")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 7
PI3K mutations in breast cancer: prognostic and therapeutic implications
Authors Mukohara T
Received 20 January 2015
Accepted for publication 18 March 2015
Published 15 May 2015 Volume 2015:7 Pages 111—123
DOI https://doi.org/10.2147/BCTT.S60696
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Toru Mukohara
Cancer Center and Division of Medical Oncology/Hematology, Kobe University Hospital, Kobe, Japan
Abstract: The PI3K pathway is the most frequently enhanced oncogenic pathway in breast cancer. Among mechanisms of PI3K enhancement, PIK3CA mutations are most frequently (~30%) observed, along with protein loss of PTEN. Since the first discovery of PIK3CA mutations in solid malignancies in 2004, numerous studies have revealed the prognostic and therapeutic implications of these mutations. Although many issues remain unconfirmed, some have been carved in stone by the level of consistency they have shown among studies: 1) PIK3CA mutations are most likely to be observed in ER-positive/HER2-negative tumors, and are associated with other good prognostic characters; 2) PIK3CA mutations can coexist with other PI3K-enhancing mechanisms, such as HER2 amplification and PTEN protein loss; 3) PIK3CA mutations are potentially a good prognostic marker; 4) PIK3CA may predict a poorer tumor response to trastuzumab-based therapies, but its impact on disease-free survival and overall survival is uncertain; and 5) based on reports of early clinical trials, PIK3CA mutations do not guarantee a dramatic response to PI3K inhibitors. Collectively, there is currently no sufficient evidence to recommend routine genotyping of PIK3CA in clinical practice. Given that PIK3CA-mutant breast cancer appears to have a distinct tumor biology, development of more individualized targeted therapies based on the PIK3CA genotype is awaited.
Keywords: PI3K, PIK3CA, prognostic factor, predictive factor, trastuzumab
Introduction
Molecularly based choice of care for solid tumors was pioneered in the treatment of breast cancer. In addition to classic pathological information, such as tumor size, number of involved lymph nodes, presence or absence of metastasis, and histological grade, treatment selection today requires information about the expression of ER, PgR, and HER2. Expression of ER and/or PgR is not only a good prognostic factor but a well-characterized predictive factor for benefit from endocrine therapies. In contrast, HER2 overexpression is a poor prognostic factor, but does predict benefit from HER2-targeting drugs. The new concept of intrinsic subtypes created based on complementary deoxyribonucleic acid (cDNA) microarray data has been introduced to clinical care.1 Nevertheless, subtype classification is actually done by the protein-expression pattern of ER, PgR, HER2, and Ki67 in combination, mainly measured with immunohistochemistry, because this method provides high concordance with authentic microarray-determined intrinsic subtype and is much less expensive.2 It can therefore be said that molecularly based choice of care has seen little progression in the 21st century, and that patients with breast cancer require more sophisticated molecular markers able to deliver more individualized and effective care.
The PI3K pathway is one of the most characterized signaling pathways with relevance to oncogenic properties in a variety of malignancies. In particular, breast cancer tumorigenesis is believed to depend on the PI3K pathway. This is based on the fact that the majority of cases of this disease harbor at least one molecular mechanism that potentially enhances the pathway. These PI3K-enhancing mechanisms include mutations of the PI3K gene, more specifically PIK3CA gene mutations. First discovered in 2004 in various solid tumors, including breast cancer,3 these mutations have the potential to become a clinically useful biomarker, because they 1) are gain-of-function mutations of molecules located on an important signaling pathway, 2) are found at high frequency, and 3) are easy to measure (present or absent).
In this review, we focus on the many studies that have explored the prognostic value and therapeutic relevance of PIK3CA mutations since their discovery.
Physiology of PI3K
Structure of PI3K
PI3K is grouped into three classes (I–III) based on their structure and substrate specificity. Class I PI3K is further categorized into class IA and IB (Figure 1). Class IA PI3K is the class most closely implicated in cancer, and is referred to in this review simply as “PI3K” (Figure 1). PI3K is constituted of a p110 catalytic domain and p85 regulatory domain. There are three isoforms of p110, namely p110α (encoded by PIK3CA), p110β, and p110δ. While p110δ is expressed almost exclusively in leukocytes, p110α and p110β are expressed ubiquitously in all types of cells.4 In humans, PI3K regulatory subunit 1 (PIK3R1), PIK3R2, and PIK3R3 code p85α (or its splicing variant p55α or p50α), p85β, and p55γ, respectively.4
