")
Back to Journals » International Journal of Nanomedicine » Volume 14
Physical PEGylation Enhances The Cytotoxicity Of 5-Fluorouracil-Loaded PLGA And PCL Nanoparticles
Authors Ashour AE , Badran M, Kumar A, Hussain T, Alsarra IA , Yassin AEB
Received 16 July 2019
Accepted for publication 15 October 2019
Published 28 November 2019 Volume 2019:14 Pages 9259—9273
DOI https://doi.org/10.2147/IJN.S223368
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Abdelkader E Ashour,1,2 Mohammad Badran,3 Ashok Kumar,4 Tajamul Hussain,5 Ibrahim A Alsarra,3 Alaa Eldeen B Yassin3,6,7
1Department of Pharmacology and Toxicology, College of Pharmacy, King Saud University, Riyadh, Saudi Arabia; 2Department of Basic Medical Sciences, Kulliyyah of Medicine, International Islamic University Malaysia, Kuantan 25200, Pahang Darul Makmur, Malaysia; 3Department of Pharmaceutics, College of Pharmacy, King Saud University, Riyadh, Saudi Arabia; 4Vitiligo Research Chair, College of Medicine, King Saud University, Riyadh, Saudi Arabia; 5Center of Excellence in Biotechnology Research, King Saud University, Riyadh, KSA; 6Pharmaceutical Sciences Department, College of Pharmacy-3163, King Saud Bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia; 7King Abdullah International Medical Research Center, Ministry of National Guard, Health Affairs, Riyadh, Saudi Arabia
Correspondence: Alaa Eldeen B Yassin
College of Pharmacy-3163 King Saud bin Abdulaziz University for Health Sciences PO Box: 3660, Riyadh 11481, Saudi Arabia
Tel +966509426323
Fax +966114295058
Email [email protected]
Purpose: The main goal of this study is to evaluate the impact of physical incorporation of polyethylene glycol (PEG) into 5-fluorouracil (5-FU)-loaded polymeric nanoparticles (NPs).
Methods: The 5-FU-loaded NPs were prepared utilizing a simple double emulsion method using polycaprolactone (PCL) and polylactic-co-glycolic acid (PLGA) with or without PEG 6000. The surface charge, particle size, and shape of NPs were evaluated by standard procedures. Both Fourier Transform Infrared Spectroscopy and X-ray diffraction spectra of the 5-FU loaded NPs were compared against the pure 5-FU. The in vitro release profile of 5-FU from the NPs was monitored by the dialysis tubing method. Cell death and apoptosis induction in response to 5-FU NP exposure were measured by MTT and Annexin–V/7-amino-actinomycin D (7-AAD) assays, respectively, in Daoy, HepG2, and HT-29 cancer cell lines.
Results: The 5-FU loaded NPs were found to be spherical in shape with size ranging between 176±6.7 and 253.9±8.6 nm. The zeta potential varied between −7.13± 0.13 and −27.06±3.18 mV, and the entrapment efficiency was between 31.96% and 74.09%. The in vitro release of the drug followed a two-phase mode characterized by rapid release in the first 8 hrs followed by a period of slow release up to 72 hrs with composition-based variable extents. Cells exposed to NPs demonstrated a significant cell death which correlated with the ratio of PEG in the formulations in Daoy and HepG2 cells but not in HT-29 cells. Formulations (F1–F3) significantly induced early apoptosis in HT-29 cell lines.
Conclusion: The physical PEGylation significantly enhanced the entrapment and loading efficiencies of 5-FU into NPs formulated with PLGA and PCL. It also fostered the in vitro cytotoxicity of 5-FU-loaded NPs in both Daoy and HepG2 cells. Induction of early apoptosis was confirmed for some of the formulations.
Keywords: hepatocellular carcinoma HepG2, emulsification-solvent evaporation technique, colorectal carcinoma HT-29, MTT assay, apoptosis
Introduction
Cancer nanomedicine is considered a relatively recent interdisciplinary research area cutting across chemistry, engineering, pharmaceutical drug delivery, and medicine with the goal to drive major enhancement in both diagnosis and treatment of cancer.1,2 During the last decade, many research articles were published presenting smart targeted nano-drug delivery particulate systems for specific ways of enhanced tumor therapy and imaging where many reviews have tried to define and classify those NPs systems.3,4 They all agreed to describe such systems as nano-sized composites able to incorporate drug(s) and/or contrast agent(s) and they can be either nanocarriers or nanovectors.5 They were particularly composed of a scaffold mainly made from a corona of polymers able to enhance many biopharmaceutical and pharmacokinetic properties of incorporated drugs or contrast agents. They can also have a ligand attached to their surfaces that provides targeting for a specific cancer biomarker overexpressed in certain cancer cells.6
Nanoparticulate drug delivery is an evolving research field aiming to apply the beneficial attributes of nanotechnology in developing new advantageous therapeutic systems including polymeric biodegradable nanoparticles,7 ceramic nanoparticles,8 polymeric micelles,9 liposomes,10 dendrimers,11 and solid lipid nanoparticles.12
Nanoparticulate systems made of biodegradable polymers are considered effective controlled release drug delivery systems in which a drug is entrapped, encapsulated, or adsorbed. Lactic acid and glycolic acid-based polymers, and their hybrid co-polymer PLGA are considered the most commonly used biodegradable polymers due to their biocompatible, nontoxic properties and their ability to prolong drug residence time at the target sites.13
The 5-FU is a pyrimidine analogue antimetabolite, which is used in the treatment of many types of cancer including anal, colorectal, stomach, pancreatic, breast, and head and neck cancers.14 It was listed by World Health Organization (WHO) among the most important drugs needed in a basic health system.15 The 5-FU is considered a narrow therapeutic index drug with very close minimum effective dose and maximum tolerated dose, in addition to its high inter-subject pharmacokinetic variability.16,17 Because of the high metabolism rate, the drug elimination half-life is usually less than 20 mins, and this requires a continuous administration of large doses and consequently resulting in multiple adverse effects such as severe nausea and vomiting, diarrhea, and severe anemia.18–20 The anticancer effects of 5-FU are highly dependent on the duration of tumor exposure to the drug and not on the blood levels of the drug.21 The incorporation of 5-FU in a nanoparticulate drug delivery would have a positive impact on the anticancer therapeutic efficacy through prolonging the drug retention time inside the body and thereby reducing the dose and all dose-related adverse effects. In addition to the high tendency of nanoparticles to accumulate in tumors as a result of the enhanced permeation and retention (EPR) effect, a tumor characteristic that has been exploited by Matsumura and Maeda22 as a mean to target anticancer agents to solid tumors.
In the present study, we investigated the impact of physical PEGylation of biodegradable polymeric nanoparticles on the anticancer activity of 5-FU NPs in medulloblastoma (Daoy), hepatocellular carcinoma (HepG2), and colorectal carcinoma (HT-29) cell lines.
Materials And Methods
Materials
Poly(D,L-lactide-coglycolide) (50:50) (Mw: 12,000 g/mol (PLGA), Polycaprolactone (PCL; Mw: 42,000 Da), MTT (3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide), Poly (ethylene glycol) PEG Mn average 6000, and 5-FU were purchased from Sigma-Aldrich Chemical Co. (St Louis, MO). Polyvinyl alcohol (PVA) with a molecular weight of 16,000 and dichloromethane (DCM) were obtained from Acros Organics (Geel, Belgium).
DMEM and DMEM/F12, FBS, L-glutamine, and penicillin/streptomycin were obtained from Gibco Inc. (NY, USA). Annexin V-Phycoerythrin (PE) kit/7-amino-actinomycin D (7-AAD) was obtained from Invitrogen (CA, USA). All other reagents and chemicals were of analytical grade.
Preparation Of 5-FU-Loaded Nanoparticles
The 5-FU NPs were prepared with 6 different formulation compositions described as F1, F2, F3, F4, F5, and F6 to optimize high drug entrapment efficiency and low particle size, and to evaluate the impact of physical incorporation of PEG. Table 1 shows the composition of each of the six formulations.
