")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
Phosphorylation of Tau and α-Synuclein Induced Neurodegeneration in MPTP Mouse Model of Parkinson’s Disease
Authors Hu S, Hu M, Liu J, Zhang B, Zhang Z, Zhou FH, Wang L , Dong J
Received 22 October 2019
Accepted for publication 9 February 2020
Published 4 March 2020 Volume 2020:16 Pages 651—663
DOI https://doi.org/10.2147/NDT.S235562
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yuping Ning
Shanshan Hu, 1,* Meigui Hu, 2,* Jian Liu, 3 Bei Zhang, 4 Zhen Zhang, 5 Fiona H Zhou, 6 Liping Wang, 5, 6 Jianghui Dong 5, 6
1Good Clinical Practice Center, Affiliated Hospital of Zunyi Medical University, Zunyi 563003, Guizhou, People’s Republic of China; 2The Second School of Clinical Medicine, Zhuhai Campus of Zunyi Medical University, Zhuhai 519041, Guangdong, People’s Republic of China; 3Department of Anatomy, Zunyi Medical University, Zunyi 563000, Guizhou, People’s Republic of China; 4Department of Stomatology, The First People’s Hospital of Zunyi, Zunyi 563099, Guizhou, People’s Republic of China; 5Department of Hand Surgery, Department of Plastic Reconstructive Surgery, Ningbo No. 6 Hospital, Ningbo 315040, People’s Republic of China; 6School of Pharmacy and Medical Sciences, and UniSA Cancer Research Institute, University of South Australia, Adelaide, SA 5001, Australia
*These authors contributed equally to this work
Correspondence: Jianghui Dong; Liping Wang
Department of Hand Surgery, Ningbo No. 6 Hospital, Ningbo 315040, People’s Republic of China
Tel +618 8302 2715
Fax +618 8302 1087
Email [email protected]; [email protected]
Purpose: Parkinson’s disease (PD) is the second most common neurodegenerative disease. The α-Synuclein is a major component of Lewy bodies and Lewy neurites, the pathologic hallmark of PD. It is known that α-Synuclein is phosphorylated (p-α-Synuclein) in PD and tau-hyperphosphorylation (p-Tau) is also a pathologic feature of PD. However, the relationship between p-Synuclein and p-Tau in PD is not clear, in particular in the MPTP model of PD. The purpose of this study was to reveal their relationship in the mouse MPTP model.
Methods: Firstly, the p-α-Synuclein, α-Synuclein, p-Tau and Tau protein levels were analyzed. Then, GSK3β activation was determined using immunoblot and immunohistochemical staining. Finally, the dopaminergic neurodegeneration was assessed using Tyrosine Hydroxylase (TH) staining and retrograde labeling and microglial marker were labeled. Microglial activation and nigrostriatal pathway degeneration were observed.
Results: The results showed that p-α-Synuclein, α-Synuclein, p-Tau and Tau were upregulated in both hippocampus and substantia nigra of the PD mouse model. Furthermore, p-α-Synuclein and p-Tau were localized in the same regions of substantial nigra (SN) and dentate gyrus (DG) of hippocampus (Hippo). The activated form of GSK3β (phosphor GSK3β Y216) was increased in multiple brain areas. The GSK3β inhibitor AZD1080 injected in MPTP mice suppressed the expression of p-Tau and p-GSK3β and improved motor functions.
Conclusion: These findings revealed that p-α-Synuclein and p-Tau proteins are key pathological events leading to neurodegeneration and motor dysfunctions in the mouse MPTP model of PD. Our data suggest that the interference with the GSK3β activity may be an effective approach for the treatment of PD.
Keywords: α-Synuclein phosphorylation, Tau phosphorylation, Parkinson’s disease
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease and is characterized clinically by movement disorder, tremor, rigidity, as well as bradykinesia.5,30 The clinical pathophysiological changes of PD are the nigrostriatal system degeneration, which is characterized by loss of dopaminergic neurons in substantial nigra compacta (SNc), leading to dopamine depletion in the caudate putamen (CPu).14 The pathological hallmark of PD is Lewy bodies and Lewy neurites appeared in neurons in the brain.44 Studies have shown that Lewy bodies are composed of p-α-Synuclein and other aberrant proteins.34 Various neurotoxic chemicals including 1-methyl-4-phenyl-1,2,3,6-tetra hydropyridine (MPTP), 6-hydroxydopamine, rotenone and paraquat could induce PD in animals and human.10,18,26
Microtubule-associated protein Tau is a neuronal protein that plays important roles in microtubule assembly and stabilization.41 The phosphor Tau (p-Tau) shows its tendency of aggregation which occurs in Alzheimer disease (AD), Pick disease and frontotemporal dementia with parkinsonism-17.37 The phosphorylation of Tau reduces its affinity for microtubules and accumulation in the brain and causing related Tauopathies,17 and also impact on microtubule binding and induces microtubule instability.4 Although p-Tau links to the pathogenesis of AD and other Tauopathies, recent studies have revealed that p-Tau has a relationship with PD.46 MAPT (encoded Tau protein) gene mutation in humans increases the risk of PD, and Tau aggregation and filaments have been observed in familial PD. P-Tau is also a component of Lewy bodies, which suggests that p-α-Synuclein and p-Tau have shared pathways or synergistic effect in the PD progression.13 The relationship between p-α-Synuclein and p-Tau remains unclear.
A recent report shows that α-Synuclein binds to phosphor Ser214 Tau to promote Tau phosphorylation at Ser262 in vitro.27 Glycogen synthase kinase 3β (GSK3β), cyclin-dependent kinase 5 (CDK5), mitogen-activated protein kinase (MAPK) target threonine and proline motifs of Tau to induce Tau phosphorylation.25 However, the role of p-Tau and the kinase GSK3β that phosphorylates Tau in PD remains unclear. Therefore, the aim of the current study was to reveal the relationship between p-α-Synuclein and p-Tau in the mouse MPTP model of PD.
Materials and Methods
The research was conducted in accordance with the Declaration of Helsinki and with the Guide for Care and Use of Laboratory Animals as adopted and promulgated by the United National Institutes of Health. All experimental protocols were approved by the Review Committee for the Use of Animal Subjects of Zunyi Medical University.
Animals and Treatments
Six-week-old male C57BL/6 mice (n=36), weighting between 18 g and 20 g, were purchased from Zunyi Medical University (Zunyi, Guizhou, China). All mice were housed under standard conditions (22 ± 3°C, 50–55% humidity, 12 h light/dark cycles with food and water ad libitum) and randomly divided into three groups: Saline, MPTP and MPTP+AZD 1080 group. Mice in MPTP group were injected intraperitoneally 30 mg/kg body weight MPTP (M 0896, Sigma-Aldrich, St Louis, MO, USA) daily for 5 consecutive days.18 Mice in Saline group were injected intraperitoneally equal volume of saline. Mice in MPTP + AZD 1080 group were orally given 20 mg/kg body weight AZD 1080 (T 1741, Topscience Co Ltd, Shanghai, China) and MPTP injection. Twenty-four hours after the last injection, mice were euthanized and perfused with saline from the left ventricles. For Western blot analysis, the protein of six mice brains tissue was extracted for antibody incubation. For immunohistochemical staining, six mice brains were fixed with paraformaldehyde for 24 h.