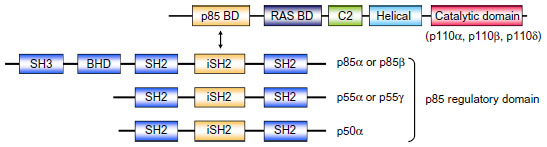 | Figure 1 Structure of class IA PI3K. Class IA PI3Ks are heterodimers consisting of p110 and p85 subunits. There are three p110 catalytic isoforms: p110α, p110β, and p110δ. The p110 isoforms share five distinct domains: an amino-terminal p85-binding domain (p85 BD), an RAS-binding domain (RAS BD), a putative membrane-binding domain (C2), the helical domain, and the carboxy-terminal kinase catalytic domain. There are also three p85 isoforms: p85α (and its splice variants p55α and p50α), p85β, and p55γ. They share three core domains, including a p110-binding domain called the inter-Src homology 2 (iSH2) domain, along with two SH2 domains. The two longer isoforms, p85α and p85β, have an SH3 domain and a BCR homology domain (BHD) located in their extended N-terminal regions. |
PI3K signalling
On RTK activation, p85 interacts directly with RTK or via adaptor proteins, and the resulting PI3K is recruited to the membrane (Figure 2).4 In addition to RTKs, RAS, which triggers MAPK pathways, can also directly bind to and activate PI3K (Figure 2).5 On the cell membrane, inhibitory regulation of p85 to 110 is canceled, and PI3K becomes active as a kinase. Subsequently, PI3K catalyzes the conversion of PIP2 to PIP3.4,5 In physiological conditions, the intracellular concentration of PIP3 is meticulously regulated by PTEN, which catalyzes the conversion of PIP3 to PIP2.4,5 As a result, PTEN functions as a negative regulator of PI3K. PIP3 is further recognized by AKT and PDPK1.4,5 Connection of PIP3 to PDPK1 and AKT allows the physical interaction of PDPK1 and AKT, which leads to activation of AKT by phosphorylation of the T308 residue.4 Maximal activation of AKT requires phosphorylation of the S473 residue by PDPK2, and mTORC2 mainly works as PDPK2.4 AKT phosphorylates several cellular proteins, GSK3, FOXO1, MDM2, and BAD (Figure 2).5 In addition, AKT phosphorylates and inactivates TSC2, which allows RHEB to activate mTORC1 (Figure 2).5 These AKT signalings result in enhanced growth, antiapoptosis, cell-cycle progression, and translation (Figure 2).4,5
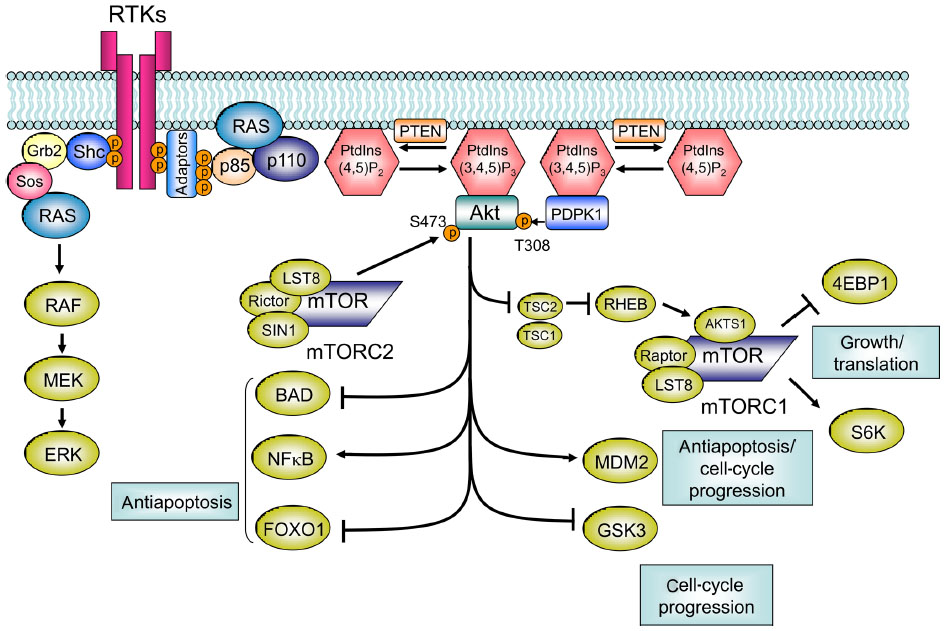 | Figure 2 Class I PI3K pathway. RTK activation allows p85 to interact with RTK directly or via adaptor proteins, which recruits PI3K to the membrane. On the cell membrane, inhibitory regulation of p85 to 110 is canceled, and PI3K becomes active as a kinase. Subsequently, PI3K catalyzes the conversion of PIP2 to PIP3. PTEN catalyzes the conversion of PIP3 to PIP2. PIP3 is further recognized by AKT and PDPK1. The connection of PIP3 to PDPK1 and AKT allows the physical interaction of PDPK1 and AKT, which leads to activation of AKT by phosphorylation of the T308 residue. For maximal activation of AKT, phosphorylation of the S473 residue by mTORC2 is required. AKT phosphorylates GSK3, FOXO1, MDM2, BIM, and BAD. AKT also phosphorylates and inactivates TSC2, which subsequently allows RHEB to activate mTORC1. |
PI3K-enhancing mechanisms in breast cancer
PI3K alteration
PIK3CA mutations
Somatic mutations of PIK3CA coding p110α in various solid malignancies were first reported in 2004.3 Although the initial study reported that the frequency of mutations was relatively low in breast cancer (10%), later studies revealed that breast cancer was in fact among the most frequently affected cancers (~30%) (Table 1). The majority of PIK3CA somatic mutations are located in two “hot spots”: E542K or E545K in exon 9, and H1047R or H1047L in exon 20.3 Both types of mutation were shown to be gain-of-function mutations and to have transforming capacity.6,7 Exon 9 mutations are located in the helical domain of p110α, and are considered to enable p110α to escape the inhibitory effect of p85 via the Src-homology 2 (SH2) domain. Exon 20 mutations are located near the activation loop in the kinase domain, but the mechanism by which they promote constitutive PI3K signaling remains unclear.8,9
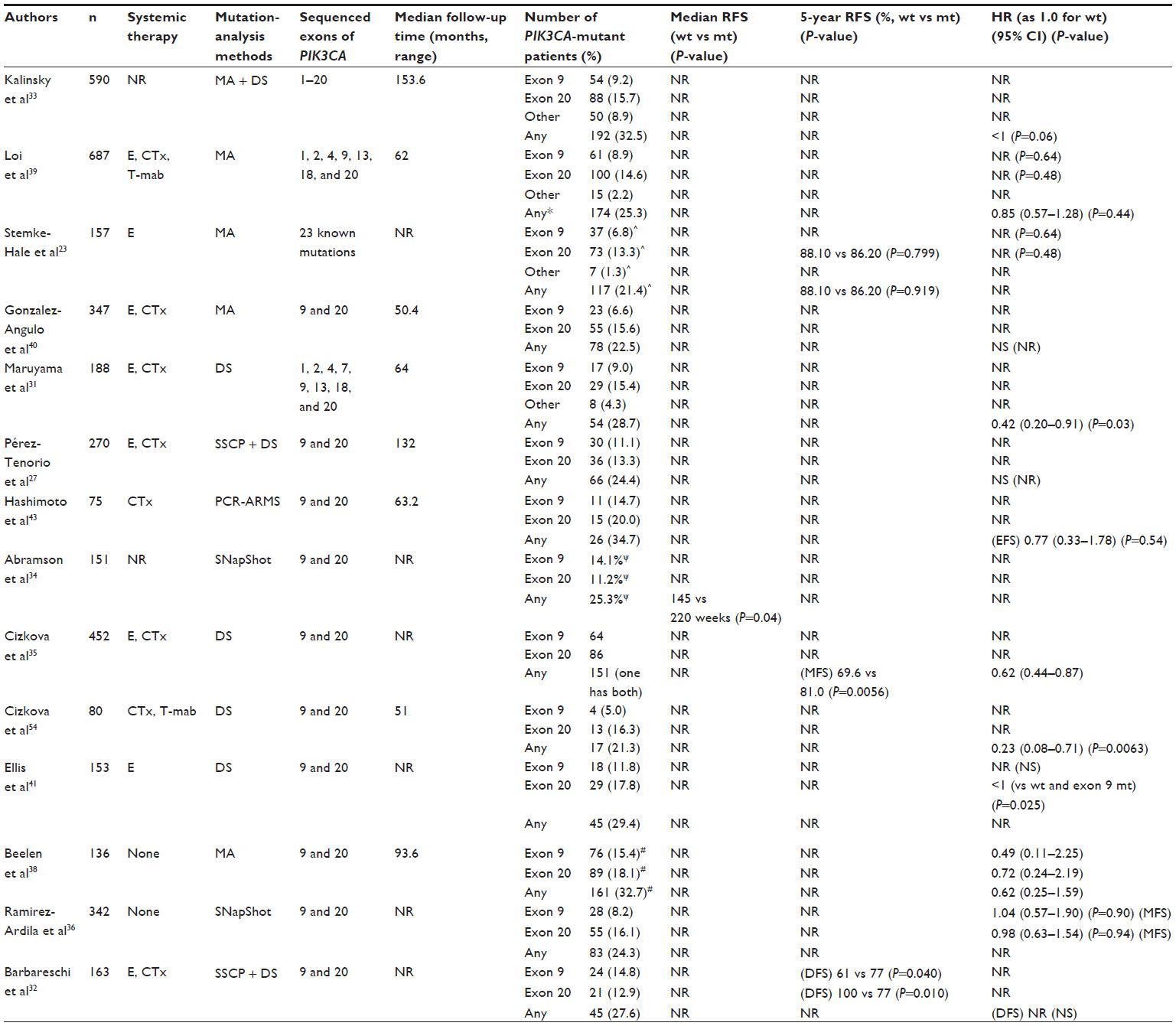 | Table 1 Studies evaluating prognostic impact of PIK3CA mutations with recurrence-free survival (RFS) as end point |
PIK3CA amplification
Preceding the discovery of PIK3CA mutations, gene amplification of PIK3CA was reported in various malignancies, including approximately 10% of cases of breast cancer.4,10 Like PIK3CA mutations, PIK3CA amplification has been shown to lead to increased PI3K activity.11
PIK3R1 mutation/low expression
The PIK3R1 gene product p85α appears to play a tumor-suppressor role by stabilizing p110α.12 PIK3R1 mutations have been found in breast cancer, although with much lower occurrence than PIK3CA mutations (~3%).12,13 PIK3R1 mutations are located at the inter-SH2 domain, which is required for connection of p85α to p110α. They are believed to cancel the inhibitory effect of p85α on p110α, eventually leading to PI3K hyperactivation.5 In contrast with the rarity of PIK3R1 mutations in breast cancer, reduced PIK3R1 messenger ribonucleic acid (RNA) expression, defined as <0.5-fold of normal breast tissue, is reported to be frequent (183 of 458 cases [61.8%]).12
Other PI3K-enhancing mechanisms
HER2 amplification
As described earlier, PI3K is activated by being connected with RTK directly or indirectly via adaptor proteins. Gene-amplified HER2 is the best-characterized RTK with regard to breast cancer tumorigenesis. While HER2 can theoretically form four different types of dimer (with HER1, HER2, HER3, or HER4), the HER2/HER3 heterodimer is thought to be the most mitogenic and transforming.14–17 HER3 is distinguished from other HER family members by two peculiar characteristics: it lacks tyrosine-kinase activity on its own, and it contains at least six docking domains for p85 of PI3K.18 These properties allow HER3 to function as a scaffold protein to efficiently trigger the PI3K pathway. In fact, a study has suggested that breast cancer cell lines expressing both HER2 and HER3 appear to have a higher degree of AKT phosphorylation.19 Other studies have suggested that the HER2/HER3/PI3K complex and subsequent PI3K–AKT signaling pathway play central roles in cell proliferation in HER2-amplified cells.20,21