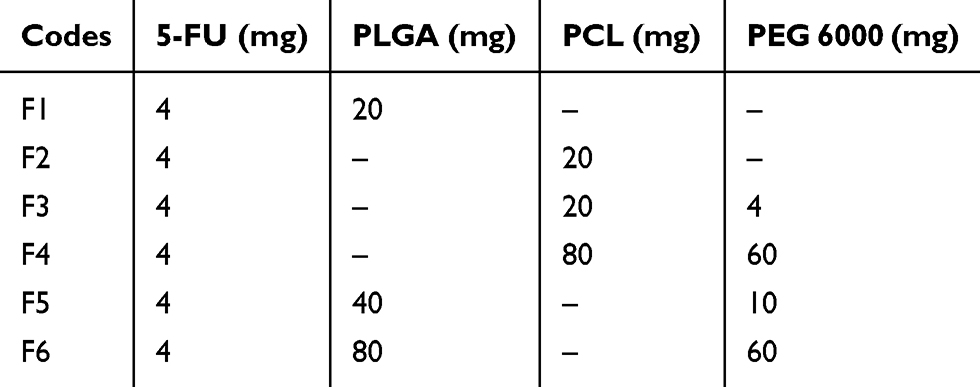 |
Table 1 The Composition Of 5-Fluorouracil (5-FU)-Loaded Nanoparticles |
The double emulsion-solvent evaporation method was employed for loading 5-FU into polymeric NPs a reported previously.23
Briefly, an aqueous solution of 5-FU (4mg/mL) was prepared in purified water by vortex mixing. The organic phase was prepared by dissolving the required weights of polymer(s) in 10 mL of DCM. One mL of 5-FU solution was emulsified in 10 mL of DCM using a probe-sonicator for 1 min at 40% power under ice bath. The secondary emulsion was formed by emulsifying the primary emulsion into 40 mL of an aqueous solution of 1% PVA using another 3-min probe-sonication period. The mixture was stirred at room temperature (22°C) and at 500 rpm under hood for at least 3 hrs to remove the DCM. The NPs dispersion was subjected to three cycles of centrifugation at 10,000 rpm for 10 mins followed by decantation of the supernatant solution and re-dispersion in double-distilled water. The supernatant solutions were analyzed for drug contents by HPLC analysis and used to calculate % entrapment efficiency (EE%). The residue was dispersed in 1.2% sodium lauryl sulfate solution by vortexing for 5 mins and lyophilized using a freeze dryer (Alpha 1–4 LD Plus, Martin Christefriertrocknugsanlagen GmbH, Osterode am Harz, Germany) at −60°C for 3 days. All formulations were prepared in triplicate.
Particle Size And Zeta Potential
A Zetasizer Nano ZS (Malvern Instruments, UK) was utilized for monitoring the mean particle size and polydispersity index (PDI) of each formulation at the 25°C after proper dilution. The same instrument was also used for measuring the zeta potential by applying the Laser Doppler Velocimetry (LDV) mode at 25°C. All experiments were performed in triplicate. Each value reported is the average of five measurements.
Entrapment Efficiency And Drug Loading
The efficiency of 5-FU entrapment into NP formulations was indirectly obtained by determining the concentration of 5-FU in the supernatant obtained after subjecting them to ultracentrifugation for 30 mins at 40,000 rpm in OptimaTM Max-E, Ultra Centrifuge (Beckman Coulter, Pasadena, CA) at 4°C. The amount of non-entrapped 5-FU in the supernatant was determined by an HPLC method.
The following Equations 1 and 2 were used to calculate both entrapment efficiency (EE%) and drug loading (DL%) respectively:
(1) (2)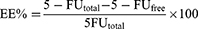

The Particle Morphology
The particle shape and surface topography of the 5-FU-loaded NP formulations were examined by scanning electron microscopy (JSM-6360 LV, JEOL, Tokyo, Japan). The dried samples were first fixed on carbon tape and then subjected to gold coating under argon atmosphere applying a gold sputter module in a high-vacuum evaporator (JFC-1100 fine coat ion sputter; JEOL). The gold-coated samples were then examined at different magnification levels. Photomicrographs were taken at an acceleration voltage of 10 kV.
X-Ray Diffraction
Powder X-ray diffraction (PXRD) was utilized to monitor any changes in the crystalline nature of 5-FU-loaded NP in comparison to pure powder. Wide-angle X-ray diffractometer (Rigaku Ultima IV, Tokyo, Japan) was employed to monitor PXRD patterns using CuKα radiation. A 2 range of 5–60° was set for sample scanning.
Fourier Transform Infrared Spectroscopy (FTIR)
FTIR (Thermo Scientific, Nicolet 380, USA) was employed for monitoring any changes in the FTIR spectra of NPs loaded 5-FU from the pure powder using the potassium bromide (KBr) disc technique. Simply, a small portion of each sample was mixed with KBr and discs were obtained by compression using a hydraulic press. Scanning of discs was done from 5000 to 400 cm−1 using transmission model.
In Vitro Release Study
Dialysis membrane bag method was employed to monitor in vitro release profile of 5-FU from NP formulations. Phosphate-buffered saline (PBS, pH 7.4) was used as the receptor compartment. Briefly, freeze-dried NPs equivalent to 5 mg of the 5-FU were dispersed in 1 mL distilled water and instilled into the dialysis bag (molecular weight cut off: 5 kDa, Livingstone, NSW, Australia) which were placed in a beaker containing 40 mL of preheated PBS, pH 7.4. The beakers were kept in a thermostatic shaker water bath at 37°C and 100 rpm. Five mg of pure 5-FU processed similarly was used as a control. Three beakers were used for each formulation. At certain time intervals, 3 mL samples were withdrawn from each beaker and replaced by the same volume of fresh media pre-heated at 37°C to maintain sink condition. The amount of 5-FU in each sample was determined using an HPLC method.
HPLC Analysis
A reverse-HPLC method reported earlier by Alanzi et al24 was used for the analysis of 5-FU in each sample. The HPLC system (WatersTM 600 controller Milford, MA, USA) containing a dual λ UV detector (WatersTM 2487 detector Milford, MA, USA). The system contains a binary pump (WatersTM 1252 pump Milford, MA, USA) and an automating sampling system (WatersTM 717 Plus Milford, MA Autosampler, USA). A mobile phase composed of 40 mM phosphate buffer adjusted to pH 7.0 by 10% w/v potassium hydroxide was used and samples were pumped through C18 column (BondapakTM, 4.6 × 150 mm, 10 m particle size) at a rate of 1 mL/min. The injection volume was 20 μL and the UV detector was adjusted at λ=260 nm. All the operations were carried out at ambient temperature.
Cell Lines, Cell Culture, And Chemicals
Three cancer cell lines were utilized in this study, namely the hepatocellular carcinoma HepG2, the human colorectal cancer HT-29, and medulloblastoma Daoy cells. These cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). HepG2 and HT-29 cells were aseptically cultured in DMEM/high glucose supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine, and 1% penicillin–streptomycin. Daoy cells were aseptically grown in DMEM/F12 medium, supplemented with 1% non-essential amino acids, 0.04% HEPES, in addition to the above-mentioned additives. All cells were maintained in a humidified atmosphere of 5% C02 at 37°C. Cells were kept in the logarithmic growth phase by routine passage every 3–4 days utilizing 0.025% trypsin-EDTA treatment. 5-FU was purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). 5-FU and 5FU-loaded NPs were dispersed in phosphate-buffered saline (PBS, pH 7.4) and used in the in vitro cytotoxicity and apoptosis studies at 5-FU concentration of 2.5 µg/mL which is equal to 19.22 µM. NP dispersions were prepared by weighing the equivalent weights from the freeze-dried powders under aseptic condition and dispersed in the required volume of PBS previously syringe-filtered using 0.45 µm membrane filters to ensure sterility. The dispersion was further sonicated for 30 mins using a bath sonicator.
In Vitro Cytotoxicity Studies
The cytotoxicity of 5-FU-loaded NPs was assessed by MTT assay in Daoy, HepG2, and HT-29 cells.25 Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) and DMEM/F12 at a density of 1–2 × 104 cells per well in 96-well plates in a humidified atmosphere of 5% CO2 at 37°C for 24 hrs. Following this, culture media were replaced with fresh DMEM media containing 5% FBS and cells were exposed to different 5-FU NP formulations containing 5-FU (2.5 μg/mL which is equivalent to 19.22 μM), pure 5-FU (2.5 μg/mL) or unloaded NPs for 24, 48, or 72 hrs. The culture media were replaced with 100 μL/well of MTT solution [0.5 mg/mL in phosphate-buffered saline (PBS)] and cells were further incubated for 3 hrs. Following this, MTT solution was removed and 100 μL of isopropyl alcohol was added to each well and the plates were left on a shaker for 2 hr at room temperature. The absorbance was measured at 549 nm in an ELISA reader (ELX 800; Bio-Tek Instruments, Winooski, VT, USA). The resulting absorbance was compared with the mean absorbance of the control wells containing only buffer in order to determine the cell viability. The following equation was used to calculate the cell viability.26
(3)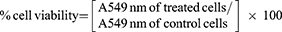
Detection Of Apoptosis By Flow Cytometry
Annexin-V-PE/7-AAD assay was employed to monitor the early apoptosis induced by 5-FU-loaded NPs as described earlier.27,28 Briefly, cells were grown as described above. Culture media were removed from the wells and cells were washed once with PBS, binding buffer (500 μL/well)(Invitrogen, CA, USA) and Annexin-V-PE (7 μL/well) (Invitrogen, CA, USA) were added to each well and the plate was incubated at room temperature for 10 mins. After this, 7 μL of 7-AAD was added to each well, and plates were further incubated at room temperature for another 10 mins. Excess label was washed with binding buffer and cells were harvested by scraping in binding buffer (700 μL/well). Cells were immediately analyzed by flow cytometry on Cytomics FC 500 (Beckman Coulter, Fullerton, CA, USA), and CXP acquisition and analysis software was used for data processing. The fluorescence of Annexin V-PE was analyzed on FL2 (575 nm) after exciting cells with a 488 nm argon laser line whereas fluorescence of 7-AAD was analyzed on FL4 (608 nm), as reported by Merkel et al (2008).29
Statistical Data Analysis
The software package Microsoft Excel, Version 2010, Origin software, version 6.1 and GraphPad InStat version 3 was used for data analysis. Results are expressed as mean ± standard error (n = 3–8 independent samples).