Retrograde Labeling the Nigrostriatal Pathway
Six mice were used for retrograde labeling. A recombinant adeno-associated virus serotype 2 expressing mCherry under the control of the hSyn promoter was constructed. 1.0 × 109 genome copies in a volume of 300 nL were slowly injected at CPu (anterior/posterior (AP) + 0.74 mm, medial/lateral (ML) ± 1.5 mm, dorsal/ventral (DV) −3 mm), according to the Mouse Brain in Stereotaxic Coordinates. The wounds were conducted for anti–inflammatory treatment. Two weeks after the virus injection, mouse brains were acquired. MPTP was injected during the last five days. After the final MPTP injection, brain sections (40 µm) containing the SN area were cut using a cryostat microtome (Leica CM1950, Germany). Images were acquired using a LSM880 confocal microscope (Zeiss, Wurttemberg, Germany). The numbers of mCherry positive cells were calculated using the NeuronStudio.
Western Blot
The hippocampus (Hippo) and substantia nigra (SN) tissues were dissected and lysed in radioimmunoprecipitation assay buffer with protease inhibitors at 4°C for 30 min. Protein concentration of the samples was measured with the BCA quantitative analysis kit (Biocolors, Shanghai, China). Equal amount of protein samples was separated by 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA, USA). The membranes were blocked for 1h at room temperature using 5% BSA buffer, and then incubated with primary antibodies. Phosphor α-Synuclein (#23706, Cell Signaling Technology, Danvers, MA, USA) was used to test the α-Synuclein when phosphorylated at Ser129. AT8 (#MN1020, Thermo Fisher Scientific, MA, USA) was used to test the Tau when phosphorylated at Ser202/Thr205. α-Synuclein (#2647, Cell Signaling Technology, Danvers, MA, USA) and Tau5 (#ab80579, Abcam plc, Cambridge, UK) were used to test the total α-Synuclein and Tau protein, respectively. Membranes were washed with TBST and then incubated with the horseradish peroxidase conjugated secondary antibodies. Bands were visualized by ECL Plus chemiluminescent Western blot detection reagents and then quantified using Gel-Pro analyzer (Media Cybernetics Inc., Rockville, MD, USA). The amount of proteins was analyzed after adjusting for the loading control β-actin. The bands intensity was analyzed using Image J version 1.46 (NIH. MA. USA).
Immunofluorescence
Brain specimens were collected from mice in saline and MPTP groups. Coronal sections (40um) containing the SN, cortex, CA3 area of hippocampus, amygdala area and VTA were cut using a cryostat microtome. Brain sections were incubated with an anti-p-α-Synuclein (#23706, Cell Signaling Technology, Danvers, MA, USA), anti-Tyrosine Hydroxylase (#58844, Cell Signaling Technology, Danvers, MA, USA), anti- AT8 (#MN1020, Thermo Fisher Scientific, MA, USA) or anti-Iba1 (#ab178846, Abcam plc, Cambridge, UK). Second Alexa Fluor 568 or 488 conjugated antibody (#A-11031, #A-11001, Thermo Fisher Scientific, MA, USA) were incubated for 1 h followed by mounting in VECTASHIELD Antifade Mounting Medium containing DAPI (H-1200, Vector Laboratories, CA, USA). Images were obtained using a confocal LSM 880 microscope (Zeiss, Wurttemberg, Germany).
Immunohistochemistry
Brain samples were fixed followed by embedding in paraffin wax. Brain sections (3um) containing the middle brain (MB), Hipp and SN were cut using a microtome (RM2235, Leica, Wetzlar, Germany). Antigen retrieval was performed by heating the sections at 120°C for 5 min and blocked using normal goat serum. Brain sections were incubated with anti- AT8 (#MN1020, Thermo Fisher Scientific, MA, USA) or phospho GSK3β (Y216) (#ab75745, Abcam plc, Cambridge, UK) overnight at 4°C followed by incubating with the second antibody. A 3, 3ʹ-diaminobenzidine (DAB) kit (CW2069S, CWBIO, Beijing, China) was used to protein visualization and couterstained with hematoxylin. Images were captured with a microscope equipped with a camera (Eclipse80, Nikon, Tokyo, Japan). The immune-reactive cells were counted using Image J version 1.46 (NIH. MA. USA).
Transmission Electron Microscope (TEM)
Brain specimens were collected from mice in saline and MPTP group. After fixation and processing, the tissues were embedded in resin and were cut using an ultratome UC7 (Leica, Wurttemberg, Germany). The sections were stained with lead citrate and then observed using an electron microscope (Tecnai G2 F20, FEI, La Jolla, CA, USA).
Behavioral Tests
Rotarod test and static rod test were performed following the standard protocol.8 For rotarod test, the rotarod was set at a start speed of 5 rpm, and acceleration rate of 20 rpm. Animals were placed on the rod and the rod start acceleration after 10 sec. The time taken by a mouse to fall from the rod was recorded and the test was repeated three times. Before the test, the mice were trained for three days. For static rod test, the 60 cm long rod with diameter of 15 mm was used. The rod was horizontally placed 30 cm above the ground and attached to the platform. Animals were placed at the end of the rod facing away from the platform. Orientation time (the time taken to turn the body around) and transition time (the time taken to touch the platform) were recorded. Pole test was performed at the second day after the PD model was accomplished. Animals were placed on the top of a metal rod (80 cm long with 9 mm diameter) facing up. The time taken by animals to reach the base of the rod was recorded. Before the test, animals were trained for three days.
Statistical Analysis
All analysis was performed using the Graphpad Prism version 7.0 (Graphpad Software Inc., San Diego, CA, USA). Differences among means were assessed by Student’s t-test for the saline and MPTP group samples. When comparing Saline group, MPTP group and MPTP + AZD 1080 group, one-way ANOVA followed by Bonferroni post hoc was used. The null hypothesis was rejected at 0.05.
Results
Phospho α-Synuclein and Phospho Tau Protein Level Evaluation
The immunoblotting, immunofluorescence and immunohistochemical staining were conducted to test the effect of MPTP on the levels of phospho α-Synuclein and Tau after 8 days of intoxication. As shown in Figure 1A and B, MPTP treatment increased the levels of phospho α-Synuclein, phospho-Tau, total α-Synuclein and total Tau protein in Hippo and SN. Moreover, immunofluorescence analysis showed comparable P-α-Synuclein positive cell number in cortex, CA3 area of hippocampus, amygdala area and VTA (Figure 2A and B). P-α-synuclein was mainly localized in the nucleus and cytoplasm of the cells. Immunohistochemical analyses revealed that P-Tau positive cell number in MPTP group was increased in DG and CA1 area of hippocampus and EC than that in saline group. AZD 1080, a selective GSK3β inhibitor, reversed the increased number of P-Tau positive cell induced by MPTP (Figure 2C and D). P-Tau was localized in the cytoplasm around the nucleus and neurites with anomalous shapes.