PTEN dysfunction
Malignant tumors frequently show dysfunction of PTEN. While PTEN mutations are relatively uncommon in breast cancer (<5%), PTEN protein loss is frequent (~30%).22,23 This loss is reported to be caused by various mechanisms, such as promoter methylation, loss of heterozygosity, and regulation at the RNA or protein level.22,23
AKT1 mutation
AKT1 mutations (E17K) have been found in 1.4%–8% of breast cancers.12,23,24 Although this low frequency precludes the drawing of any definitive answers, a large-scale genotyping effort (547 breast tumor and 41 breast cancer cell lines) revealed that AKT1 mutations were exclusively observed in tumors expressing both ER and PgR.23
Relationship between PI3K-enhancing mechanisms in breast cancer
Molecular changes on the same signaling axis can be mutually exclusive in malignant tumors. If two genes are mutated in a mutually exclusive fashion in a certain type of cancer, it is likely that they provide the same selective pressure for clonal expansion. As an example, somatic mutations of EGFR and K-RAS and ALK rearrangements have been consistently shown to be mutually exclusive in lung adenocarcinoma.25 As shown later in this review, however, PIK3CA mutations and HER2 amplification often coexist. An early study demonstrated that PIK3CA mutations and PTEN loss, another PI3K-enhancing change frequently observed in breast cancer, were present in a nearly mutually exclusive fashion.26 More recent studies, however, denied this mutual exclusivity of the two molecular changes.27,28 PIK3CA mutations and gain of copy number of PIK3CA and PTEN loss and PTEN mutations have also been reported to coexist.11,23,28,29 On the other hand, PIK3CA, PTEN, AKT1, and PIK3R1 mutations are reported to be mutually exclusive, although the low frequency of the latter three limits the reliability of the finding.12,23 One study showed that PIK3CA mutations and reduced PIK3R1 messenger RNA expression are mutually exclusive.12
Collectively, while mutual exclusivity is maintained between some PI3K-enhancing molecular changes, three major types – HER2 amplification, PIK3CA mutations, and PTEN loss – appear to coexist, indicating that none of these three mechanisms may be sufficient to keep PI3K-pathway activity at a high oncogenic level.
PIK3CA mutations and clinicopathological factors
Correlation with hormone receptors and HER2
Among studies, the correlation of PIK3CA mutations with hormone-receptor status has received the most extensive investigation. A meta-analysis involving 26 studies found significant association between PIK3CA mutations and ER and PgR expressions (for ER, odds ratio [OR] 1.92, 95% confidence interval [CI] 1.65–2.23; for PgR, OR 1.88, 95% CI 1.61–2.20).30
In terms of HER2 status, an early study showed a positive correlation between PIK3CA mutation and HER2 overexpression.26 However, subsequent studies demonstrated no association of PIK3CA with HER2 status,31,32 or rather an association with HER2-negativity.27,33–36
Further, some studies found associations of PIK3CA mutations with good prognostic features, such as lower histological grade,33–35,37–39 non-triple-negative subtype,40 luminal A subtype,37 smaller tumor size,27,35,39 and lower levels of Ki67.37
Overall, it appears safe to say that PIK3CA mutations are most likely found in luminal-type (HR-positive/HER2-negative) tumors, in particular those with markers indicating less aggressive tumor characteristics.
Correlation of PIK3CA mutations with phosphorylated downstream proteins
Hot-spot PIK3CA mutations were found to be gain-of-function mutations, and resulted in higher phosphorylation of downstream signaling molecules on the PI3K pathway in preclinical studies.6,7 Several studies tried to address the clinical relevance of this higher phosphorylation using tumors obtained from patients. However, results of the correlation of PIK3CA mutations and level of phosphorylated AKT (Ser473 or Thr308), the most frequently used biomarker of PI3K-pathway activity, are controversial, with some studies showing positive correlation31,38,41 and others showing no correlation.27,42,43 Results of studies correlating PIK3CA mutations with phosphorylated mTOR (p-mTOR) expression have also been inconsistent, with positive correlations in some42 but not others.38 A study utilizing a reverse-phase protein array, which enables more comprehensive protein analysis, failed to show a difference between PIK3CA-mutant tumors and PIK3CA wild-type tumors in the phosphorylation of AKT, GSK3, mTOR, or p70S6K.23 Of note, phosphorylation of AKT, mTOR, and p70S6K was significantly higher in PTEN-low than PTEN-high tumors.23
Based on these inconsistent results, correlation between PIK3CA mutations and downstream protein activation must be considered inconclusive, at least in clinical specimens. These inconsistent results may be due to the lack of standardization in detection methods for the phosphorylated proteins and possible variation in the amount of phosphorylated residue preserved depending on sample condition.