Results
Particle Size And Zeta Potential Measurements
Six different formulations of 5-FU-loaded NPs were prepared to evaluate the effect of physical PEGylation using two different polymers; PLGA and PCL. The exact composition of each formulation is shown in (Table 1). Particle size, PDI, and zeta potential of NPs for each of the prepared formulations are shown in Table 2. It was observed that all prepared NP formulations exhibited very close low particle sizes between 176.3±6.7 nm for F3 to 253.9±8.6 nm for F4. They also exhibited a narrow range of particle size distribution indicated by low PDI (<0.25) except for F4 (0.317). These results indicate that the method of preparation was well optimized to produce 5-FU loaded NPs of small uniform sizes.
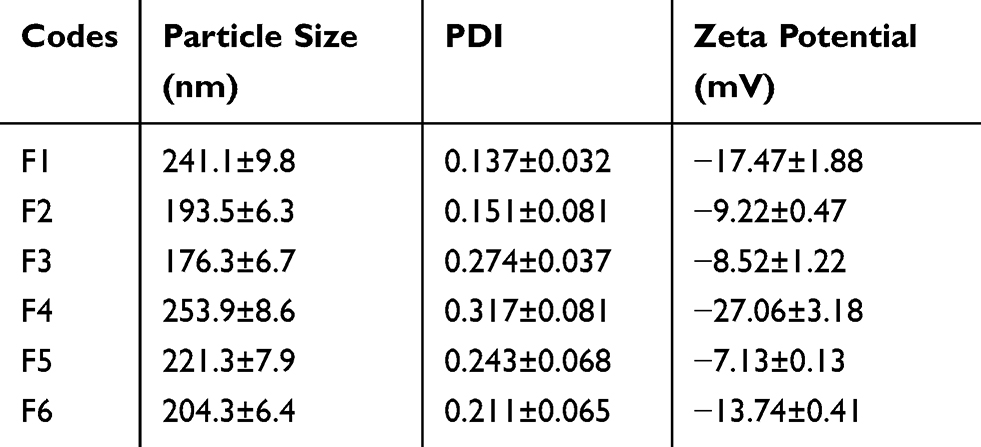 |
Table 2 The Physicochemical Characterization Of 5-FU-Loaded Nanoparticles |
The zeta potential values were −17.47±1.88, −9.22±0.47, −8.52±1.22, −27.06±3.18, −7.13±0.13, and −13±0.41 for F1, F2, F3, F4, F5, and F6, respectively. Such negative surface charge of all NP formulations is attributed to the existence of numerous free carboxylic acid groups within the chemical structure of the PLGA and PCL polymers present on NP surfaces.
Yield, Entrapment Efficiency, Drug Loading, And Particle Morphology
Table 3 shows the effect of the polymer and copolymer nature on the percent yield, entrapment efficiency (EE), and drug loading (DL) of 5-FU into NPs. Generally, all formulations showed relatively high percent yield values indicating the robustness of preparation method. It is clear that the incorporation of PEG 6000 in the composition of NPs significantly enhanced both percent EE and DL. They were found to be dependent on the nature of the polymer and the magnitude of PEG 6000. An increase in EE% from 32 to 74 and DL% from 7 to 32 were recorded for F1and F5, respectively. These results revealed that both F5 and F6 exhibited a significant higher EE % and DL% than other NP formulations. They are both composed of different ratios of PLGA and PEG 6000. Increasing the polymers portion in the ratio of drug: PCL: PEG6000 was found to significantly enhance both the %EE and %DL as indicated from the higher %EE (68.29%) and drug loading (21.27%) achieved with F4 (1:20:15) compared with lower values (44.24% for %EE and 12.72% for %DL) obtained with F3 (1:5:1).
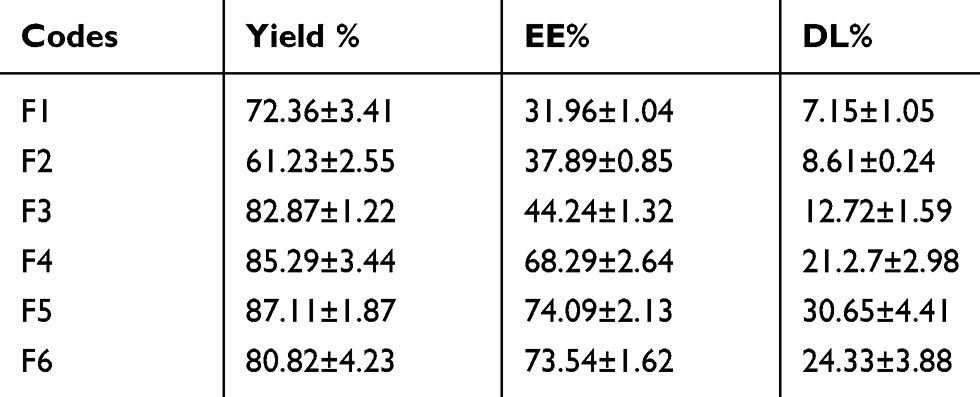 |
Table 3 The Yield %, Entrapment Efficiency (EE %) And Drug Loading (DL%) Of 5-FU-Loaded Nanoparticles (Mean ± S. D., N = 3) |
The SEM images depict almost spherical shape for all of 5-FU-loaded NPs with smooth surfaces (Figure 1). Some of the images showed aggregation of NPs which is probably attributed to the high surface activity and conditions of the drying process.
 |
Figure 1 SEM micrographs of 5-FU loaded nanoparticles. Notes: F1 (composed of 1:5:0, drug:PLGA:PEG6000), F2 (composed of 1:5:0, drug:PCL:PEG6000), F3 (composed of 1:5:1, drug:PCL:PEG6000), F4 (composed of 1:20:15, drug:PCL:PEG6000), F5 (composed of 1:5:1, drug:PLGA:PEG6000), and F6 (composed of 1:20:15, drug:PLGA:PEG6000). |
X-Ray Diffraction (XRD) And Fourier Transform Infrared Spectroscopy (FTIR)
Figure 2 depicts the XRD spectra of the six 5-FU-loaded NP formulations in comparison to pure 5-FU powder. The spectrum of pure 5-FU shows a peak near 30° at the 2Ө, which can be ascribed to its crystalline nature. The appearance of the broad peaks in 2Ө = 10°–25° is due to the amorphous nature of PLGA polymer. But in the presence of PCL polymer, crystalline peaks at 21.1° and 24.0° were observed (Figure 2). The XRD of 5-FU-loaded NP formulation indicates the disappearance of the characteristic peak for 5-FU.
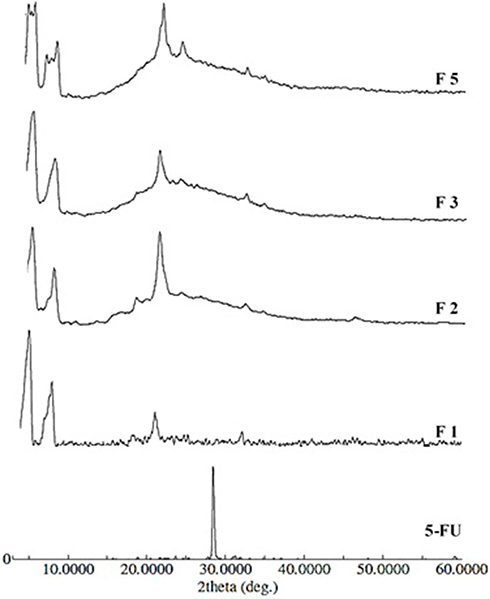 |
Figure 2 XRD diffractograms of pure 5-FU and 5-FU-loaded nanoparticles. Notes: F1 (composed of 1:5:0, drug:PLGA:PEG6000), F2 (composed of 1:5:0, drug:PCL:PEG6000), F3 (composed of 1:5:1, drug:PCL:PEG6000), and F5 (composed of 1:5:1, drug:PLGA:PEG6000). |
Figure 3 shows FTIR spectra of the six 5-FU-loaded NP formulations compared with pure 5-FU. The characteristic bands of pure 5-FU are detected at 1722, 1430, 1246, 811, and 551 cm−1 resulting from the vibration of imide stretch (amide II and amide III) and aromatic ring, 1348 cm−1 associated with the vibration of the pyrimidine ring, 1181 cm−1 assigned to the C=O vibrations, and 1246 cm−1 associated with C-N vibrations. While the spectra of NP formulations have shown the amide group bands in the range between 1470 and 1760 cm−1 and the carbonyl band observed at 1733 cm−1. It also depicts -CH2 stretching bands in the range of 2851–2920 cm−1 and C=O stretching in the range of 1081–1252 cm−1. The C-H stretching band was demonstrated as a characteristic peak in the range of 3136–2830 cm−1. Generally, the spectrum of NP formulations kept the sharpest characteristic peaks of 5-FU with slight shifting.