 |
Figure 1 MPTP induced α-syn and Tau phosphorylation in hippocampus and SN. (A) Immunoblot analysis with anti-α-Synuclein and anti-p-Tau antibody of hippocampal tissues. Analysis of immunoblotting bands showed that MPTP increased the expression of p-α-Synuclein and p-Tau protein in hippocampus. (B) Immunoblot analysis with anti-p-α-synuclein and anti-p-Tau antibody of SN tissues. Analysis of immunoblotting bands showed that MPTP increased the expression of P-α-syn and P-Tau protein in SN. Data were shown as mean ± SEM. Statistical analysis was performed using Student’s t-test. *p<0.05. n=6. |
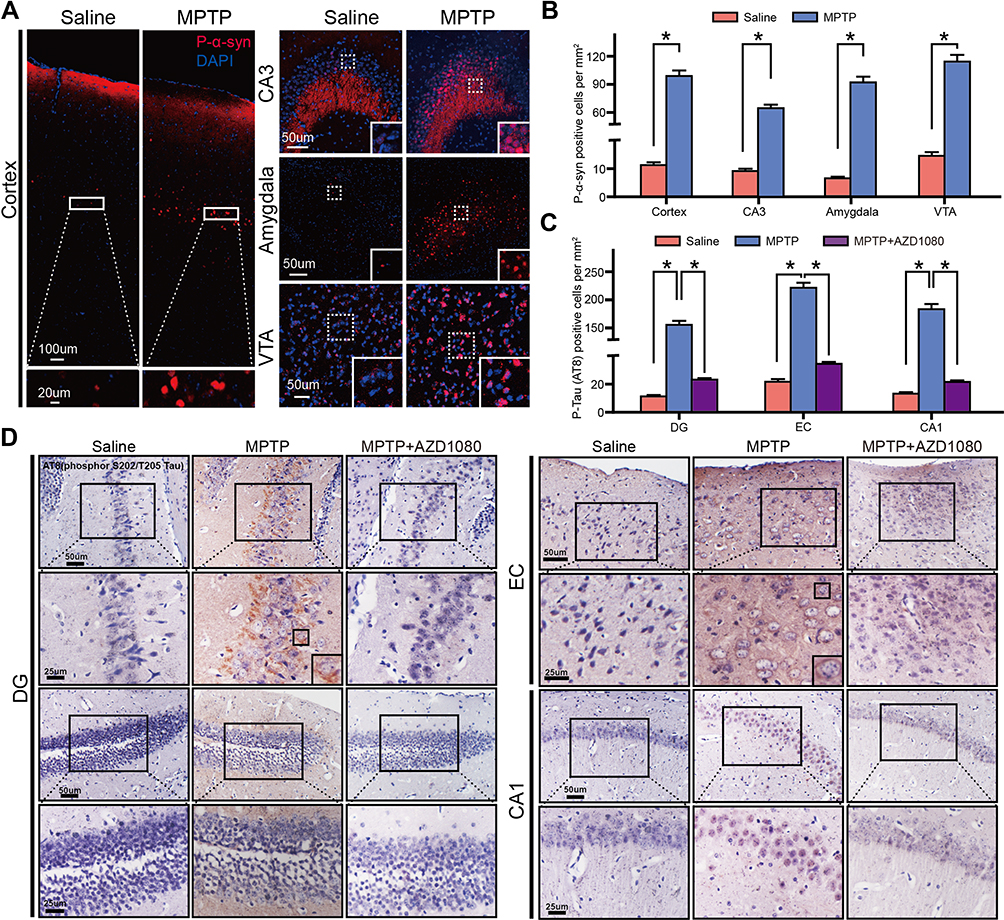 |
Figure 2 MPTP induced α-syn and Tau phosphorylation. (A) Representative images of P-α-syn staining in cortex, CA3 area of hippocampus, amygdala area and VTA. (B) Statistical analysis showed MPTP treatment increased the P-α-syn positive cell number in MPTP group was higher than that in saline group in cortex, CA3 area of hippocampus, amygdala area and VTA. (C) Statistical analysis showed P-Tau positive cell number in MPTP group was higher than that in saline group in DG, EC and CA1. AZD 1080 treatment reverse the p-Tau positive cell number increasing induced by MPTP in DG, EC and CA1. (D) Representative images of p-Tau staining in DG and CA1 area of hippocampus, and EC. Data were shown as mean ± SEM. Statistical analysis was performed using Student’s t-test or one-way ANOVA followed by Bonferroni post hoc. *p<0.05. n=6. |
The Locational Relationship Between p-α-Synuclein and p-Tau
Double immunofluorescence staining showed that p-α-Synuclein and p-Tau were localized in SN and hippocampal DG after MPTP treatment (Figure 3A–C). P-α-Synuclein and p-Tau localized in the polymorph layer and molecular layer of DG in the saline group, respectively. P-α-Synuclein was upregulated and localized in polymorph layer, granule layer and molecular layer after MPTP treatment. P-Tau was also upregulated and localized in polymorph layer and molecular layer after MPTP treatment. It is notable that the two phosphor proteins were not localized in the same structures of polymorph layer and molecular layer of DG in Hippo (Figure 3D). P-tau protein formed plaques whereas p-α-synuclein was in the granule structures in SN (Figure 3A).
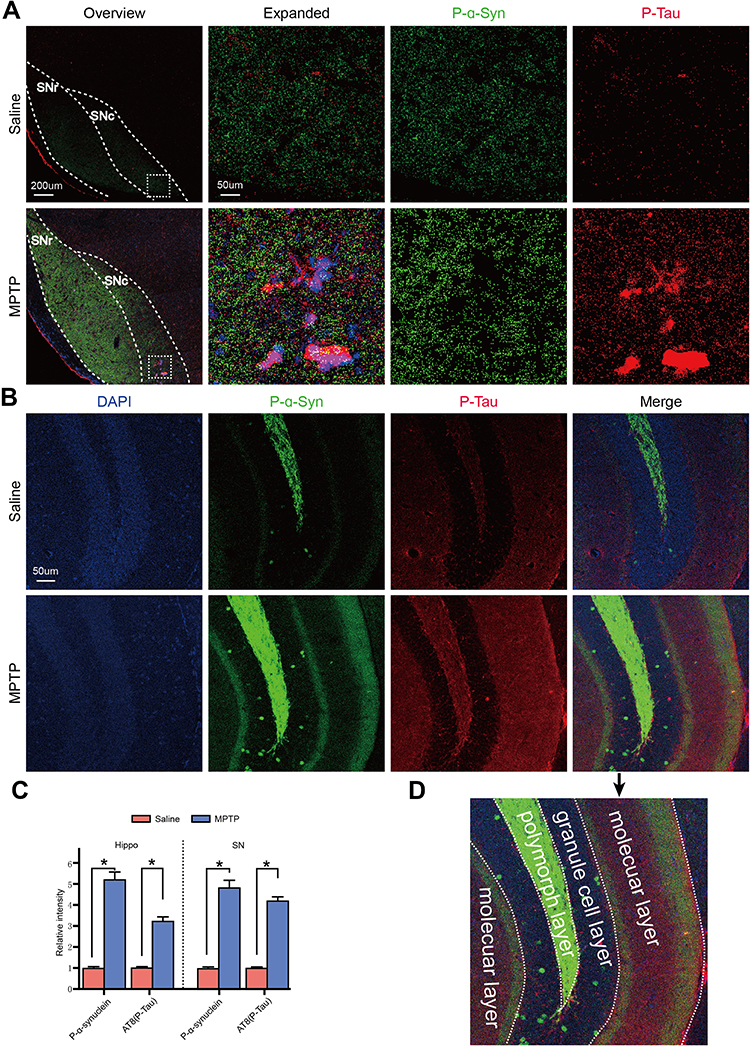 |
Figure 3 MPTP induced p-α-syn and p-Tau colocalization in SN and hippocampus. (A) Frozen sections contain SN area were co-immunostained for p-α-syn and p-Tau. MPTP induced the p-α-syn p-Tau protein increasing in SN compared to the saline group. Overlap of p-α-syn and p-Tau was observed in the expanded images. (B) Frozen sections contain hippocampal DG area were co-immunostained for p-α-syn and p-Tau. MPTP induced the p-α-syn p-Tau protein increasing in DG compared to the saline group. (C) Statistical analysis showed p-α-syn and p-Tau protein expression in SN and Hippo. (D) Schematic of the DG area showed the layers of p-α-syn and p-Tau colocalization. Overlap of p-α-syn and p-Tau was colocalized in polymorph layer and molecular layer of DG. Statistical analysis was performed using Student’s t-test. *p<0.05. n=6. |
GSK3β Activation in PD Mice Model
Immunohistochemical staining against phosphor GSK3β Y216, the activated form of GSK3β, was performed in order to investigate the kinase that may phosphorylate Tau.3 The phosphor GSK3β positive cell number in MPTP group was increased in MB, SN, CA3 and DG than that in saline group. AZD 1080 reversed the increased number of GSK3β positive cell induced by MPTP (Figure 4A–C). The Western blot analysis showed the similar results (Figure 4D and E). The phosphor GSK3β localized in the cytoplasm and neurites of the neurons. Furthermore, about 80% of cells expressed high amount of phosphor GSK3β protein in MB and, DG and CA3 of Hippo. Similar to the immunohistochemical analysis, AZD 1080 reversed the increasing level of phosphor GSK3β/Total GSK3β induced by MPTP (Figure 4D and E) in the hippocampus. As shown in Figure 4B, there was a significant difference in two groups.
