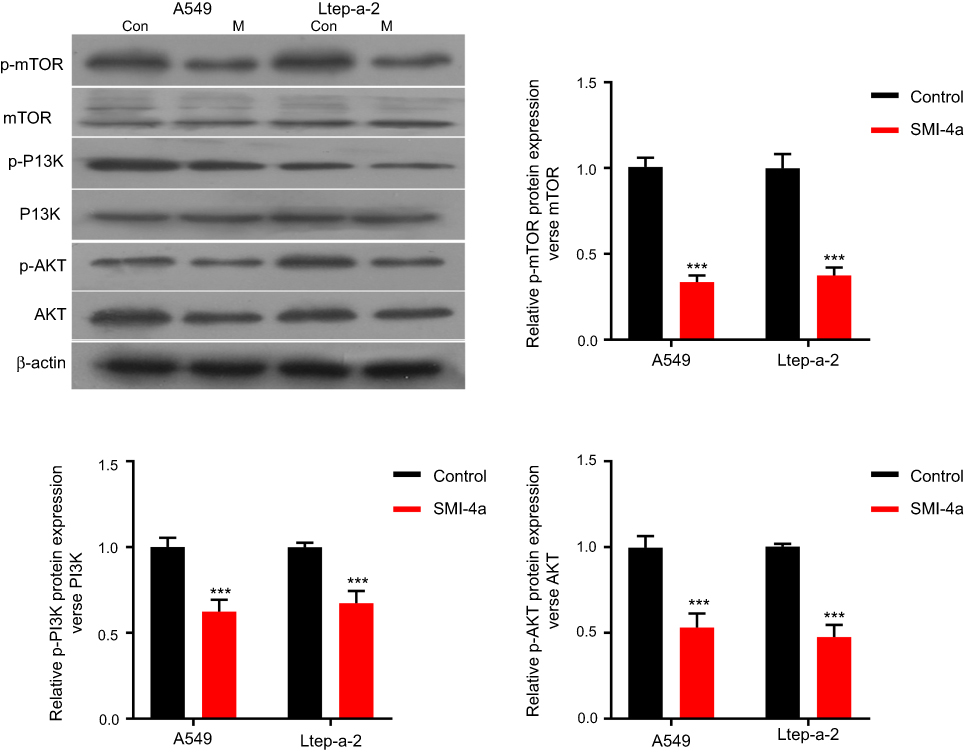 | Figure 4 SMI-4a inhibits PI3K/AKT/mTOR pathway in vitro. The protein levels of PI3K/AKT/mTOR pathway in A549 and Ltep-a-2 cells were detected at 48h after treatment with 80 μmol/L of SMI-4a. Representative images and quantitative data are shown. Each column is shown as the mean of triplicate experiments; bars, SD. Student’s t-test; ***P<0.001. |
SMI-4a suppresses tumor growth in vivo
Mouse models with NSCLC were established to validate the suppressive effect of SMI-4a on tumor growth. The mice were grouped according to the dose of SMI-4a. Compared with the control group (0 mg/kg), groups treated with 5/10/20/40 mg/kg of SMI-4a had lower tumor growth rates and lighter tumor weight (P<0.05, Figure 5). The tumor suppressive effect of SMI-4a in vivo was also in a dose-dependent manner.