Prognostic and predictive values of PIK3CA mutations
Prognostic factor vs predictive factor
A number of studies have evaluated the clinical relevance of PI3K mutations, some of which asked if they have prognostic or predictive value, or both. Prognostic and predictive factors are often confused. The original definition of a prognostic biomarker is a marker that provides information on the likely course of the cancer disease in an untreated individual. In contrast, predictive biomarkers are defined as markers that can be used to identify subpopulations of patients who are most likely to respond to a given therapy. Distinguishing these terms thus requires the use of biomarker-positive and -negative subgroups and treated and untreated subgroups (Figure 3). However, if time-independent end points such as response rate (RR) and pathologic complete response (p-CR) rate are considered, a cohort in which all patients are treated with a given therapy will provide a predictive biomarker, because p-CR rate as an example in untreated patients would have been 0, regardless of the biomarker status.
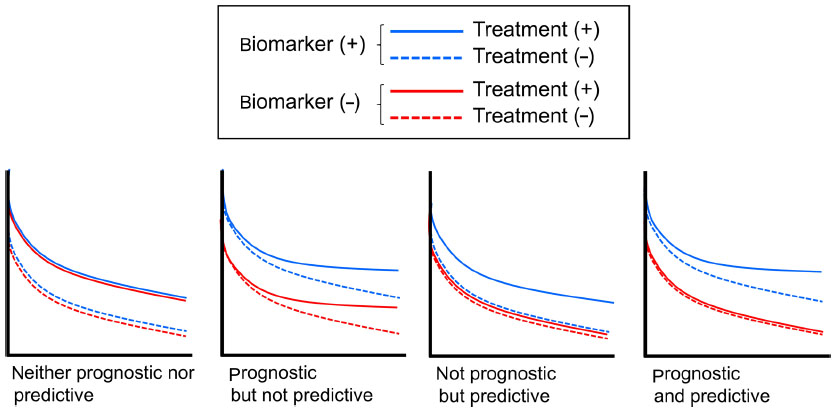 | Figure 3 Difference between prognostic and predictive biomarkers. |
Prognostic value of PIK3CA mutations
Prognostic value for early breast cancer: recurrence-free survival
Although few studies solely showed prognostic value of PIK3CA mutations for recurrence-free survival (RFS) with statistical significance, many of these indicated the tendency for patients with mutant PIK3CA to have better RFS (Table 1). In fact, one meta-analysis involving five studies listed in Table 127,31,33,39,40 demonstrated a significant correlation of PIK3CA mutation with better RFS (hazard ratio [HR] 0.76, 95% CI 0.59–0.98; P=0.03).30 Another meta-analysis involving six studies31,33,35,39,44,45 reached essentially the same conclusion (HR for disease-free survival [DFS] 0.72, 95% CI 0.57–0.91; P=0.006).46
It remains unclear, however, whether PIK3CA mutation itself biologically affects tumor character or is just a product of confounding effects between PIK3CA mutations and other good prognostic factors, such as ER positivity, HER2 negativity, lower Ki67, and lower histological grade, as described earlier. In addition, the great majority of patients involved in these meta-analyses received perisurgical treatment. Because any treatments could potentially affect the natural consequence of the disease, their involvement precludes evaluation of “pure” prognostic value of PIK3CA mutations. In fact, a recent study that collected tumor samples from ER-positive postmenopausal patients who had participated in randomized trials comparing adjuvant use of tamoxifen for 1–3 years with observation failed to show any impact of PIK3CA mutations on RFS among patients assigned to the observation arm (HR 0.62, 95% CI 0.25–1.59; P=0.32) (Table 1).38 Similarly, another study that analyzed PIK3CA genotype in 342 tumors obtained from nonmetastatic lymph node-negative patients who had not had adjuvant therapy did not find any difference in metastasis-free survival between PIK3CA-mutant tumors and wild-type tumors (Table 1).36
Prognostic value for early breast cancer: overall survival
Study results show significantly more variation for OS than RFS. The wide variety of results may reflect the influence of postrecurrence treatment, which varies astronomically. As a reference, a meta-analysis of seven studies indicated that PIK3CA mutations had no prognostic impact on OS (HR 1.14, 95% CI 0.72–1.82; P=0.57).30
Prognostic value for advanced breast cancer
Unlike the case for early breast cancer, the prognostic value of PIK3CA mutations for advanced cases has not been studied extensively. One study evaluated the prognostic and predictive value of PIK3CA mutations in patients who received paclitaxel alone or in combination with lapatinib in a prospective randomized trial setting.47 This study concluded that PIK3CA mutations were an adverse prognostic factor for survival, but were not predictive for lapatinib benefit.47 This result should be interpreted with caution, because postprogression therapies with other anti-HER2 drugs might have affected subsequent patient outcome.
Potentially different prognostic value between exon 9 and 20 PIK3CA mutations
Although both exon 9 and exon 20 hot-spot PIK3CA mutations were initially found to be gain-of-function and transforming mutations,6,7 more recent studies have noted differences in the protein partners required for PI3K activation and different tumorigenic potential in animal models between exon 9 and exon 20 mutations.48,49
The possibility that clinical implications differ between these mutation sites remains controversial. One study demonstrated that patients with exon 9-mutant tumors were independently associated with early recurrence and death, whereas patients with exon 20-mutant tumors were associated with better prognosis than those with PIK3CA wild-type tumors.32 Consistently, a recent study found that more exon 9-mutant patients experienced recurrence than exon 20-mutant patients (recurrence rate for exon 9 vs exon 20, 89% [39 of 44] vs 63% [22 of 35], P=0.007), while there was no difference in patient characteristics between the two sites of mutations.34 Another study also showed that the RFS Kaplan–Meier curve for exon 20-mutant patients runs above that for exon 9-mutant patients, though the difference did not achieve statistical significance.39 Collectively, these data suggest that exon 20 mutations might be a more favorable prognostic factor than exon 9 mutations, although further data collection is required to draw a definitive answer.