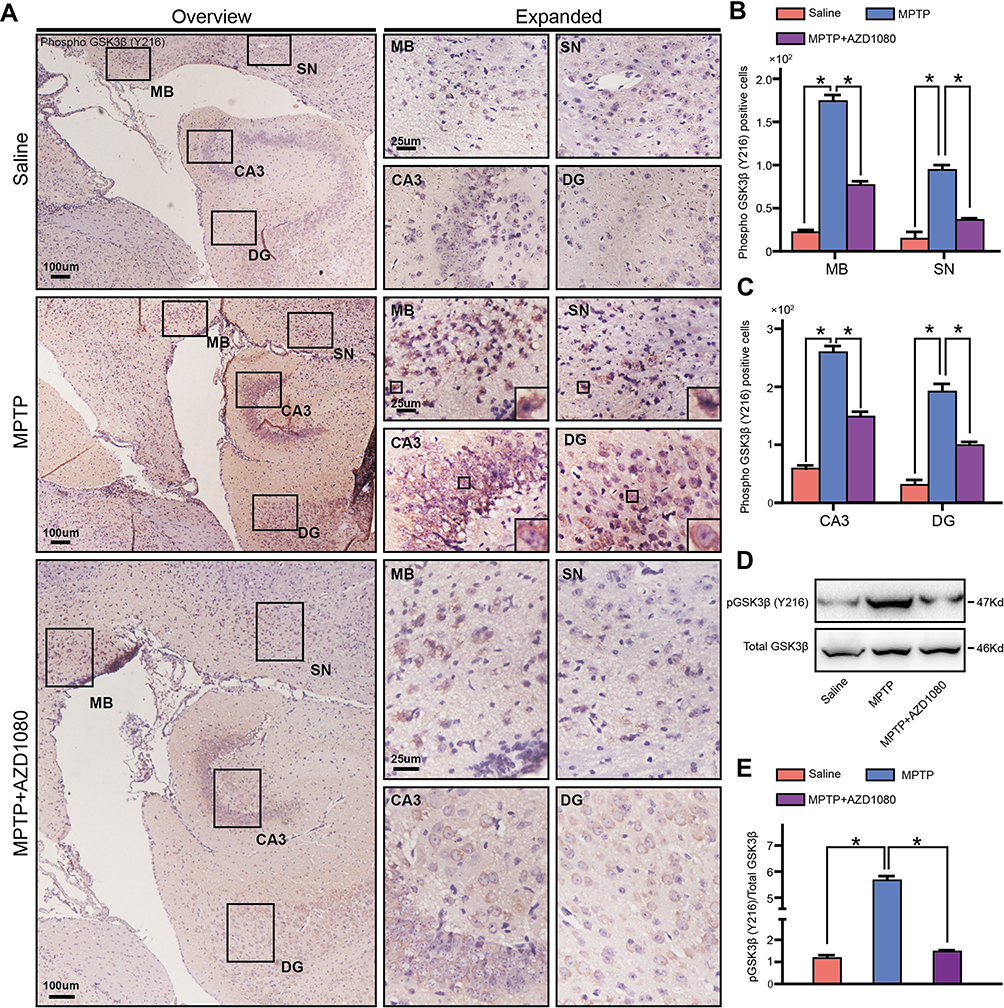 |
Figure 4 MPTP induced phosphor GSK3β in MB, SN and Hippo. (A) Paraffin sections containing MB, SN and hippocampus were staining for phospho GSK3β (Y216). (B) Statistical analysis showed MPTP treatment increased the phosphor GSK3β protein expression in MB and SN. AZD 1080 inhibit GSK3β phosphorylation level. (C) Statistical analysis showed MPTP treatment increased the phosphor GSK3β protein expression in CA3 and DG. AZD 1080 inhibit GSK3β phosphorylation level. (D) Immunoblot analysis with anti-phosphor GSK3β and anti-Total GSK3β antibody of Hippo tissues. (E) Statistical analysis showed relative phosphor GSK3β/Total GSK3β of Hippo tissues. Statistical analysis was performed using one-way ANOVA followed by Bonferroni post hoc. *p<0.05. n=6. |
Dopaminergic Neurodegeneration Evaluation
TH antibody immunofluorescence staining was conducted to test the dopaminergic neurodegeneration of SN in MPTP-treated mice. As shown in Figure 5A and C, MPTP reduced the number of dopaminergic neurons in SNc and VTA. Specifically, MPTP caused an approximately 56% and 62% reduction in the number of dopaminergic neurons in SNc and VTA, respectively. Next, we injected AAV-retro-hSyn-mCherry virus into CPu, the innervating target of SNc neurons, to retrograde label dopaminergic neurons in the SNc. Figure 5B and D showed that MPTP reduced number of retrogradely labelled neurons in the SNc when compared with saline group.