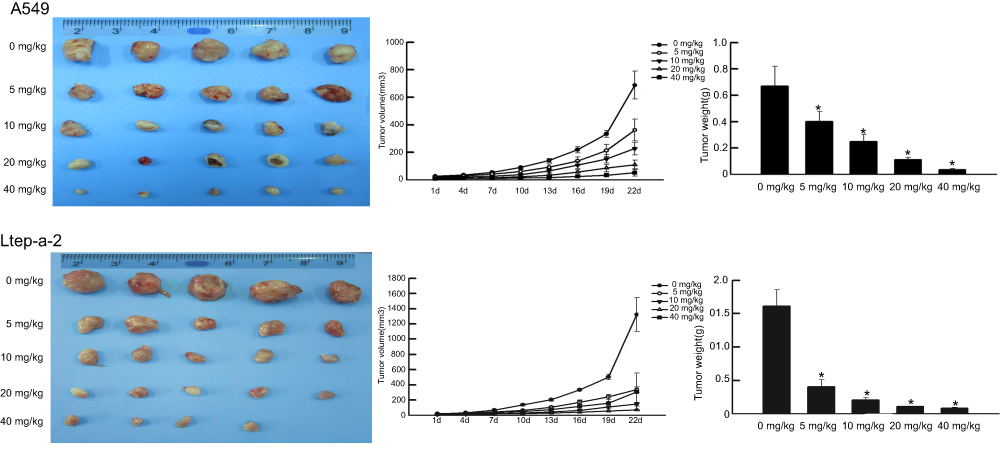 | Figure 5 SMI-4a suppresses tumor growth in mouse models NSCLC. The tumor volume in A549 and Ltep-a-2 mouse models treated with various concentrations of SMI-4a (0, 5, 10, 20, or 40 mg/kg) was measured every three days. At 22 d, the tumor weight was measured. Each value represents the mean tumor volume of five mice; bars, SD. Each column represents the mean tumor weight of five mice; bars, SD. ANOVA; *P<0.05, vs 0 mg/kg. |
To further confirm the role of PI3K/AKT/mTOR pathway in the anti-tumor process induced by SMI-4a, we collected the mouse tumor tissues to detect the expression levels of associated proteins in the pathway. The results showed that the phosphorylation of PI3K, AKT and mTOR was downregulated by SMI-4a in a dose-dependent manner (P<0.05, Figure 6), while the total expression of PI3K, AKT and mTOR was still not altered by SMI-4a (P>0.05, Figure 6).
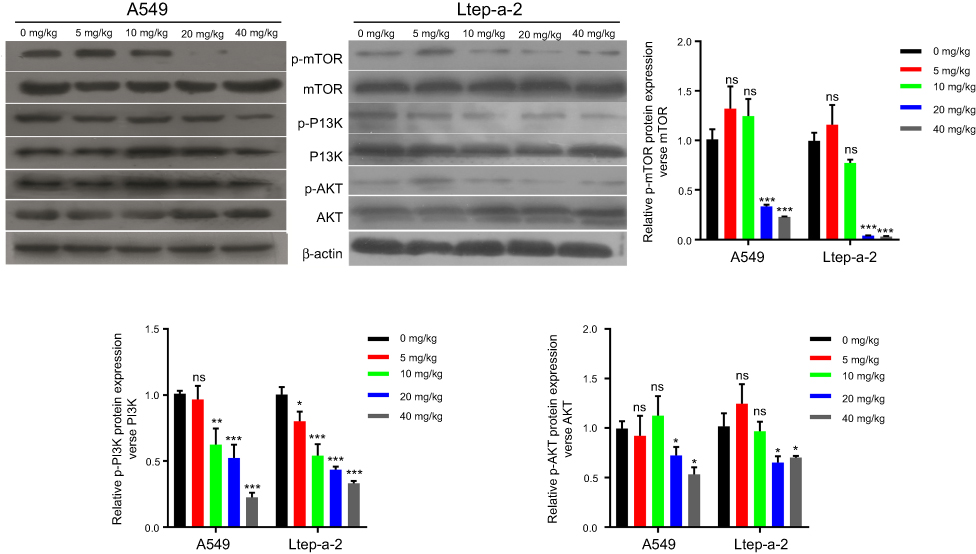 | Figure 6 SMI-4a inhibits PI3K/AKT/mTOR pathway in vivo. At 22 d, the tumors of A549 and Ltep-a-2 mouse models treated with various concentrations of SMI-4a (0, 5, 10, 20, or 40 mg/kg) were lysed for Western blotting analysis. The protein levels of PI3K/AKT/mTOR pathway were examined. Representative images and quantitative data are shown. Each column is shown as the mean of triplicate experiments; bars, SD. ANOVA; *P<0.05, **P<0.01, ***P<0.001, vs 0 mg/kg. Abbreviation: ns, no significance. |
Discussion
Previous studies have confirmed the anti-tumor effects of SMI-4a in a variety of tumors, including osteosarcoma, melanoma and leukemia.18–20 Liao et al found that SMI-4a significantly downregulated the expression of anti-apoptotic protein Bcl-2, and inhibited the cell proliferation, migration, and invasion in osteosarcoma.18 Lv et al confirmed that SMI-4a inhibited tumor growth by inducing autophagy via AKT/mTOR pathway in melanoma.19 Fan et al proved that SMI-4a induced the apoptosis and inhibited the colony formation of leukemia cells.20 There findings indicated the potential of SMI-4a as a new molecular targeted drug for the treatment of tumor. The present study is the first to demonstrate the anti-tumor effects of SMI-4a in NSCLC. We found that SMI-4a inhibited the proliferation and cell cycle, and induced the apoptosis of NSCLC cells in vitro. SMI-4a also had an inhibitory effect on tumor growth in vivo in a dose-dependent manner.