PI3K mutations in predicting the efficacy of systemic therapies
Anti-HER2 therapies
Previous studies have suggested that the HER2/HER3/PI3K complex and subsequent PI3K–AKT signaling pathway was critical in HER2-amplified cells, and shown that disruption of this complex was a molecular mechanism of action of the anti-HER2 monoclonal antibody trastuzumab.20,21 Theoretically therefore, PI3K gain-of-function mutations were hypothesized to cause resistance to trastuzumab and other HER2-targeting drugs. A series of preclinical studies tested this hypothesis by transfecting wild-type and mutant forms of PIK3CA in HER2-amplified breast cancer cells.50,51 Results showed that mutant PIK3CA transfection resulted in resistance to trastuzumab or lapatinib, a small-molecule HER2 inhibitor.50,51 Consistent with these previous studies, our study using HER2-amplified breast cancer cell lines showed that cell lines with PIK3CA hot-spot mutations were significantly more resistant to trastuzumab and CL-387,785, a small-molecule HER2 inhibitor, than those without mutations.52 However, another preclinical study using HER2-amplified breast cancer cell lines provided different conclusions. It showed that while PIK3CA mutations themselves did not correlate with in vitro resistance to either trastuzumab or lapatinib, PIK3CA mutations and/or PTEN-low cell lines as a group correlated with trastuzumab resistance, while still not affecting lapatinib sensitivity.53
Trastuzumab in early breast cancer: adjuvant setting
Several retrospective studies have supported the hypothesis generated by preclinical studies. A retrospective study analyzed the PIK3CA genotype in tumor samples from 240 HER2-positive early breast cancer patients who had been treated with cyclophosphamide, epirubicin, and fluorouracil followed by 1 year’s trastuzumab in an adjuvant trial setting.45 Results showed that patients with mutant PIK3CA had a shorter OS than those with wild-type PIK3CA (multivariate HR for OS 2.14, 95% CI 1.01–4.51; P=0.046), with no difference in DFS.45 Similarly, in another study, the PIK3CA genotype was analyzed in samples obtained from 80 HER2-positive patients who participated in a Phase II study in which all patients received perisurgical chemotherapy consisting of anthracycline-based combinations and docetaxel and 1 year’s trastuzumab. Results showed better DFS in patients with PIK3CA wild-type tumors than in those with PIK3CA-mutant tumors (HR for DFS 0.23, 95% CI 0.08–0.71; P=0.0063).54
However, studies from larger Phase III trials have produced different results. These studies were randomized trials that compared trastuzumab with no trastuzumab, which enables better distinction of the PIK3CA genotype as a prognostic factor from a predictive factor with regard to trastuzumab benefit, compared to studies where all patients received trastuzumab. In a biomarker study associated with the FinHER trial, a Phase III trial in which patients with HER2-amplified tumors were randomized to either 9 weeks’ trastuzumab or control combined with chemotherapy, tumors were genotyped for 20 genes, including PIK3CA.39 The study found no clear difference in benefit from trastuzumab therapy between PIK3CA-mutant and wild-type patients,39 though the sample size was relatively small (total 157; PIK3CA wild-type 123, PIK3CA-mutant 34) and the duration of trastuzumab treatment was shorter than the current standard of 1 year.39 More recently, a biomarker study evaluating the predictive value of the PIK3CA genotype in an adjuvant setting using 672 archived tumor samples from the National Surgical Adjuvant Breast and Bowel Project (NSABP) trial B-31 was reported.55 NSABP B-31 was a randomized Phase III trial that compared doxorubicin/cyclophosphamide followed by paclitaxel with the same regimen combined with 1 year’s trastuzumab starting from the paclitaxel phase.55 Results showed that the DFS benefit from trastuzumab in PIK3CA-mutant tumors (HR 0.44, 95% CI 0.24–0.82; P=0.009) was similar to the benefit in PIK3CA wild-type tumors (HR 0.51, 95% CI 0.37–0.71; P=001),55 suggesting a lack of predictive value for PIK3CA in trastuzumab efficacy, consistent with the FinHER trial.
Overall, it should be still considered inconclusive whether PIK3CA genotype has predictive value for a gain in DFS from adjuvant trastuzumab therapy (Table 2).
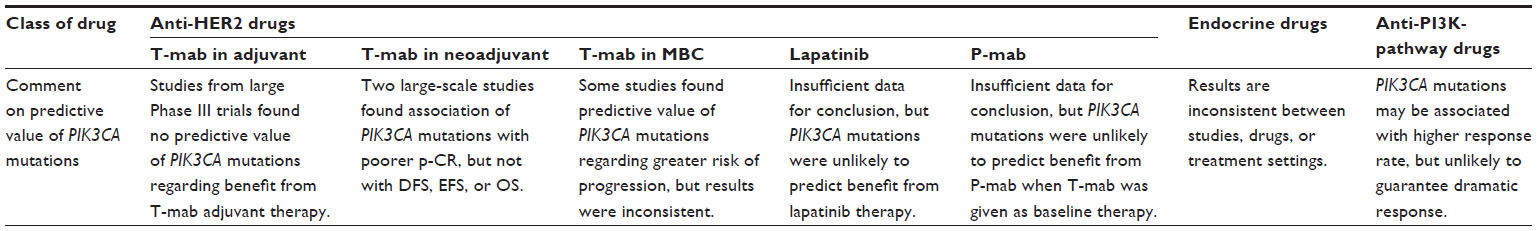 | Table 2 Summary of predictive value of PIK3CA mutations in clinical settings |
Trastuzumab in early breast cancer: neoadjuvant setting
Two recent large-scale studies evaluated the influence of PIK3CA genotypes on p-CR rate when trastuzumab was involved in neoadjuvant systemic therapy in HER2-positive patients. The first study evaluated the PIK3CA genotype in 504 tumor samples from participants in three independent neoadjuvant studies, in which all HER2-positive patients received either trastuzumab or lapatinib or this combination plus anthracycline–taxane chemotherapy.56 Results showed that PIK3CA mutation was significantly associated with a lower p-CR rate (PIK3CA-mutant vs wild-type, 19.4% vs 32.8%, OR 0.49, 95% CI 0.29–0.83; P=0.008) in the overall population.56 On classification by type of anti-HER2 therapy, p-CR rates in PIK3CA-mutant group were 16%, 24.3%, and 17.4% with lapatinib, trastuzumab, and their combination, respectively, and were 18.2%, 33.0%, and 37.1%, respectively, in the PIK3CA wild-type group. These results suggest that the influence of the PIK3CA genotype is greater in patients treated with trastuzumab-containing regimens than in those treated with lapatinib as the only HER2-targeted drug. Of note, this study did not find differences in DFS or OS between patients with mutant and wild-type PIK3CA (Table 2).56
The second biomarker study was accompanied by the NeoALTTO trial, a Phase III randomized neoadjuvant study of trastuzumab, lapatinib, or their combination in patients with early HER2-positive breast cancer.57 Patients received anti-HER2 therapy for 6 weeks. Paclitaxel was then added to the regimen for a further 12-week period until definitive surgery.57 The study produced similar results to those in the first study: a tendency for greater p-CR rates in PIK3CA-wild-type tumors than in wild-type tumors, particularly in the trastuzumab-containing group (p-CR rates with lapatinib, trastuzumab, and their combination of 14.8%, 20.0%, and 28.6%, respectively, in the PIK3CA-mutant group, and 20.4%, 28.4%, and 55.8%, respectively, in the PIK3CA wild-type group).57 In terms of event-free survival and OS, the PIK3CA genotype again had no influence (Table 2).57
Trastuzumab in metastatic breast cancer
An initial study analyzing the PIK3CA genotype in tumor samples obtained from HER2-positive metastatic breast cancer patients who had undergone trastuzumab-based therapy showed an association between the presence of PIK3CA mutations and reduced time to progression (TTP).50 Results of subsequent studies, however, were somewhat inconsistent, although not contradictory. A retrospective analysis of 137 HER2-positive breast tumor samples from patients who had been treated with trastuzumab-based therapies showed that PIK3CA mutation itself was not associated with the clinical benefit rate (CR + partial response [PR] + stable disease; PIK3CA wild-type vs mutant, 60% vs 58%; P=1.000), or OS (P=0.172).28 However, the clinical benefit rate was significantly reduced in patients with PIK3CA mutations and/or PTEN loss (P=0.047).28 In another retrospectively identified cohort of 139 patients with HER2-positive metastatic cancer treated with trastuzumab alone or in combination with chemotherapy, PIK3CA mutation was associated with increased risk of progression (HR 2.50, 95% CI 1.35–4.61; P=0.003), but not with OS. Multivariate analysis revealed PTEN loss and/or PIK3CA mutation as an independent single parameter associated with shorter TTP (HR 2.16, 95% CI 1.27–3.66; P=0.004) and reduced survival time from diagnosis of metastatic disease (HR 2.12, 95% CI 1.15–3.92; P=0.041).29
Lapatinib
The predictive value of the PIK3CA genotype for lapatinib is controversial from preclinical and clinical perspectives. In neoadjuvant lapatinib trials, as discussed earlier, the difference in clinical CR (c-CR) rate after lapatinib treatment between PIK3CA-mutant and wild-type groups was trivial.56,57 In a Phase II trial of lapatinib monotherapy in HER2-positive metastatic breast cancer, three PIK3CA-mutant patients were detected, and one durable PR and two stable disease responses were observed.58 Considered together with the inconsistent preclinical findings,53 the predictive value of the PIK3CA genotype for lapatinib may not be as high as that for trastuzumab (Table 2).