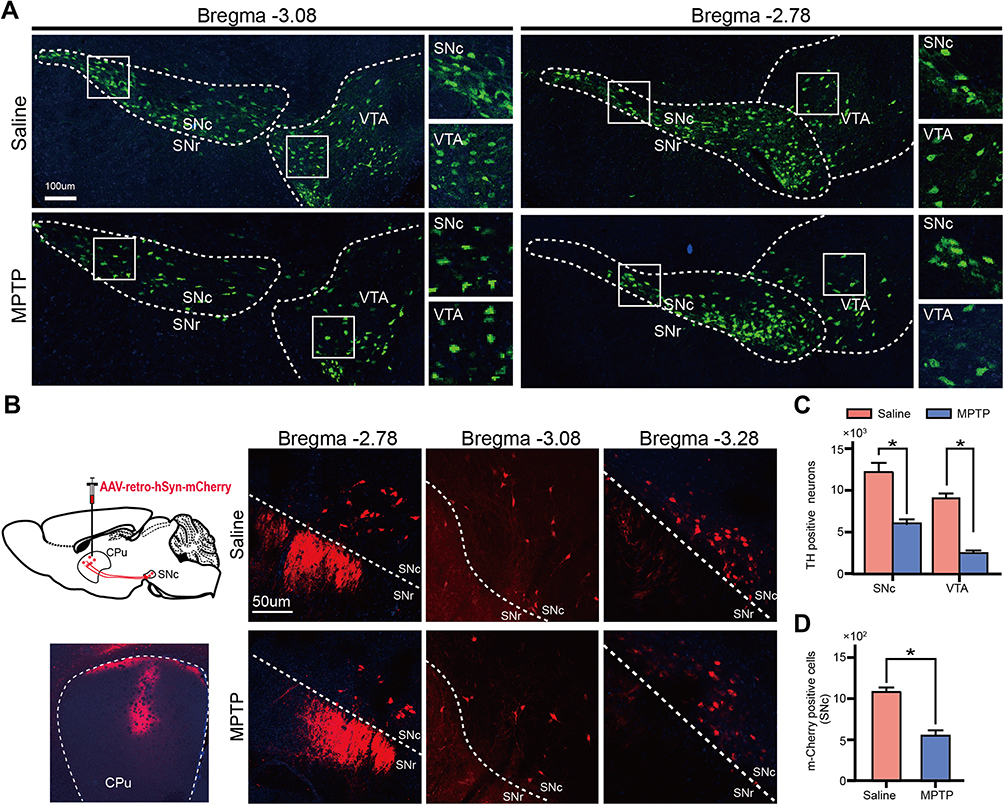 |
Figure 5 MPTP induced dopaminergic neurodegeneration. (A) Immunofluorescence of SN and VTA dopaminergic neurons by TH antibody. (B) Schematic of the retrograde labeling. AAV-retro-hSyn-mCherry virus were injected into CPu to label the dopaminergic neurons in SNc. Representative image showed AAV-retro-hSyn-mCherry virus were injected into the CPu area. The mCherry fluorescence was visualized by laser confocal microscope. (C) MPTP treatment decreased TH positive neuron number compared with the saline group in SNc and VTA. (D) Representative images showed neurons which projected to CPu in SN were labeled by AAV-retro-hSyn-td tomato virus. Data were shown as mean ± SEM. Statistical analysis was performed using Student’s t-test. *p<0.05. n=6. |
Microglia Activation in PD Mice Model
Microglial marker Iba1 antibody immunofluorescence staining was conducted in order to test the inflammation response after MPTP treatment in mice. In Figure 6, increased microglia numbers in MPTP group were observed in Hippo and SN compared with the saline group. The number of microglia in Hipp and SN of mice in the MPTP group were ten-fold and five-fold that in the saline group, respectively. Furthermore, the microglial processes were elongated and increased when compared with the saline group (Figure 6A and B).
 |
Figure 6 Microglia activation after MPTP treatment. (A) Representative photomicrographs of the Hippo and SN of frozen section immunolabeled with Iba1 (microglial marker). (B) Stereological assessment of sections showed a significant increase of the microglial number in Hippo and SN. *p<0.05, n=6. |
Microtubule Depolymerization in PD Mice Model
Immunofluorescence staining with Tau antibody was conducted to test the microtubule number of the neurons. Microtubule loss was observed in MPTP group compared with the Saline group (Figure 7A and D). Consistent with the immunofluorescence staining results, TEM results showed the number of microtubule was decreased in MPTP group compared with the Saline group (Figure 7C and G). The morphology of mitochondrial, myelin sheath, synapse was also observed using TEM. After MPTP treatment the mitochondrial cristae was disappeared and damaged (Figure 7B and E). The synaptic vesicle number was decreased in MPTP group than the Saline group (Figure 7B and F).
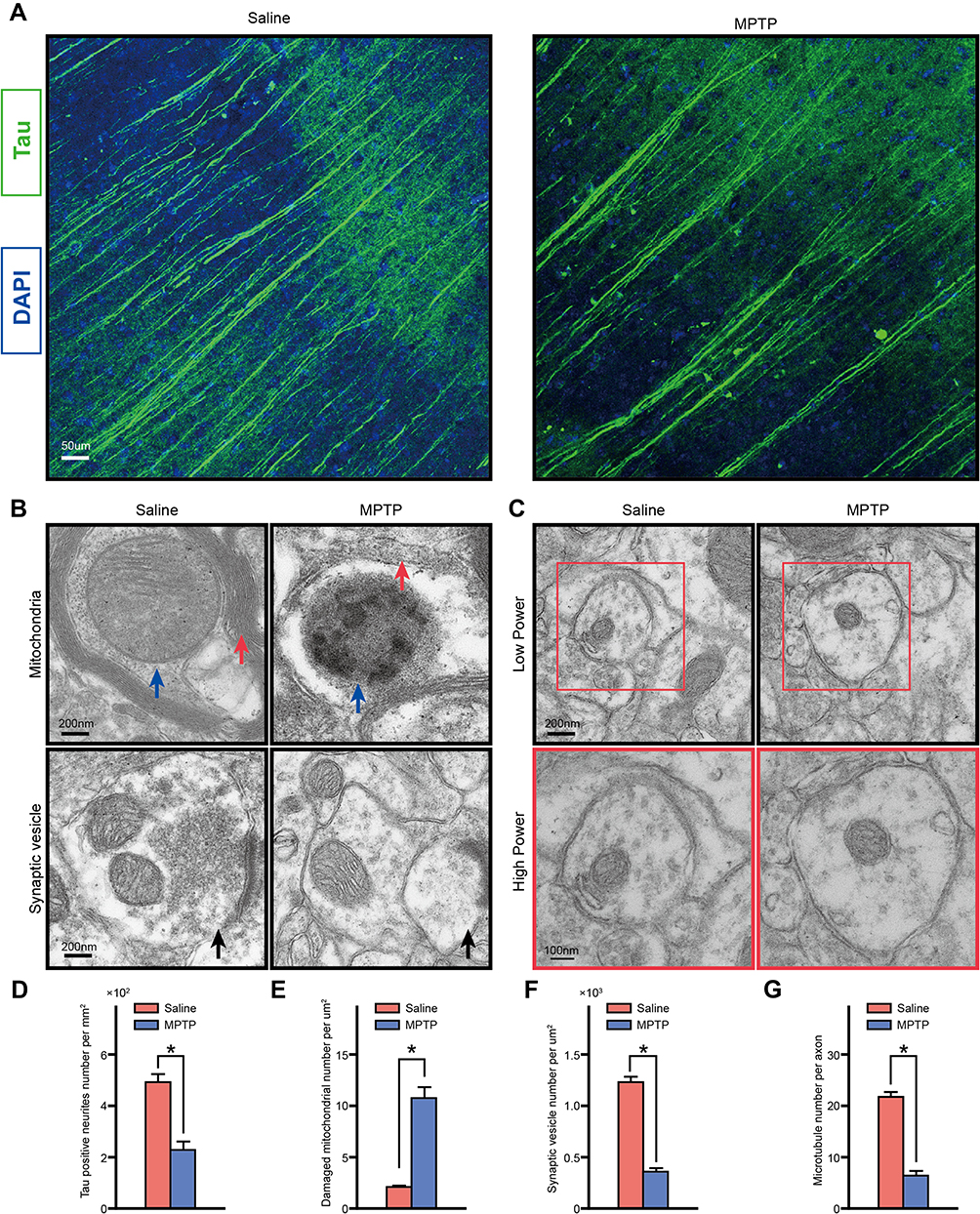 |
Figure 7 Tau phosphorylation induced neurodegeneration through microtubule depolymerization. (A) Representative photomicrographs of the Hippo of frozen section immunolabeled with Tau. (B) Electron microscopic images of the mice hippocampus showed that mitochondrial crista was disappeared and damaged, and synaptic vesicle number was decreased, and (C) microtubule loss after MPTP treatment. Red arrows indicate the myelin sheath and blue arrows indicate the mitochondrion. (D) Statistical analysis showed MPTP treatment decreased the neuritis, (E) the healthy mitochondrion number, (F) the synaptic vesicle number, and (G) the microtubule number in Hippo. *p<0.05. n=12. |
Behavioral Performance in PD Mice Model
The rotarod test, static rod test and pole were used to test the motor function of the PD mice. The rotarod test showed MPTP induced a significant loss of latency to fall from the rotarod, and AZD 1080 rescued the time loss induced by MPTP (Figure 8A). MPTP induced an increase in the orientation time and transition time in the static rod test. AZD 1080 shortened the orientation time and transition time compared to the MPTP group (Figure 8B). MPTP increased the time of moving down along the pole. AZD 1080 reversed the increase in the time of moving down induced by MPTP in the pole test (Figure 8C).