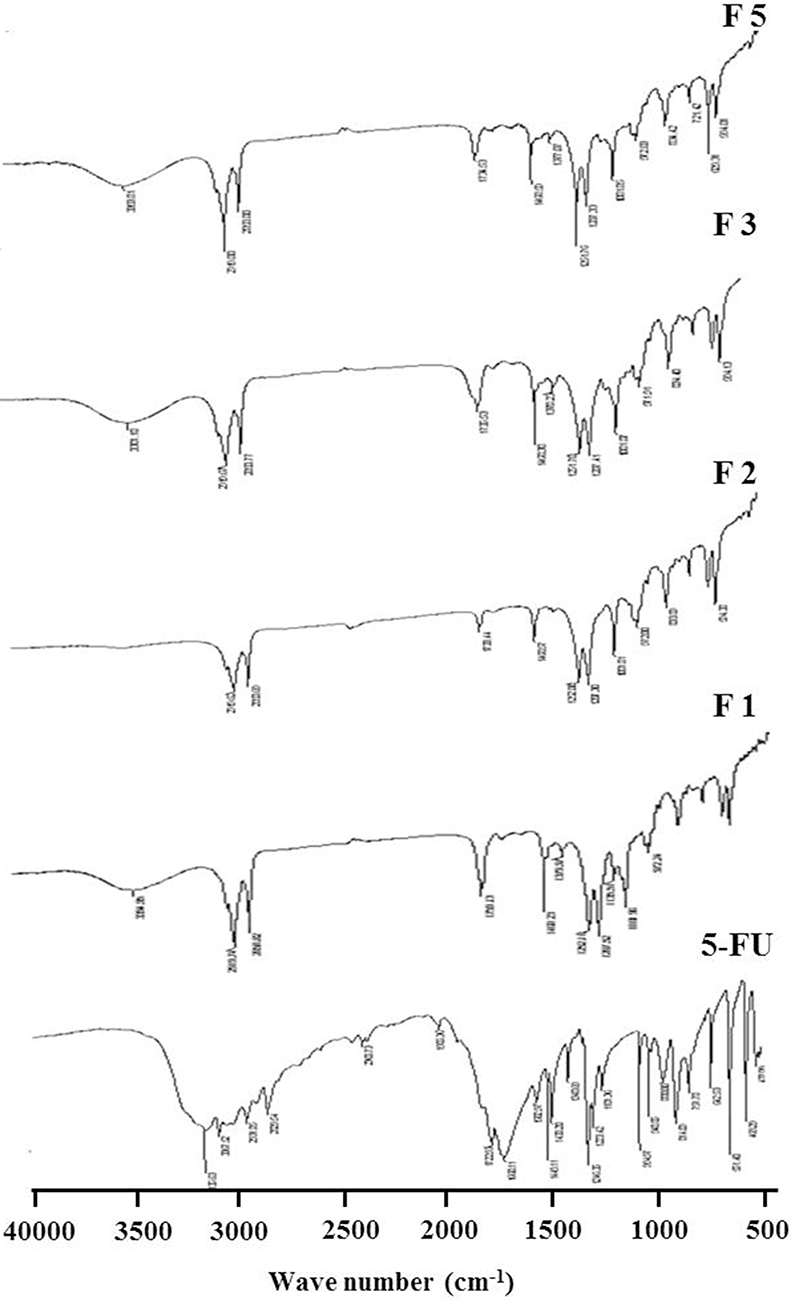 |
Figure 3 FTIR spectra of pure 5-FU and 5-FU-loaded nanoparticles. Notes: F1 (composed of 1:5:0, drug:PLGA:PEG6000), F2 (composed of 1:5:0, drug:PCL:PEG6000), F3 (composed of 1:5:1, drug:PCL:PEG6000), and F5 (composed of 1:5:1, drug:PLGA:PEG6000). |
In Vitro Release Studies
Figure 4 illustrates the 5-FU release profiles from the six NP formulations in comparison to 5-FU dispersion in distilled water as a reference. The release profile of the reference indicated instant release for most of the drug within the first hour. On the other hand, a bi-phasic pattern has been followed with all NP formulations characterized by an 8 h first rapid release phase followed by a second phase of slow-release extending until 72 hrs. The extent of this pattern was variable depending on the polymeric composition of each formulation. The cumulative % released values of 5-FU from the NPs in the first 8 hrs were about 39%, 26%, 53%, 59%, 60%, and 63% for F1, F2, F3, F4, F5, and F6, respectively, while they achieved a maximum of about 47%, 36%, 66%, 70%, 75%, and 84% after 72 hrs. The slow release could be caused by the diffusion of the drug from the NPs matrix following the slow erosion of the polymer.
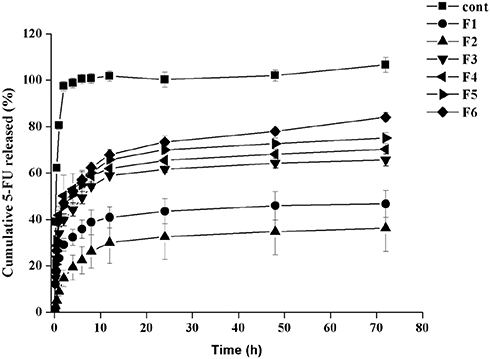 |
Figure 4 Release profiles of 5-FU from loaded nanoparticles in phosphate buffer pH 7.4 through dialysis bags with cut-off 12,000 Dalton using 5-FU aqueous solution as a control (mean ± SD, n = 3). |
The kinetics of the release data was analyzed using the DDSolver software program. The fitting of the data was assessed in multiple kinetic models including zero, first, Higuchi and Korsmeyer-Peppas. Table 4 presents the square of the regression values (R2) and the Korsmeyer-Peppas exponent (n) for all of the 6 formulations (F1–F6). The results indicated the high compliance of the 5-FU release from the six NP formulations with Korsmeyer-Peppas as shown from the significant higher R2 values. Moreover, the values of n are in the range from 0.157 to 0.282, thus far below 0.5 pointing out a Fickian diffusion kinetics.