Pertuzumab
A biomarker study associated with the TRYPHAENA study, a randomized Phase II study comparing three treatment arms involving pertuzumab and trastuzumab in combination with chemotherapy, showed that patients carrying PIK3CA mutations tended to have a lower p-CR rate compared to patients with wild-type PIK3CA, although the difference did not reach statistical significance.59 This finding indicates that the addition of pertuzumab may not be sufficient to overcome trastuzumab resistance caused by PIK3CA mutations.
The CLEOPATRA study was a randomized Phase III trial comparing docetaxel and trastuzumab with the same regimen combined with pertuzumab for first-line chemotherapy in HER2-positive recurrent or metastatic breast cancer patients.60 Results of a biomarker study analyzing tumor samples obtained from these patients were recently published.61 These showed that the addition of pertuzumab to trastuzumab resulted in an improvement in progression-free survival in both the PIK3CA-mutant and PIK3CA wild-type groups,57 suggesting that the PIK3CA genotype does not predict a benefit from the addition of pertuzumab when trastuzumab is given as baseline treatment (Table 2).
Endocrine therapies
Because PIK3CA mutations are most frequently found in ER-positive tumors, the question of whether these mutations cause resistance to endocrine therapies is of great interest. In in vitro studies, it was shown that PI3K and AKT can activate ER in the absence of estrogen, and constitutively active AKT causes tamoxifen resistance.62 One study analyzed the correlation of the PIK3CA genotype and response to neoadjuvant endocrine therapy consisting of either tamoxifen, letrozole, or exemestane in three different clinical trials (n=235). Results showed that tumors with PIK3CA mutation tended not to respond to these therapies compared to those with wild-type PIK3CA (RR, PIK3CA-mutant [exon 9] vs PIK3CA-mutant [exon 20] vs PIK3CA wild-type, 14 of 25 [56%] vs 29 of 51 [57%] vs 111 of 159 [70%]; P=0.0459).41 The aforementioned study involving tamoxifen and observation groups, however, failed to show a predictive value of PIK3CA mutation measured with RFS from adjuvant tamoxifen treatment.38 Another study analyzing PIK3CA genotypes in tumors from 447 ER-positive metastatic patients who had undergone first-line tamoxifen treatment found no association of genotypes with treatment outcome measured by TTP.36 On the other hand, patients with PIK3CA-mutant tumors treated with first-line aromatase inhibitors showed a longer TTP than patients with PIK3CA wild-type tumors, though the sample size was small (n=84).36
Anti-PI3K pathway drugs
As of today, the mTOR inhibitor everolimus is the only drug targeting the PI3K pathway, which is indicated for breast cancer. While a preclinical study demonstrated that PIK3CA mutations can sensitize cells to everolimus,63 clinical validation remains to be done. In addition to mTOR inhibitors, numerous inhibitors targeting molecules on the PI3K pathway are under preclinical and clinical development.64 One study correlated the PIK3CA genotype and tumor response in patients who participated in Phase I studies of various PI3K/AKT/mTOR inhibitors.65 Results showed that patients with a PIK3CA H1047R mutation had a higher PR rate than those with other PIK3CA mutations or wild-type PIK3CA treated under the same protocols (six of 16 [38%] vs five of 50 [10%] vs 23 of 174, [13%], respectively; P≤0.02).65 Of note, RR in patients with exon 9 mutations was a mere 13.8%.65
Discussion
Vigorous investigations of PIK3CA mutations in breast cancer have led to the carving of several matters in stone with a certain level of consistency. Namely, PIK3CA mutations 1) are most likely to be observed in ER-positive/HER2-negative tumors, 2) can coexist with other PI3K-enhancing mechanisms, such as HER2 amplification and PTEN loss, 3) may potentially be a good prognostic marker, 4) may predict resistance to trastuzumab, and 5) do not guarantee a dramatic response to PI3K inhibitors.
In the clinic, prognostic biomarkers are used to select patients who have high risk for recurrence or relapse and thus need more potent treatment. As reviewed in this paper, many studies have found PIK3CA mutations to be good prognostic markers in patients with early breast cancer. However, it has to be emphasized that at this point PIK3CA should not be used to determine the intensity of systemic therapies for individual patients. This is because most studies that reported good prognostic impact of PIK3CA mutations were retrospectively conducted in a single hospital base, which limits their reliability and generalizability. In addition, most patients involved in these studies received certain systemic therapy, which precludes evaluation of prognostic biomarkers under their strict definition as “a marker that provides information on the likely course of the cancer disease in an untreated individual”. In fact, two studies that retrospectively genotyped PIK3CA in patients untreated after surgery failed to show any prognostic impact of PIK3CA mutations.32,36
On the other hand, predictive biomarkers can be used to select patients who are likely or unlikely to benefit from a certain treatment. Among studies conducted to evaluate the PIK3CA genotype as a predictive biomarker for various therapies, those involving trastuzumab-based therapies in the neoadjuvant setting provided the most consistent results: PIK3CA was associated with a lower p-CR rate.56,57 It is unclear, however, whether or not trastuzumab can be omitted in patients with HER2-positive/PIK3CA-mutant tumors, and a definitive answer will not be available in the future, because clinical trials employing a non-trastuzumab-containing arm are in effect impossible to conduct. Rather, the development of therapies to overcome trastuzumab resistance by PIK3CA mutations is awaited. One potential solution would be the combination of anti-HER2 drugs and PI3K-targeting drugs. Numerous PI3K-targeting drugs are under preclinical and clinical development, and HER2-positive/PIK3CA-mutant tumors are among their main targets.