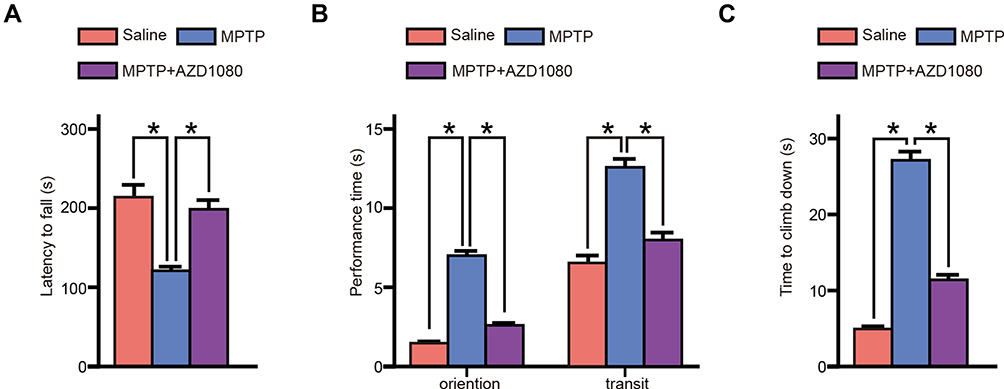 |
Figure 8 Behavioral performance in PD mice model. (A) The mice in each group were analyzed using the rotarod test. MPTP induced a significant loss of latency to fall from the rotarod, and AZD 1080 rescued the time loss induced by MPTP. (B) Statistical analysis of the static rod test showed MPTP induced an increasing time of orientation time and transit time. AZD 1080 shortened the orientation time and transit time compared to the MPTP group. (C) Statistical analysis of the pole test showed MPTP extended the time of climb down the pole. AZD 1080 reversed the increasing time induced by MPTP. Data were shown as mean ± SEM. Statistical analysis was performed using one-way ANOVA followed by Bonferroni post hoc. *p<0.05. n=6. |
Discussion
It has remained a great challenge to develop an appropriate model for PD. Despite a number of models are currently available, none of these models mimics all features of PD. MPTP model is one of the classic models and used by many researchers but it is not clear whether MPTP model is appropriate to model the main pathological hallmarks of AD such as α-Synucleinopathy and tauopathy. Previous studies have found that accumulation, phosphorylation and aggregation of Tau and α-Synuclein are features of distinct neurodegenerative diseases known as Tauopathies and Synucleinopathies, respectively.1,40 PD is characterised by α-synuclein phosphorylation and fibrillization.37,39 The hyperphosphorylation of Tau may promote its aggregation in vitro.9 However, whether Tau phosphorylation occurs in the MPTP model of PD remains unknown. In the current study, the p-α-Synuclein and p-Tau protein level, and GSK3β activation in PD mouse model were evaluated. We demonstrated that P-Tau protein was upregulated in the MPTP mouse model of PD and proposed the possible mechanism of MPTP induced neurodegeneration. Inhibiting GSK3β activation by AZD 1080 alleviated the P-Tau protein level. P-α-Synuclein and p-Tau were localized in the same brain structures such as hippocampus and SNc after MPTP treatment, forming larger plaques in PD progression. Our investigation for the first time revealed the activation of GSK3β in MPTP mouse model of PD, and revealed that both p-Tau and p-α-Syn are present together with the loss of dopaminergic neurons in the MPTP mouse model. Moreover, inhibiting GSK3β activation alleviated the motor impairment in PD model. As all these pathological changes are hallmarks of PD, our data suggest MPTP model is likely an appropriate model for PD.
The MPTP were used to produce a reliable mouse model of PD.18 Reduction of neurons in SNc which project to CPu reflects the nigrostriatal system neurodegeneration after MPTP treatment, suggesting the mouse model of PD was successful (Figure 5B and D). Consistent with the previous studies,15 after five days of MPTP injection, a 56% loss of dopaminergic cell bodies in the SNc was observed.22 The reduction in the number of retrogradely labelled SNc neurons further confirmed MPTP-induced neurodegeneration.