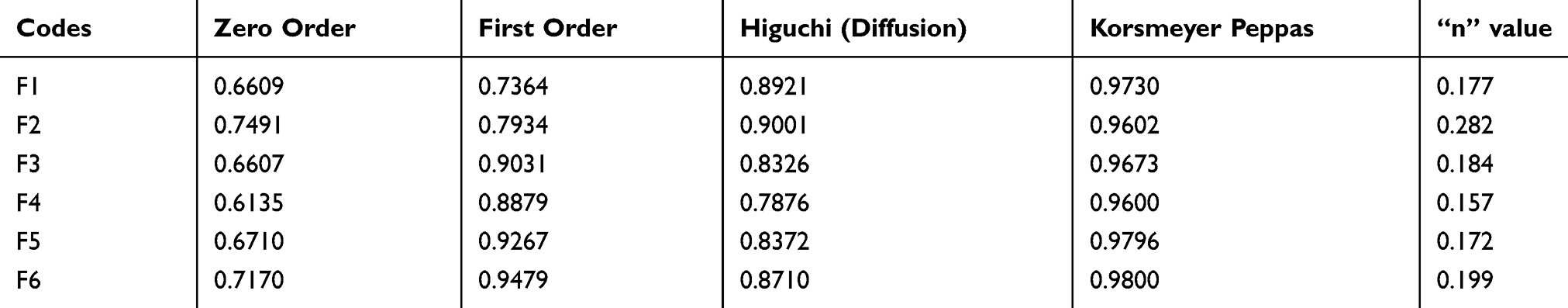 |
Table 4 Release Kinetics Of 5-FU Release From Different NP Formulations |
In Vitro Cytotoxicity
We assessed the cytotoxicity of all 5-FU-loaded NP formulations -(F1–F6) and their corresponding controls including drug-free NP formulations and pure 5-FU in HT-29, HepG2, and Daoy cancer cells by MTT assay. We used 5-FU in a concentration of 2.5 μg/mL, which is equal to 19.22 μM. This concentration is equivalent to the IC50 of 5-FU on HT-29 cells reported by Nita et al30 who concluded that the IC50 of 5-FU in HT-29 was 19.3 μM after 72 hrs of incubation. It is generally noticed that the cytotoxic activity of both 5-FU-loaded NPs and pure 5-FU is highly time-dependent. It is obvious from Figures 5–7 that loading of 5-FU into such NP formulations significantly fostered the cytotoxicity of 5-FU, as compared to pure 5-FU in the three tested cell lines, but with different potencies at different time points. This is an interesting finding, considering the incomplete and slow in vitro release of 5-FU from NPs in the 72-hr period, in contrast with the instant solubility of pure 5-FU in the cell culture medium. The results also demonstrated that significant anticancer activity was achieved for all 5-FU-loaded NP formulations as well as pure 5-FU in all the three tested cancer cell lines, as compared to the buffer (control), but with different potencies at different time points. As shown in Figure 6, the F1, F4, F5, and F6 formulations significantly enhanced the cytotoxicity of 5-FU, relative to pure 5-FU in HepG2 cells after 24 hrs of incubation. The F5 formulation significantly increased the antiproliferative activity of 5-FU, relative to pure 5-FU in HepG2 cells after 48 hrs of incubation. All 5-FU-loaded NPs, except F4, significantly enhanced the cytotoxicity of 5-FU, relative to pure 5-FU in HepG2 cells after 72 hrs of incubation. F5 (PLGA-PEG) was the most potent formula among all 5-FU-loaded NPs against HepG2 cells at all durations. The F5 formulation induced 70%, 60%, and 72% cell death after 24, 48, and 72 hrs, respectively. With regard to Daoy cells, F1, F3, and F5 significantly improved the cytotoxicity of 5-FU, relative to pure 5-FU after 24 hrs of incubation (Figure 5). All 5-FU-loaded NPs, except F4, significantly augmented the cytotoxicity of 5-FU, relative to pure 5-FU in Daoy after 48 hrs of incubation. The formulations F4, F5, and F6 significantly ameliorated the cytotoxicity of 5-FU, relative to pure 5-FU in Daoy after 72 hrs of incubation. Interestingly, F5 was also the most potent formulation among all 5-FU-loaded NPs against Daoy cells at all durations. It induced 34%, 61%, and 74% cell death after 24, 48, and 72 hrs, respectively. The formulations F1, F2, and F3 significantly enhanced the cytotoxicity of 5-FU, relative to pure 5-FU against HT-29 after 72 hrs incubation as depicted in Figure 7. These formulations induced 60% cell death after 72 hrs, whereas pure 5-FU inhibited HT-29 cell proliferation by about 51%; similar to results reported by Nita et al30 who showed that 5-FU induced 50% cell death of after 72 hrs.
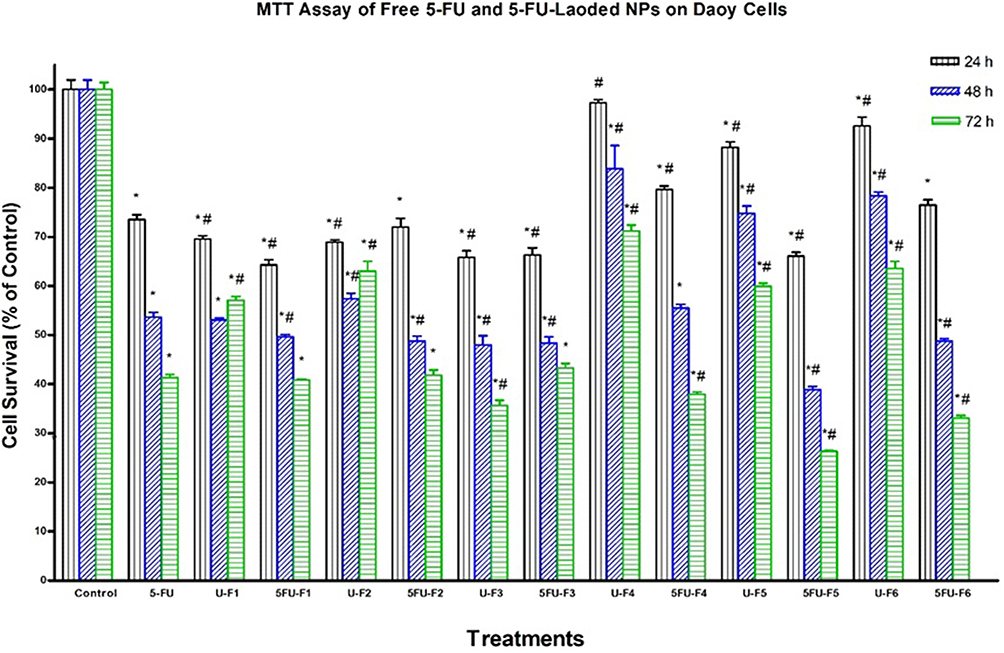 |
Figure 5 Effect of several formulas of 5-FU-loaded nanoparticles on human medulloblastoma cells. Daoy cells were treated with indicated formulas of 5-FU-loaded nanoparticles (5-FU-Fx), unloaded nanoparticles (U-Fx), pure 5-FU or buffer (control) for 24, 48, or 72 hrs. Cell viability was determined by MTT assay as indicated in Methods. At the end of the assay, the absorbance at 549 nm was read on a microplate reader. Significant differences between treatments and control and 5-FU were analyzed by ANOVA followed by unpaired t-test. *p < 0.05 compared with control (0 µM). #P<0.05 compared with 5-FU. |
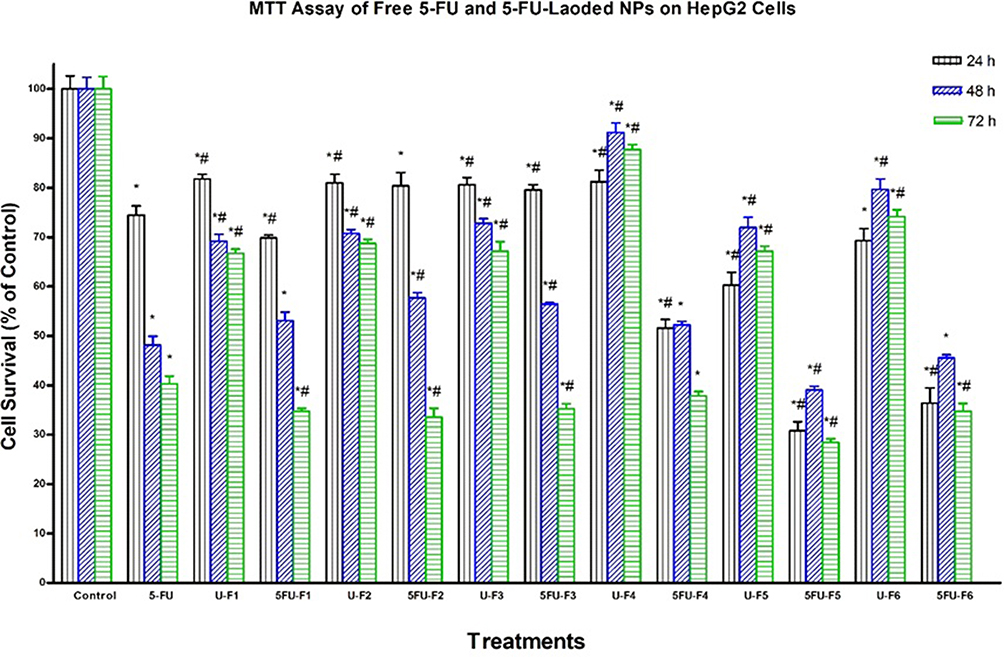 |
Figure 6 Effect of several formulas of 5-FU-loaded nanoparticles on human hepatocellular carcinoma cells. HepG2 cells were treated with indicated formulas of 5-FU-loaded nanoparticles (5-FU-Fx), unloaded nanoparticles (U-Fx), pure 5-FU or buffer (control) for 24, 48, or 72 hrs. Cell viability was determined by MTT assay as indicated in Methods. At the end of the assay, the absorbance at 549 nm was read on a microplate reader. Significant differences between treatments and control as well as 5-FU were analyzed by ANOVA followed by unpaired t-test. *p < 0.05 compared with control (0 µM). #P<0.05 compared with 5-FU. |
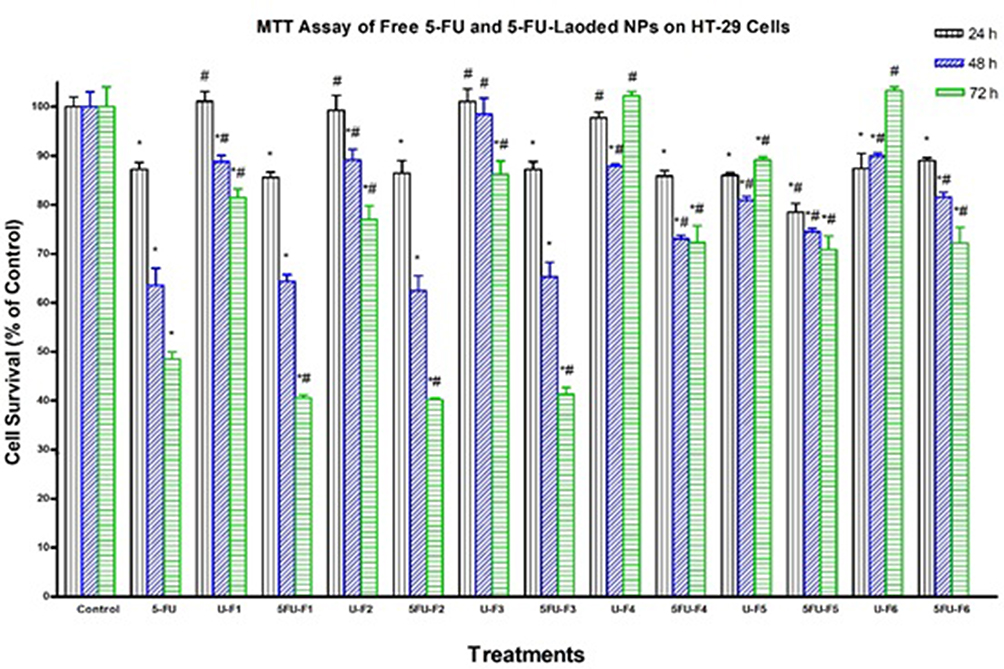 |