Despite consistently lower p-CR rates in PIK3CA-mutant HER2-positive patients treated with trastuzumab-containing neoadjuvant therapies than in PIK3CA wild-type counterparts, no difference in DFS, a true end point for perisurgical systemic therapy, was observed in large adjuvant or neoadjuvant trials between PIK3CA-mutant and wild-type patients. This is seemingly contradictory, based on two widely accepted postulations in breast oncology: that systemic therapy provides an identical impact on DFS regardless of the timing of the therapy given (neoadjuvant vs adjuvant), and that p-CR is associated with better prognosis thereafter. However, it is not definitely contradictory, for the following reasons. First, because neoadjuvant = adjuvant is a proven concept established by trials involving only chemotherapy, it may be inapplicable to targeted therapies. Recently, the ALLTO trial comparing trastuzumab and a trastuzumab plus lapatinib combination in an adjuvant setting failed to show an advantage for the combination in DFS, despite a higher p-CR rate in the identical combination arm in the NeoALTTO trial.66 Second, while p-CR is consistently associated with a better prognosis after therapy on an individual patient basis, it was recently shown that impact on p-CR rate does not necessarily correlate with impact on DFS when analyzed on a trial basis.67
At the time of discovery of frequent PIK3CA somatic mutations in 2004,3 which happened almost simultaneously with the development of clinically applicable PI3K inhibitors, it was expected that mutated PI3K would become a “home run” target, providing dramatic tumor regression when targeted, as had already been witnessed in EGFR-mutant lung adenocarcinoma and KIT-mutant gastrointestinal stromal tumors. However, the results of early clinical trials of various PI3K-targeting drugs suggested that this would not be the case (Table 2): RR was a mere 17% in PIK3CA-mutant tumors, albeit somewhat higher than in PIK3CA wild-type tumors.65 As discussed in this review, PIK3CA mutations can be coobserved with other PI3K-enhancing molecular changes, such as HER2 amplification and PTEN loss, which suggests that PIK3CA mutations themselves solely indicate a high level of cellular addiction to the pathway. In addition, adaptive compensative responses after inhibition of one pathway have been revealed to be important, so combination therapies targeting multiple signaling axes may be one way to maximize the effect.
Conclusion
In conclusion, there is presently no sufficient evidence to support the clinical usefulness of PIK3CA genotyping in daily practice. Given that PIK3CA-mutant breast cancer appears to have distinct tumor biology, the development of more individualized targeted therapies, such as a combination of two or more targeted drugs, based on PIK3CA genotype is awaited.
Disclosure
The author reports no conflicts of interest in this work.
References
Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. | |
Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thurlimann B, Senn HJ. Strategies for subtypes – dealing with the diversity of breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol. 2011;22(8):1736–1747. | |
Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. | |
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–644. | |
Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. | |
Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110α and p110β phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A. 2005; 102(51):18443–18448. | |
Isakoff SJ, Engelman JA, Irie HY, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65(23):10992–11000. | |
Miled N, Yan Y, Hon WC, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317(5835):239–242. | |
Huang CH, Mandelker D, Schmidt-Kittler O, et al. The structure of a human p110α/p85α complex elucidates the effects of oncogenic PI3Kα mutations. Science. 2007;318(5857):1744–1748. | |
Gonzalez-Angulo AM, Chen H, Karuturi MS, et al. Frequency of mesenchymal-epithelial transition factor gene (MET) and the catalytic subunit of phosphoinositide-3-kinase (PIK3CA) copy number elevation and correlation with outcome in patients with early stage breast cancer. Cancer. 2013;119(1):7–15. | |
Wu G, Xing M, Mambo E, et al. Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res. 2005;7(5):R609–R616. | |
Cizkova M, Vacher S, Meseure D, et al. PIK3R1 underexpression is an independent prognostic marker in breast cancer. BMC Cancer. 2013;13:545. | |
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. | |
Wallasch C, Weiss FU, Niederfellner G, Jallal B, Issing W, Ullrich A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995;14(17):4267–4275. | |
Zhang K, Sun J, Liu N, et al. Transformation of NIH 3T3 cells by HER3 or HER4 receptors requires the presence of HER1 or HER2. J Biol Chem. 1996;271(7):3884–3890. | |
Pinkas-Kramarski R, Lenferink AE, Bacus SS, et al. The oncogenic ErbB-2/ErbB-3 heterodimer is a surrogate receptor of the epidermal growth factor and betacellulin. Oncogene. 1998;16(10):1249–1258. | |
Alimandi M, Romano A, Curia MC, et al. Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene. 1995;10(9):1813–1821. | |
Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341–354. | |
Knuefermann C, Lu Y, Liu B, et al. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003;22(21):3205–3212. | |
Junttila TT, Akita RW, Parsons K, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15(5):429–440. | |
Lee-Hoeflich ST, Crocker L, Yao E, et al. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 2008;68(14):5878–5887. | |
Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004. | |
Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68(15):6084–6091. | |
Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007; 448(7152):439–444. | |
Gainor JF, Varghese AM, Ou SH, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res. 2013;19(15):4273–4281. | |
Saal LH, Holm K, Maurer M, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65(7):2554–2559. | |
Pérez-Tenorio G, Alkhori L, Olsson B, et al. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin Cancer Res. 2007;13(12):3577–3584. | |
Esteva FJ, Guo H, Zhang S, et al. PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol. 2010;177(4):1647–1656. | |
Razis E, Bobos M, Kotoula V, et al. Evaluation of the association of PIK3CA mutations and PTEN loss with efficacy of trastuzumab therapy in metastatic breast cancer. Breast Cancer Res Treat. 2011;128(2):447–456. | |
Pang B, Cheng S, Sun SP, et al. Prognostic role of PIK3CA mutations and their association with hormone receptor expression in breast cancer: a meta-analysis. Sci Rep. 2014;4:6255. | |
Maruyama N, Miyoshi Y, Taguchi T, Tamaki Y, Monden M, Noguchi S. Clinicopathologic analysis of breast cancers with PIK3CA mutations in Japanese women. Clin Cancer Res. 2007;13(2 Pt 1):408–414. | |