There are many other Pim-1 inhibitors besides SMI-4a. Lee et al proved that the combination of Pim-1 inhibitor AZD1208 and AKT inhibitor had synergism inhibitory effects on the progression of gastric cancer cells.21 Hu et al produced an anti-Pim-1 mAb and demonstrated that it suppressed the tumor growth by inhibiting the phosphorylation of AKT in prostate cancer.13 Kim et al found that Pim-1 inhibitors (SGI-1776, ETP-45299, and tryptanthrin) enhanced the radiosensitization of tumor cells by inhibiting radioresistant signaling pathway in mouse models with NSCLC.22 However, more studies are needed to compare the safety and effectiveness of different Pim-1 inhibitors.
Our results indicated that PI3K/AKT/mTOR pathway was significantly inhibited by SMI-4a in NSCLC, which is consistent with previous studies.13,19 In malignant tumors, the activation of PI3K/AKT/mTOR pathway breaks the balance between pro-apoptotic and anti-apoptotic proteins to exert an anti-apoptotic effect. Briefly, AKT increases the dissociative anti-apoptotic protein Bcl-2 and blocks the activation of pro-apoptotic proteins caspase-9 and caspase-3.23,24 The activation of PI3K/AKT/mTOR pathway can also promote cell proliferation. For example, AKT directly phosphorylates glucose synthase kinase-3β (GSK3β) and increases the accumulation of cyclin D1 that accelerates the G1-S phase.25 Besides, AKT inhibits the expression of p27Kip1, a cell cycle-dependent protein that blocks cell cycle in G1 phase.26 However, our study showed that SMI-4a blocked the cell cycle of NSCLC cells in G2/M phase rather than G1 phase. Fan et al also revealed that SMI-4a blocked the cell cycle of chronic myeloid leukemia cells in S-phase rather than G1 phase.20 Based on these inconsistent results, we hypothesized that in addition to D1 and p27Kip1, there are several other cell cycle regulatory factors involved in the SMI-4a-induced NSCLC cell cycle arrest. Further studies are needed to verify this hypothesis.
In recent years, EGFR-TKIs have been widely used in NSCLC patients with EGFR sensitive mutation.27 Although the clinical effect was significant in these patients, they will ultimately develop resistance to EGFR-TKIs after about 10–14 months.28 Moreover, targeted therapy with EGFR-TKIs can improve the quality of life in NSCLC patients, but its impact on overall prognosis is still not satisfactory.29 Therefore, new drugs targeting other molecules may be better choices for patients without EGFR sensitive mutation or resistant to EGFR-TKIs. SMI-4a selectively inhibiting Pim-1 kinase significantly inhibited tumor growth and was well tolerated in vivo in our study, so we surmised that SMI-4a has the potential to be the ideal alternative to EGFR-TKIs for NSCLC patients. More clinical trials are needed to prove the safety and effectiveness of SMI-4a.
In conclusion, the present study is the first research evaluating the therapeutic effect of Pim-1 inhibitor SMI-4a in NSCLC. The in vitro and in vivo experimental results showed that SMI-4a significantly suppresses the progression of NSCLC by blocking the PI3K/AKT/mTOR pathway. Pim-1 inhibitors may become new targeted drugs for NSCSL patients in the future.
Disclosure
The authors declare no potential conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi:10.3322/caac.21387
2. Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol. 2016;893:1–19. doi:10.1007/978-3-319-24223-1_1
3. Boolell V, Alamgeer M, Watkins DN, et al. The evolution of therapies in non-small cell lung cancer. Cancers (Basel). 2015;7(3):1815–1846.
4. Du L, Morgensztern D. Chemotherapy for advanced-stage non-small cell lung cancer. Cancer J. 2015;21(5):366–370.
5. Wood SL, Pernemalm M, Crosbie PA, et al. Molecular histology of lung cancer: from targets to treatments. Cancer Treat Rev. 2015;41(4):361–375.
6. Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10(11):760–774.
7. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–132.
8. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957.
9. Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11(2):121–128.
10. Wu Y-L, Lee JS, Thongprasert S, et al. Intercalated combination of chemotherapy and erlotinib for patients with advanced stage non-small-cell lung cancer (FASTACT-2): a randomised, double-blind trial. Lancet Oncol. 2013;14(8):777–786. doi:10.1016/S1470-2045(13)70254-7
11. Tursynbay Y, Zhang J, Li Z, et al. Pim-1 kinase as cancer drug target: an update. Biomed Rep. 2016;4(2):140–146. doi:10.3892/br.2015.561
12. Beharry Z, Mahajan S, Zemskova M, et al. The Pim protein kinases regulate energy metabolism and cell growth. Proc Natl Acad Sci U S A. 2011;108(2):528–533. doi:10.1073/pnas.1013214108
13. Hu XF, Li J, Vandervalk S, Wang Z, Magnuson NS, Xing PX. PIM-1-specific mAb suppresses human and mouse tumor growth by decreasing PIM-1 levels, reducing Akt phosphorylation, and activating apoptosis. J Clin Invest. 2009;119(2):362–375. doi:10.1172/JCI33216
14. Jin Y, Tong D-Y, Chen J-N, et al. Overexpression of osteopontin, αvβ3 and Pim-1 associated with prognostically important clinicopathologic variables in non-small cell lung cancer. PLoS One. 2012;7(10):e48575. doi:10.1371/journal.pone.0048575
15. Guo S, Mao X, Chen J, et al. Overexpression of Pim-1 in bladder cancer. J Exp Clin Cancer Res. 2010;29:161. doi:10.1186/1756-9966-29-17
16. Tanaka S, Kitamura T, Higashino F, et al. Pim-1 activation of cell motility induces the malignant phenotype of tongue carcinoma. Mol Med Rep. 2009;2(2):313–318. doi:10.3892/mmr_00000102
17. Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi:10.1038/nmeth.2089
18. Liao Y, Feng Y, Shen J, et al. Clinical and biological significance of PIM1 kinase in osteosarcoma. J Orthop Res. 2016;34(7):1185–1194. doi:10.1002/jor.23134
19. Lv D-L, Chen L, Ding W, et al. Ginsenoside G-Rh2 synergizes with SMI-4a in anti-melanoma activity through autophagic cell death. Chin Med. 2018;13:11. doi:10.1186/s13020-018-0168-y
20. Fan R-F, Lu Y, Fang Z-G, et al. PIM-1 kinase inhibitor SMI-4a exerts antitumor effects in chronic myeloid leukemia cells by enhancing the activity of glycogen synthase kinase 3β. Mol Med Rep. 2017;16(4):4603–4612. doi:10.3892/mmr.2017.7215
21. Lee M, Lee K-H, Min A, et al. Pan-Pim kinase inhibitor AZD1208 suppresses tumor growth and synergistically interacts with Akt inhibition in gastric cancer cells. Cancer Res Treat. 2018. doi:10.4143/crt.2017.341
22. Kim W, Youn H, Kwon T, et al. PIM1 kinase inhibitors induce radiosensitization in non-small cell lung cancer cells. Pharmacol Res. 2013;70(1):90–101. doi:10.1016/j.phrs.2013.01.005
23. Henshall DC, Araki T, Schindler CK, et al. Activation of Bcl-2-associated death protein and counter-response of Akt within cell populations during seizure-induced neuronal death. J Neurosci. 2002;22(19):8458–8465.
24. Altieri DC. The molecular basis and potential role of survivin in cancer diagnosis and therapy. Trends Mol Med. 2001;7(12):542–547.
25. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12(22):3499–3511.
26. Liang J, Zubovitz J, Petrocelli T, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8(10):1153–1160. doi:10.1038/nm761
27. Zhou C, Yao LD. Strategies to improve outcomes of patients with EGRF-mutant non-small cell lung cancer: review of the literature. J Thorac Oncol. 2016;11(2):174–186. doi:10.1016/j.jtho.2015.10.002
28. Shukla S, Chen ZS, Ambudkar SV. Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance. Drug Resist Updat. 2012;15(1–2):70–80.
29. Lin Y, Wang X, Jin H. EGFR-TKI resistance in NSCLC patients: mechanisms and strategies. Am J Cancer Res. 2014;4(5):411–435.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.