Figure 7 Effect of several formulas of 5-FU-loaded nanoparticles on human colorectal cancer cells. HT-29 cells were treated with indicated formulas of 5-FU-loaded nanoparticles (5-FU-Fx), unloaded nanoparticles (U-Fx), pure 5-FU or buffer (control) for 24, 48, or 72 hrs. Cell viability was determined by MTT assay as indicated in Methods. At the end of the assay, the absorbance at 549 nm was read on a microplate reader. Significant differences between treatments and control and 5-FU were analyzed by ANOVA followed by unpaired t-test. *p < 0.05 compared with control (0 µM). #P<0.05 compared with 5-FU. |
Induction Of Apoptosis In HT-29 Cells Caused By 5-FU NPs
The most potent 5-FU NP formulations (F1–F3) which showed the highest antiproliferative effects as shown in Figure 7 were selected for measuring apoptosis. Unloaded nanoparticles (F1–F3) and pure 5-FU were used as negative and positive controls respectively. The results showed that the three formulations significantly increased the percent of Annexin V-PE positive/7-AAD negative (early apoptotic) cells, compared to control as well as to 5-FU which induced only modest pro-apoptotic effects as shown in Figure 8A and B. These data indicated that 5-FU NP formulations 1, 2, and 3 are capable of inhibiting HT-29 human colon cancer cell growth, at least in part, by stimulating early apoptosis.
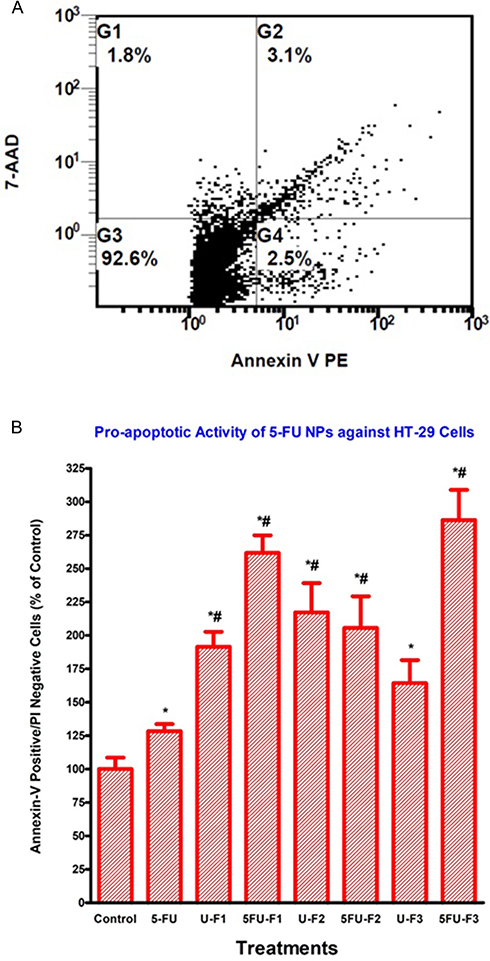 |
Figure 8 5-FU-loaded nanoparticles induce apoptosis of human colorectal cancer cells. HT-29 cells were treated with indicated formulas of 5-FU-loaded nanoparticles (5-FU-Fx), unloaded nanoparticles (U-Fx), pure 5-FU or buffer (control) for 48 hrs. Cells were then labeled with Annexin V and 7-AAD, and analyzed by flow cytometry. (A) A representative dot plot from the assay results, illustrating the discrimination between viable, early apoptotic and late apoptotic/necrotic cells. (B) Pro-apoptotic activity of 5-FU NP formulations F 1, 2 and 3, as well as pure 5-FU against HT-29 cells. Annexin V+/7-AAD: early apoptotic cells. Significant differences between treatments and control as well as 5-FU were analyzed by ANOVA followed by unpaired t-test. *p < 0.05 compared with control (0 µM). #P<0.05 compared with 5-FU. |
Discussion
The entrapment of 5-FU into polymeric NPs was intended to prolong the residence time of the drug inside the body and thereby to improve the cellular uptake of drugs by cancer cells. The above strategy was aimed to significantly enhance the antitumor activity and the overall therapeutic efficacy of 5-FU. The use of PLGA and PCL-based NPs as targeted drug delivery systems has been reported to be successful in different cancer types.31,32 Among the used polymeric NPs, PCL NPs have shown lower particle size than that of PLGA due to their higher flexibility.33 Consistently, in the present study, the particle size of F2 (PCL-based formulation) was lower than that of F1 (PLGA-based formulation). The existence of many free carboxylic acid groups on the surface of both PLGA and PCL indicates negative surface charge on all of the prepared NP formulations. Moreover, it was observed that the incorporation of PEG in the NPS composition has resulted in a reduction in the magnitude of zeta potential value as a result of the PEG induced shielding effect on the NP surfaces.34 The reproducible low particle sizes and PDI are indicative of high physical stability and reduced aggregation of NPs. The significantly higher %EE observed with both F5 and F6 formulations could be attributed to the presence of PEG 6000 together with PLGA, which have the ability to create an irregular crystalline network. This may promote more space for the drug molecules.35 On the other hand, the lower %EE and %DL obtained for F1 might be ascribed to higher precipitation of polymers during NP preparation.35 Moreover, it is clear from our results that PEG has a valuable effect on the EE of the drug. This action might be explained by the amphiphilic nature of the polymer blends PEG/PLGA and PEG/PCL.36,37
Commonly higher particle sizes obtained by DLS in contrast to those obtained by SEM might be related to the tendency of NPs to swell in aqueous media.38 In addition, DLS measures the diameter of stagnant solvent layer adjacent to the NPs, while the SEM measures the exact diameters of NPs in the dry state.
The absence of the characteristic peaks of 5-FU from the XRD spectra in NP formulations indicates the existence of the polymer matrix entrapped 5-FU in the amorphous state. The XRD analysis suggests that the majority of 5-FU molecules were deeply incorporated within the NPs matrix rather than adsorbed on their surfaces. This was also confirmed by the FTIR by the slight shifting of the characteristic peaks of 5-FU observed in NP formulations and the appearance of C-H and C=O stretching peaks. These findings indicated that slight chemical interaction between the drug and the polymer, which revealed that the drug was encapsulated inside the polymer.