Barbareschi M, Buttitta F, Felicioni L, et al. Different prognostic roles of mutations in the helical and kinase domains of the PIK3CA gene in breast carcinomas. Clin Cancer Res. 2007;13(20):6064–6069. | |
Kalinsky K, Jacks LM, Heguy A, et al. PIK3CA mutation associates with improved outcome in breast cancer. Clin Cancer Res. 2009;15(16):5049–5059. | |
Abramson VG, Cooper Lloyd M, Ballinger T, et al. Characterization of breast cancers with PI3K mutations in an academic practice setting using SNaPshot profiling. Breast Cancer Res Treat. 2014;145(2):389–399. | |
Cizkova M, Susini A, Vacher S, et al. PIK3CA mutation impact on survival in breast cancer patients and in ERα, PR and ERBB2-based subgroups. Breast Cancer Res. 2012;14(1):R28. | |
Ramirez-Ardila DE, Helmijr JC, Look MP, et al. Hotspot mutations in PIK3CA associate with first-line treatment outcome for aromatase inhibitors but not for tamoxifen. Breast Cancer Res Treat. 2013;139(1):39–49. | |
López-Knowles E, Segal CV, Gao Q, et al. Relationship of PIK3CA mutation and pathway activity with antiproliferative response to aromatase inhibition. Breast Cancer Res. 2014;16(3):R68. | |
Beelen K, Opdam M, Severson TM, et al. PIK3CA mutations, phosphatase and tensin homolog, human epidermal growth factor receptor 2, and insulin-like growth factor 1 receptor and adjuvant tamoxifen resistance in postmenopausal breast cancer patients. Breast Cancer Res. 2014;16(1):R13. | |
Loi S, Michiels S, Lambrechts D, et al. Somatic mutation profiling and associations with prognosis and trastuzumab benefit in early breast cancer. J Natl Cancer Inst. 2013;105(13):960–967. | |
Gonzalez-Angulo AM, Stemke-Hale K, Palla SL, et al. Androgen receptor levels and association with PIK3CA mutations and prognosis in breast cancer. Clin Cancer Res. 2009;15(7):2472–2478. | |
Ellis MJ, Lin L, Crowder R, et al. Phosphatidyl-inositol-3-kinase alpha catalytic subunit mutation and response to neoadjuvant endocrine therapy for estrogen receptor positive breast cancer. Breast Cancer Res Treat. 2010;119(2):379–390. | |
Lazaridis G, Lambaki S, Karayannopoulou G, et al. Prognostic and predictive value of p-Akt, EGFR, and p-mTOR in early breast cancer. Strahlenther Onkol. 2014;190(7):636–638, 640–645. | |
Hashimoto K, Tsuda H, Koizumi F, et al. Activated PI3K/AKT and MAPK pathways are potential good prognostic markers in node-positive, triple-negative breast cancer. Ann Oncol. 2014;25(10):1973–1979. | |
Bozhanov SS, Angelova SG, Krasteva ME, et al. Alterations in p53, BRCA1, ATM, PIK3CA, and HER2 genes and their effect in modifying clinicopathological characteristics and overall survival of Bulgarian patients with breast cancer. J Cancer Res Clin Oncol. 2010;136(11):1657–1669. | |
Jensen JD, Knoop A, Laenkholm AV, et al. PIK3CA mutations, PTEN, and pHER2 expression and impact on outcome in HER2-positive early-stage breast cancer patients treated with adjuvant chemotherapy and trastuzumab. Ann Oncol. 2012;23(8):2034–2042. | |
Liu YR, Jiang YZ, Zuo WJ, Yu KD, Shao ZM. PIK3CA mutations define favorable prognostic biomarkers in operable breast cancer: a systematic review and meta-analysis. Onco Targets Ther. 2014;7:543–552. | |
Xu B, Guan Z, Shen Z, et al. Association of phosphatase and tensin homolog low and phosphatidylinositol 3-kinase catalytic subunit alpha gene mutations on outcome in human epidermal growth factor receptor 2-positive metastatic breast cancer patients treated with first-line lapatinib plus paclitaxel or paclitaxel alone. Breast Cancer Res. 2014;16(4):405. | |
Bader AG, Kang S, Vogt PK. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci U S A. 2006;103(5):1475–1479. | |
Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110α of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A. 2008;105(7):2652–2657. | |
Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. | |
Eichhorn PJ, Gili M, Scaltriti M, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68(22):9221–9230. | |
Kataoka Y, Mukohara T, Shimada H, Saijo N, Hirai M, Minami H. Association between gain-of-function mutations in PIK3CA and resistance to HER2-targeted agents in HER2-amplified breast cancer cell lines. Ann Oncol. 2010;21(2):255–262. | |
O’Brien NA, Browne BC, Chow L, et al. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol Cancer Ther. 2010;9(6):1489–1502. | |
Cizkova M, Dujaric ME, Lehmann-Che J, et al. Outcome impact of PIK3CA mutations in HER2-positive breast cancer patients treated with trastuzumab. Br J Cancer. 2013;108(9):1807–1809. | |
Pogue-Geile KL, Song N, Jeong JH, et al. Intrinsic Subtypes, PIK3CA Mutation, and the Degree of Benefit From Adjuvant Trastuzumab in the NSABP B-31 Trial. J Clin Oncol. Epub January 5, 2015. | |
Loibl S, von Minckwitz G, Schneeweiss A, et al. PIK3CA mutations are associated with lower rates of pathologic complete response to anti-human epidermal growth factor receptor 2 (HER2) therapy in primary HER2-overexpressing breast cancer. J Clin Oncol. 2014;32(29):3212–3220. | |
Majewski IJ, Nuciforo P, Mittempergher L, et al. PIK3CA mutations are associated with decreased benefit to neoadjuvant human epidermal growth factor receptor 2-targeted therapies in breast cancer. J Clin Oncol. Epub January 5, 2015. | |
Toi M, Iwata H, Fujiwara Y, et al. Lapatinib monotherapy in patients with relapsed, advanced, or metastatic breast cancer: efficacy, safety, and biomarker results from Japanese patients phase II studies. Br J Cancer. 2009;101(10):1676–1682. | |
Schneeweiss A, Chia S, Hegg R, et al. Evaluating the predictive value of biomarkers for efficacy outcomes in response to pertuzumab- and trastuzumab-based therapy: an exploratory analysis of the TRYPHAENA study. Breast Cancer Res. 2014;16(4):R73. | |
Baselga J, Cortés J, Kim SB, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366(2):109–119. | |
Baselga J, Cortes J, Im SA, et al. Biomarker analyses in CLEOPATRA: a phase III, placebo-controlled study of pertuzumab in human epidermal growth factor receptor 2-positive, first-line metastatic breast cancer. J Clin Oncol. 2014;32(33):3753–3761. | |
Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. 2001;276(13):9817–9824. | |
Mohseni M, Park BH. PIK3CA and KRAS mutations predict for response to everolimus therapy: now that’s RAD001. J Clin Invest. 2010;120(8):2655–2658. | |
Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13(6):224. | |
Janku F, Wheler JJ, Naing A, et al. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013;73(1):276–284. | |
Baselga J, Bradbury I, Eidtmann H, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2012;379(9816):633–640. | |
Berruti A, Amoroso V, Gallo F, et al. Pathologic complete response as a potential surrogate for the clinical outcome in patients with breast cancer after neoadjuvant therapy: a meta-regression of 29 randomized prospective studies. J Clin Oncol. 2014;32(34):3883–3891. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.