P-Tau protein was upregulated in the MPTP mouse model of PD. In the current study, p-α-Synuclein and α-Synuclein protein were increased in SN and Hippo areas (Figure 1A and B), which is consistent with former studies.23,33 In addition, the upregulated p-α-Synuclein mainly localized in the limbic systems such as cortex, polymorph layer of hippocampal, amygdala and VTA (Figure 2A and B). In the cellular level, the p-α-Synuclein localized in the nucleus and cytoplasm in cortex, amygdala, VTA and pyramidal layer of hippocampal CA3, and neurites in polymorph layer of hippocampal CA3. This is due to the α-Synuclein is originally localized in the nucleus and presynaptic terminals, and polymorph layer of hippocampus is rich of synapses (Figure 2A and B). A-Synuclein is mostly located in presynaptic terminals that are involved in maintaining synaptic vesicle trafficking homeostasis.19,28 The upregulation, posttranslational modification such as sumoylation and phosphorylation, and polymerization of α-Synuclein is thought to the hallmarks of PD.20,42 The fibril form of α-Synuclein in the Lewy body is the most toxic in central nervous system.11,38 Moreover, Lewy body is rich in phosphorylated α-Synuclein.16,36
It is well known that GSK3β is one of the kinases that phosphorylate Tau. Phosphor GSK3β Y216, the activated form of GSK3β, was increased in multiple brain areas including MB, SN, hippocampal DG and CA3 (Figure 4A–E). Thus, Tau phosphorylation in PD mouse model might be GSK3β -dependent. Tau phosphorylation and Aβ aggregation are pathological characteristics of AD.41 Phosphorylation of Tau including Thr231, Ser202/Thr205 and Ser396 sites occur in the AD progression.7 However, Tau phosphorylation at Ser202/Thr205 site was observed in our PD mouse model (Figure 2D), suggesting that Tau phosphorylation is also a pathological feature of PD. Using AZD 1080, a specific GSK3β inhibitor, alleviated the Tau phosphorylation level in this PD model, suggested that Tau phosphorylation in this PD model might through GSK3β pathway. Furthermore, p-α-Synuclein partially colocalized with p-Tau in Hippo and SN, and p-α-Synuclein and p-Tau formed plaques in SN (Figure 3A, C and D). This is likely due to p-α-Synuclein aggregate with p-Tau in this PD model. The total Tau protein was increased either in Hippo and SN. Previous studies showed that Tau and α-Synuclein had a synergistic fibrillization in vitro and in vivo.13,31 α-Synuclein binds to phosphorylated Ser214 Tau to promote Tau phosphorylation at Ser262 in vivo, and knockout of α-Synuclein suppressed Tau phosphorylation at Ser262.27 Multiple kinases are account for Tau phosphorylation including PKA, PP2A and GSK3β.24,41,43 Among these, GSK3β is a threonine and serine kinase, and activation of GSK3β could phosphorylate Tau in AD progression.12 Moreover, the α-Synuclein deficiency mice were resistance to the MPTP.6
In this study, the microglial processes were elongated and microglia number was increased (Figure 6A and B). Consistent with the previous studies, microglial activation was observed in the Hipp and SN after MPTP treatment.35 Microglial activation is a characteristic of neurodegenerative diseases, and the function of microglia is phagocytosing the excessive toxic protein.32,45
The microtubule number was decreased (Figure 7A, C, D and G) after MPTP treatment. In the current study, p-Tau induced neurodegeneration through microtubule depolymerization. Consistent with previous studies, Tau is thought to stabilize axonal microtubules and Tau phosphorylation leads to microtubule depolymerization.29
The motor dysfunction was observed in animal models and PD patients.2 Here, behavioral tests were used to evaluate the motor function of the mice in each group including rotarod test, static rod test and pole test. MPTP induced a severe motor impairment using different behavioral tests. Inhibiting GSK3β activation attenuated the motor impairment in PD model (Figure 8A–C). The results are consistent with previous studies, regulating GSK3β signaling might ameliorate motor deficit in early state PD.21
Conclusion
Taken together, in the PD progress, MPTP induced the phosphorylation of α-Synuclein, which leads to mitochondrial morphology damage and mitochondrial dysfunction, synaptic vesicle reduction, and motor impairment. MPTP induced the Tau phosphorylation, likely through GSK3β activation. Tau phosphorylation induced a decrease in the number of microtubules and destruction of axonal myelin sheath. These two pathways contribute to the neuronal death and neurodegeneration conjointly. These findings provide insights into mechanisms of PD, and suggest the inhibiting the GSK3β and Tau phosphorylation might be effective on PD.
Abbreviations
PD, Parkinson’s disease; TH, Tyrosine Hydroxylase; SN, substantial nigra; DG, dentate gyrus. Hippo, hippocampus; GSK3β, glycogen synthase kinase 3β; CDK5, cyclin-dependent kinase 5; MAPK, mitogen-activated protein kinase.
Acknowledgments
This work was supported by the project of National Health and Medical Research Council (NHMRC) Fellowship (1158402) and Zunyi Science and Technology Plan (2019-67).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Aksnes H, Ree R, Arnesen T. Co-translational, post-translational, and non-catalytic roles of N-terminal acetyltransferases. Mol Cell. 2019;73:1097–1114. doi:10.1016/j.molcel.2019.02.007
2. Artusi CA, Dwivedi AK, Romagnolo A, et al. Association of subthalamic deep brain stimulation with motor, functional, and pharmacologic outcomes in patients with monogenic Parkinson disease: a systematic review and meta-analysis. JAMA Netw Open. 2019;2:e187800. doi:10.1001/jamanetworkopen.2018.7800
3. Cao LL, Guan PP, Liang YY, Huang XS, Wang P. Cyclooxygenase-2 is essential for mediating the effects of calcium ions on stimulating phosphorylation of tau at the sites of Ser 396 and Ser 404. J Alzheimers Dis. 2019;68:1095–1111. doi:10.3233/JAD-181066
4. Carnwath T, Mohammed R, Tsiang D. The direct and indirect effects of alpha-synuclein on microtubule stability in the pathogenesis of Parkinson’s disease. Neuropsychiatr Dis Treat. 2018;14:1685–1695. doi:10.2147/NDT.S166322
5. Charvin D, Medori R, Hauser RA, Rascol O. Therapeutic strategies for Parkinson disease: beyond dopaminergic drugs. Nat Rev Drug Discov. 2018;17:804–822. doi:10.1038/nrd.2018.136
6. Dauer W, Kholodilov N, Vila M, et al. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci U S A. 2002;99:14524–14529. doi:10.1073/pnas.172514599
7. Davila-Bouziguet E, Targa-Fabra G, Avila J, Soriano E, Pascual M. Differential accumulation of Tau phosphorylated at residues Thr231, Ser262 and Thr205 in hippocampal interneurons and its modulation by Tau mutations (VLW) and amyloid-beta peptide. Neurobiol Dis. 2019;125:232–244. doi:10.1016/j.nbd.2018.12.006
8. Deacon RM. Measuring motor coordination in mice. J Vis Exp. 2013;75:e2609.
9. Despres C, Byrne C, Qi H, et al. Identification of the Tau phosphorylation pattern that drives its aggregation. Proc Natl Acad Sci U S A. 2017;114:9080–9085. doi:10.1073/pnas.1708448114
10. Dodiya HB, Forsyth CB, Voigt RM, et al. Chronic stress-induced gut dysfunction exacerbates Parkinson’s disease phenotype and pathology in a rotenone-induced mouse model of Parkinson’s disease. Neurobiol Dis. 2018.
11. Froula JM, Castellana-Cruz M, Anabtawi NM, et al. Defining alpha-synuclein species responsible for Parkinson disease phenotypes in mice. J Biol Chem. 2019;294:10392–10406. doi:10.1074/jbc.RA119.007743
12. Gao C, LIU Y, Jiang Y, Ding J, Li L. Geniposide ameliorates learning memory deficits, reduces tau phosphorylation and decreases apoptosis via GSK3beta pathway in streptozotocin-induced alzheimer rat model. Brain Pathol. 2014;24:261–269. doi:10.1111/bpa.12116
13. Giasson BI, Forman MS, Higuchi M, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–640. doi:10.1126/science.1082324
14. Goedert M, Compston A. Parkinson’s disease - the story of an eponym. Nat Rev Neurol. 2018;14:57–62. doi:10.1038/nrneurol.2017.165
15. Gong J, Szego EM, Leonov A, et al. Translocator protein ligand protects against neurodegeneration in the MPTP mouse model of Parkinsonism. J Neurosci. 2019;39:3752–3769. doi:10.1523/JNEUROSCI.2070-18.2019
16. Gonzalez N, Arcos-Lopez T, Konig A, et al. Effects of alpha-synuclein posttranslational modifications on metal binding. J Neurochem. 2019;150:507–521. doi:10.1111/jnc.v150.5
17. Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133:665–704.