The slower release obtained with PCL-based NP formulations can be attributed to the higher crystalline nature of PCL than PLGA.39 In addition, PCL has much less water permeability, which possibly led to very slow degradation rate.40 Therefore, the improved 5-FU release from PCL polymer was dependent on the incorporated PEG 6000 in the case of formulations F3 and F4. The increased release of 5-FU from NPs in the presence of PEG 6000 might be attributed to a higher degree of water permeability imparted by the surface PEG.32,41
In addition, the higher release rate of 5-FU from PLGA containing NPs (F1, F5, and F6) likely attributed to the hydrophilic glycolide units resulting in improved water permeability, thereby increasing drug release rate.42,43 The significant increase in the 5-FU release rate observed in the case of F5 and F6 compared to F1 can be explained by the much more hydrophilicity and water permeability induced by the incorporation of PEG in the NP formulations. This was also concluded by Sanna et al.35 The kinetic analysis of the drug release profile from the NP formulations denoted a Fickian diffusion model for all of the six formulations; therefore, a combination of diffusion and erosion which better explains the bi-phasic release pattern with faster initial followed by a much slower release rate. This is in agreement with the interpretation introduced by a number of previous reports.44,45
It is generally noticed that the cytotoxic activity of 5-FU-loaded NPs and pure 5-FU was time-dependent. This corroborates previous findings where prolonged exposure of colon, glioma, and breast cancer cells has been reported to enhance 5-FU cytotoxicity.46,47 In the present study, the level of cell death obtained with 5-FU NPs is much greater than 58% growth inhibition induced by 5-FU amphiphilic nano-micelles in HepG2 cells48 and by porous 5-FU-loaded hydrogel microparticles which induced only 6%, 31% and 33% HepG2 cell death after 24, 48, and 72 hrs, respectively.49
The significant enhancement of 5-FU cytotoxicity against Daoy cells due to its incorporation into NPs confirms our previous study showing increased cytotoxic effects of 5-FU NPs in Daoy cells.50 To the best of our knowledge, this is the first study to investigate the cytotoxicity of pure 5-FU and 5-FU NPs using medulloblastoma cells. In the case of HT-29 cells, F5 was the only formulation that significantly increased the cytotoxicity of 5-FU, relative to pure 5-FU after 24-hr incubation. Even though the F1 and F3 formulations did not significantly enhance 5-FU cytotoxicity against HT-29 after 48-hr incubation, both pure 5-FU as well as F1 and F3 caused significant cytotoxicity with 37% cell death. This is much higher, compared to 25% and 30% HT-29 cell death induced by 5-FU-loaded liposome and 5-FU-loaded folate-liposomal NPs with a 25 μM concentration of 5-FU as reported by Handali et al.51 Importantly, the 25 μM concentration of 5-FU used in their study was higher than 19.22 μM 5-FU used in the present study. Apparently our three formulations (F1-F3) are much more potent in killing HT-29 cells than 5-FU-PLGA NPs used in a previous study.46 Although they achieved higher cell death (68%) as compared to 60% in our study, they used 250 μM 5-FU which is a much higher concentration than 19.22 μM used in this study. Interestingly, our three formulations, F1, 2, and 3, showed a decent and similar time-dependent cytotoxicity amounting to about 14%, 36%, and 60% after 24, 48, and 72 hrs incubation, respectively, in HT-29 cells, further confirming the time-dependent cytotoxicity of 5-FU as reported by other researchers.44,45 With regard to the overall sensitivity, our findings showed that Daoy cells were the most sensitive cell line to the cytotoxic actions of 5-FU-loaded NPs, followed by HepG2 and HT-29 cells.
Many chemotherapeutic agents, including 5-FU, are shown to exert their anticancer effects through the induction of apoptosis. Numerous studies confirm that 5-FU induces apoptosis in colon cancer cells, including HT-29 cells, as well as in extra-colonic cells.52 5-FU has been the mainstay of CRC therapy since the late 1950;53 thus we sought to determine the extent to which our 5-FU NP formulas can induce apoptosis of the CRC HT-29 cell line. The ability of our formulations F1–F3 to stimulate early apoptosis in HT-29 human colon cancer is also consistent with the cell anti-proliferative effects found in MTT assay data, which illustrated that the three formulations inhibited HT-29 cell viabilities within 48 hrs, compared to control. (See MTT Figures). Our results corroborate previous reports showing that 5-FU NPs stimulate apoptosis of colon cancer cells and that 5-FU NPs were superior to free 5-FU.54,55
Conclusion
In this study, the physical incorporation of PEG 6000 in the formulation 5-FU-loaded PLGA and PCL NPs has been successfully achieved using a simple yet robust method with high percent yield. The generated NPs exhibited relatively small nanoparticle sizes, uniform spherical-shape, reasonable EE%, and DL%. The in vitro release profiles of these NPs indicated a prolonged release which extended over 3 days. The in vitro cytotoxicity studies indicated a highly significant activity of 5-FU when loaded into polymeric NPs than the pure drug. Some of the formulations have demonstrated ability to stimulate apoptosis in HT-29 human colon cancer cells. Physical PEGylation appeared to improve multiple characteristics of NPs including the drug loading capacity and enhance both drug release profile and in vitro cytotoxicity. Therefore, encapsulation of 5-FU into physically PEGylated PCL/PLGA NPs can be considered a beneficial approach for enhancing the efficacy of 5-FU and can be extended for many other drugs to enhance their anticancer effects. The in vivo studies using physically PEGylated PCL/PLGA NPs may yield additional information on use of these NPs in cancer therapy.
Acknowledgments
The abstract and part of this work were presented at the 44th Controlled Release Society (CRS) Annual meeting, Boston, Massachusetts, July 16–19, 2017, as a poster presentation. The poster’s abstract was published in “poster abstracts” in CRS website. This project was funded by the National Plan for Science, Technology and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology, Kingdom of Saudi Arabia, Award number (MED-1768-02).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Nie S, Xing Y, Kim GJ, et al. Nanotechnology applications in cancer. Annu Rev Biomed Eng. 2007;9:257–288. doi:10.1146/annurev.bioeng.9.060906.152025
2. Wang MD, Shin DM, Simons JW, et al. Nanotechnology for targeted cancer therapy. Expert Rev Anticancer Ther. 2007;7:833–837. doi:10.1586/14737140.7.6.833
3. Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACSNano. 2009;3:16–20.
4. Youan BB. Impact of nanoscience and nanotechnology on controlled drug delivery. Nanomedicine. 2008;3:401–406. doi:10.2217/17435889.3.4.401
5. Peer D, Karp JM, Hong S, et al. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–760. doi:10.1038/nnano.2007.387
6. Cho K, Wang X, Nie S, et al. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14:1310–1316. doi:10.1158/1078-0432.CCR-07-1441
7. Soppimath KS, Aminabhavi TM, Kulkarni AR, et al. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70:1–20. doi:10.1016/S0168-3659(00)00339-4
8. Cherian AK, Rana AC, Jain SK. Self-assembled carbohydrate-stabilized ceramic nanoparticles for the parenteral delivery of insulin. Drug Dev Ind Pharm. 2000;4:459–463. doi:10.1081/DDC-100101255
9. Jones MC, Leroux JC. Polymeric micelles – a new generation of colloidal drug carriers. Eur J Pharm Biopharm. 1999;48:101–111. doi:10.1016/S0939-6411(99)00039-9
10. Barenholz Y. Liposome: application: problems and prospects. Curr Opin Colloid Interface Sci. 2001;6:66–77. doi:10.1016/S1359-0294(00)00090-X
11. Caminad AM, Laurent R, Majora JP. Characterization of dendrimers. Adv Drug Deliv Rev. 2005;57:2130–2146. doi:10.1016/j.addr.2005.09.011
12. Yassin AEB, Anwer MK, Mowafy HA, et al. Optimization of 5-flurouracil solid-lipid nanoparticles: a preliminary study to treat colon cancer. Int J Med Sci. 2010;6:398–408. doi:10.7150/ijms.7.398
13. Bala I, Hariharan S, Kumar MR. PLGA nanoparticles in drug delivery: the state of the art. Crit Rev Ther Drug Carrier Sys. 2004;21(5):387–422. doi:10.1615/CritRevTherDrugCarrierSyst.v21.i5.20
14. Rossi S, ed. Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust; 2013.
15. World Health Organization (WHO) Model List of Essential Medicines. World Health Organization; October 2013.
16. Felici A, Verweij J, Sparreboom A. Dosing strategies for anticancer drugs: the good, the bad and body-surface area. Eur J Cancer. 2002;38(13):1677–1684. doi:10.1016/S0959-8049(02)00151-X
17. Baker SD, Verweij J, Rowinsky EK, et al. Role of body surface area in dosing of investigational anticancer agents in adults 1991–2001. J Natl Cancer Inst. 2002;94(24):1883–1888. doi:10.1093/jnci/94.24.1883
18. He YC, Chen JW, Cao J, et al. Toxicities and therapeutic effect of 5-fluorouracil controlled release implant on tumor-bearing rats. World J Gastroenterol. 2003;9:1795–1798. doi:10.3748/wjg.v9.i8.1795
19. Arias JL, Ruiz MA, López-Viota M, et al. Poly(alkylcyanoacrylate) colloidal particles as vehicles for antitumour drug delivery: a comparative study. Colloids Surf B. 2008;62:64–70. doi:10.1016/j.colsurfb.2007.09.018
20. Zhang N, Yin Y, Xu SJ, et al. 5-Fluorouracil: mechanisms of resistance and reversal strategies. Molecules. 2008;13:1551–1569. doi:10.3390/molecules13081551
21. Tanaka F, Fukuse T, Wada H, et al. The history, mechanism and clinical use of oral 5-fluorouracil derivative chemotherapeutic agents. Curr Pharm Biotechnol. 2000;1:137–164. doi:10.2174/1389201003378979
22. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–6392.