18. Jackson-Lewis V, Przedborski S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc. 2007;2:141–151. doi:10.1038/nprot.2006.342
19. Koprich JB, Kalia LV, Brotchie JM. Animal models of alpha-synucleinopathy for Parkinson disease drug development. Nat Rev Neurosci. 2017;18:515–529. doi:10.1038/nrn.2017.75
20. Ledeen RW, Wu G. Gangliosides, alpha-synuclein, and Parkinson’s Disease. Prog Mol Biol Transl Sci. 2018;156:435–454.
21. Leikas JV, Kohtala S, Theilmann W, Jalkanen AJ, Forsberg MM, Rantamaki T. Brief isoflurane anesthesia regulates striatal AKT-GSK3beta signaling and ameliorates motor deficits in a rat model of early-stage Parkinson’s disease. J Neurochem. 2017;142:456–463. doi:10.1111/jnc.14066
22. Liang Y, Chen C, Xia B, et al. Neuroprotective effect of echinacoside in subacute mouse model of Parkinson’s disease. Biomed Res Int. 2019;2019:4379639. doi:10.1155/2019/4379639
23. Matsuo K, Cheng A, Yabuki Y, Takahata I, Miyachi H, Fukunaga K. Inhibition of MPTP-induced alpha-synuclein oligomerization by fatty acid-binding protein 3 ligand in MPTP-treated mice. Neuropharmacology. 2019;150:164–174. doi:10.1016/j.neuropharm.2019.03.029
24. Miao J, Shi R, Li L, et al. Pathological Tau from Alzheimer’s brain induces site-specific hyperphosphorylation and SDS- and reducing agent-resistant aggregation of Tau in vivo. Front Aging Neurosci. 2019;11:34. doi:10.3389/fnagi.2019.00034
25. Nisbet RM, Gotz J. Amyloid-beta and Tau in Alzheimer’s disease: novel pathomechanisms and non-pharmacological treatment strategies. J Alzheimers Dis. 2018;64:S517–S527. doi:10.3233/JAD-179907
26. Oo TF, Siman R, Burke RE. Distinct nuclear and cytoplasmic localization of caspase cleavage products in two models of induced apoptotic death in dopamine neurons of the substantia nigra. Exp Neurol. 2002;175:1–9. doi:10.1006/exnr.2002.7881
27. Qureshi HY, Paudel HK. Parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and alpha-synuclein mutations promote Tau protein phosphorylation at Ser262 and destabilize microtubule cytoskeleton in vitro. J Biol Chem. 2011;286:5055–5068. doi:10.1074/jbc.M110.178905
28. Rovere M, Powers AE, Jiang H, et al. E46K-like alpha-synuclein mutants increase lipid interactions and disrupt membrane selectivity. J Biol Chem. 2019;294:9799–9812. doi:10.1074/jbc.RA118.006551
29. Safinya CR, Chung PJ, Song C, Li Y, Ewert KK, Choi MC. The effect of multivalent cations and Tau on paclitaxel-stabilized microtubule assembly, disassembly, and structure. Adv Colloid Interface Sci. 2016;232:9–16. doi:10.1016/j.cis.2015.11.002
30. Schapira AH, Olanow CW. Neuroprotection in Parkinson disease: mysteries, myths, and misconceptions. JAMA. 2004;291:358–364. doi:10.1001/jama.291.3.358
31. Singh B, Covelo A, Martell-Martinez H, et al. Tau is required for progressive synaptic and memory deficits in a transgenic mouse model of alpha-synucleinopathy. Acta Neuropathol. 2019;138:551–574. doi:10.1007/s00401-019-02032-w
32. Smolders SM, Kessels S, Vangansewinkel T, Rigo JM, Legendre P, Brone B. Microglia: brain cells on the move. Prog Neurobiol. 2019;178:101612. doi:10.1016/j.pneurobio.2019.04.001
33. Song C, Zhang J, QI S, et al. Cardiolipin remodeling by ALCAT1 links mitochondrial dysfunction to Parkinson’s diseases. Aging Cell. 2019;18:e12941. doi:10.1111/acel.12941
34. Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci. 2017;18:101–113. doi:10.1038/nrn.2016.178
35. Tamtaji OR, Behnam M, Pourattar MA, Jafarpour H, Asemi Z. Aquaporin 4: a key player in Parkinson’s disease. J Cell Physiol. 2019;234:21471–21478. doi:10.1002/jcp.v234.12
36. Tanji K, Miki Y, Mori F, et al. Phosphorylated NUB1 distinguishes alpha-synuclein in Lewy bodies from that in glial cytoplasmic inclusions in multiple system atrophy. Brain Pathol. 2019;29:803–812. doi:10.1111/bpa.v29.6
37. Tapia-Rojas C, Cabezas-Opazo F, Deaton CA, Vergara EH, Johnson GVW, Quintanilla RA. It’s all about tau. Prog Neurobiol. 2019;175:54–76. doi:10.1016/j.pneurobio.2018.12.005
38. Twohig D, Nielsen HM. alpha-synuclein in the pathophysiology of Alzheimer’s disease. Mol Neurodegener. 2019;14:23. doi:10.1186/s13024-019-0320-x
39. Ugalde CL, Lawson VA, Finkelstein DI, Hill AF. The role of lipids in alpha-synuclein misfolding and neurotoxicity. J Biol Chem. 2019;294:9016–9028. doi:10.1074/jbc.REV119.007500
40. Walsh DM, Selkoe DJ. A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat Rev Neurosci. 2016;17:251–260. doi:10.1038/nrn.2016.13
41. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17:5–21. doi:10.1038/nrn.2015.1
42. Wegrzynowicz M, Bar-On D, Calo L, et al. Depopulation of dense alpha-synuclein aggregates is associated with rescue of dopamine neuron dysfunction and death in a new Parkinson’s disease model. Acta Neuropathol. 2019;138:575–595. doi:10.1007/s00401-019-02023-x
43. Wicinski M, Socha M, Malinowski B, et al. Liraglutide and its neuroprotective properties-focus on possible biochemical mechanisms in Alzheimer’s disease and cerebral ischemic events. Int J Mol Sci. 2019;20:1050.
44. Wong YC, Krainc D. alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23:1–13. doi:10.1038/nm.4269
45. Yao L, Zhu Z, Wu J, et al. MicroRNA-124 regulates the expression of p62/p38 and promotes autophagy in the inflammatory pathogenesis of Parkinson’s disease. FASEB J. 2019;33:fj201900363R.
46. Zhang X, Gao F, Wang D, et al. Tau pathology in Parkinson’s Disease. Front Neurol. 2018;9:809. doi:10.3389/fneur.2018.00809
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.