23. Hirenkumar K, Makadia SSJ. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377–1397. doi:10.3390/polym3031377
24. Burnier M, Fricker AF, Hayoz D, et al. Pharmacokinetic and pharmacodynamic effects of YM087, a combined V1/V2 vasopressin receptor antagonist in normal subjects. Eur J Clin Pharmacol. 1999;55:633–637. doi:10.1007/s002280050685
25. Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89(2):271–277. doi:10.1016/0022-1759(86)90368-6
26. Elumalai P, Gunadharini DN, Senthilkumar K. Induction of apoptosis in human breast cancer cells by nimbolide through extrinsic and intrinsic pathway. Toxicol Lett. 2012;215:131–142. doi:10.1016/j.toxlet.2012.10.008
27. van Engeland M, Ramaekers FC, Schutte B, et al. A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry. 1996;24(2):131–139. doi:10.1002/(SICI)1097-0320(19960601)24:2<131::AID-CYTO5>3.0.CO;2-M
28. Clarke RG, Lund EK, Johnson IT, et al. Apoptosis can be detected in attached colonic adenocarcinoma HT29 cells using annexin V binding, but not by TUNEL assay or sub-G0 DNA content. Cytometry. 2000;39(2):141–150. doi:10.1002/(ISSN)1097-0320
29. Merkel O, Heyder C, Asslaber D, et al. Arsenic trioxide induces apoptosis preferentially in B-CLL cells of patients with unfavorable prognostic factors including del17p13. J Mol Med (Berl). 2008;86(5):541–552. doi:10.1007/s00109-008-0314-6
30. Nita ME, Nagawa H, Tominaga O, et al. 5-Fluorouracil induces apoptosis in human colon cancer cell lines with modulation of Bcl-2 family proteins. Br J Cancer. 1998;78(8):986–992. doi:10.1038/bjc.1998.617
31. Kunii R, Onishi H, Machida Y. Preparation and antitumor characteristics of PLA/(PEG-PPG-PEG) nanoparticles loaded with camptothecin. Eur J Pharm Biopharm. 2007;67:9–17. doi:10.1016/j.ejpb.2007.01.012
32. Ocal H, Arica-Yegin B, Vural I, et al. 5-Fluorouracil-loaded PLA/PLGA 33. PEG-PPG-PEG polymeric nanoparticles: formulation, in vitro characterization and cell culture studies. Drug Dev Ind Pharm. 2014;40(4):560–567. doi:10.3109/03639045.2013.775581
33. Mundargi RC, Srirangarajan S, Agnihotri SA, et al. Development and evaluation of novel biodegradable microspheres based on poly (d, llactide-co-glycolide) and poly(epsilon-caprolactone) for controlled delivery of doxycycline in the treatment of human periodontal pocket: in vitro and in vivo studies. J Control Release. 2007;119(1):59–68. doi:10.1016/j.jconrel.2007.01.008
34. Song Z, Feng R, Sun M, et al. Curcumin loaded PLGA-PEG-PLGA tri-block copolymeric micelles: preparation, pharmacokinetics and distribution in vivo. J Colloid Interface Sci. 2011;2011(354):116–123. doi:10.1016/j.jcis.2010.10.024
35. Sanna V, Roggio AM, Posadino AM, et al. Novel docetaxel-loaded nanoparticles based on poly (lactide-co-caprolactone) and poly(lactideco- glycolide-co-caprolactone) for prostate cancer treatment: formulation, characterization, and cytotoxicity studies. Nanoscale Res Lett. 2011;6(260):1–9. doi:10.1186/1556-276X-6-260
36. Gryparis EC, Mattheolabakis G. Effect of conditions of preparation on the size and encapsulation properties of PLGA-mPEG nanoparticles of cisplatin. Drug Deliv. 2007;14(6):371–380. doi:10.1080/10717540701202937
37. Gref R, Luck M, Quellec P, et al. Stealth corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the corecomposition on phagocytic uptake and plasma protein adsorption. Colloids Surf B. 2000;(18):301–313. doi:10.1016/S0927-7765(99)00156-3
38. Manchanda R, Fernandez-Fernandez A, Nagesetti A, et al. Preparation and characterization of a polymeric (PLGA) nanoparticulate drug delivery system with simultaneous incorporation of chemotherapeutic and thermo-optical agents. Colloids Surf B Biointerfaces. 2010;75:260–267. doi:10.1016/j.colsurfb.2009.08.043
39. Shenguo W, Hongli C, Qing C, et al. Degradation and 5-fluorouracil release behavior in vitro of polycaprolactone/poly(ethylene oxide)/polylactide tri-component copolymer. Polymer Adv Tech. 2001;12(3–4):253–258. doi:10.1002/pat.138
40. Makoto M, Akiko K, Jun’ichi O, et al. Effect of polymer crystallinity on papaverine release from poly (l-lactic acid) matrix. J Control Release. 1997;49(2–3):207–215. doi:10.1016/S0168-3659(97)00081-3
41. Torchilin VP, Omelyanenko VG, Papisov MI, et al. Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim Biophys Acta. 1994;1195:11–20. doi:10.1016/0005-2736(94)90003-5
42. Mainardes RM, Evangelista RC. PLGA nanoparticles containing praziquantel: effect of formulation variables on size distribution. Int J Pharm. 2005;290:137–144. doi:10.1016/j.ijpharm.2004.11.027
43. Zhang W, Li Y, Liu L, et al. Amphiphilic tooth brush like copolymers based on poly(ethylene glycol) and poly(epsilon-caprolactone) as drug carriers with enhanced properties. Biomacromolecules. 2010;1:1331–1338. doi:10.1021/bm100116g
44. Peppas NA, Sahlin JJ. A simple equation for the description of solute release. III. Coupling of diffusion and relaxation. Int J Pharm. 1989;57:169–172.
45. Modi S, Anderson BD. Determination of drug release kinetics from nanoparticles: overcoming pitfalls of the dynamic dialysis method. Mol Pharm. 2013;10:3076–3086. doi:10.1021/mp400154a
46. Tawfik E, Ahamed M, Almalik A, et al. Prolonged exposure of colon cancer cells to 5-fluorouracil nanoparticles improves its anticancer activity. Saudi Pharm J. 2017;25(2):206–213. doi:10.1016/j.jsps.2016.05.010
47. Nair KL, Jagadeeshan S, Nair SA, et al. Biological evaluation of 5-fluorouracil nanoparticles for cancer chemotherapy and its dependence on the carrier, PLGA. Int J Nanomedicine. 2011;6:1685–1697. doi:10.2147/IJN.S20165
48. Zhou JJ, Chen RF, Tang QB, et al. Preparation of 5-fluorouracil encapsulated in amphiphilic polysaccharide nano-micelles and its killing effect on hepatocarcinoma cell line HepG2. Ai Zheng. 2006;25(12):1459–1463.
49. Wen Y, Liu Y, Zhang H, et al. A responsive porous hydrogel particle-based delivery system for oncotherapy. Nanoscale. 2019;11(6):2687–2693. doi:10.1039/C8NR09990A
50. Ashour AE, Badran MM, Kumar A, et al. Di-block PLCL and tri-block PLCLG matrix polymeric nanoparticles enhanced the anticancer activity of loaded 5-fluorouracil. IEEE Trans Nanobioscience. 2016;15(7):739–747. doi:10.1109/TNB.2016.2612340
51. Handali S, Moghimipour E, Rezaei M, et al. A novel 5-Fluorouracil targeted delivery to colon cancer using folic acid conjugated liposomes. Biomed Pharmacother. 2018;108:1259–1273. doi:10.1016/j.biopha.2018.09.128
52. Shchepotin IB, Soldatenkov V, Buras RR, et al. Apoptosis of human primary and metastatic colon adenocarcinoma cell lines in vitro induced by 5-fluorouracil, verapamil, and hyperthermia. Anticancer Res. 1994;14(3A):1027–1031.
53. Henderson R, French D, Sullivan R, et al. Molecular biomarkers and precision medicine in colorectal cancer: a systematic review of health economic analyses. Oncotarget. 2019;10(36):3408–3423. doi:10.18632/oncotarget.v10i36
54. Jiang H, Shi X, Yu X, et al. Hyaluronidase enzyme-responsive targeted nanoparticles for effective delivery of 5-fluorouracil in colon cancer. Pharm Res. 2018;35(4):73. doi:10.1007/s11095-017-2302-4
55. Liu W, Zhu Y, Wang F, et al. Galactosylated chitosan-functionalized mesoporous silica nanoparticles for efficient colon cancer cell-targeted drug delivery. R Soc Open Sci. 2018;5(12):181027. doi:10.1098/rsos.181027
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.