")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Phosphocreatine attenuates Gynura segetum-induced hepatocyte apoptosis via a SIRT3-SOD2-mitochondrial reactive oxygen species pathway
Authors Li DP, Chen YL, Jiang HY, Chen Y, Zeng XQ , Xu LL, Ye Y, Ke CQ, Lin G, Wang JY, Gao H
Received 30 January 2019
Accepted for publication 15 May 2019
Published 27 June 2019 Volume 2019:13 Pages 2081—2096
DOI https://doi.org/10.2147/DDDT.S203564
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Dong-Ping Li,1,* Ying-Ling Chen,1,* Hong-Yue Jiang,1,* Yun Chen,1 Xiao-Qing Zeng,1 Li-Li Xu,1 Yang Ye,2 Chang-Qiang Ke,2 Ge Lin,3 Ji-Yao Wang,1,4 Hong Gao1,4
1Department of Gastroenterology and Hepatology, Zhongshan Hospital, Fudan University, Shanghai, People’s Republic of China; 2State Key Laboratory of Drug Research & Natural Products Chemistry Department, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Pudong, People’s Republic of China; 3School of Biomedical Sciences, Faculty of Medicine, The Chinese University of Hong Kong, Hong Kong, Hong Kong SAR; 4Evidance-based Medicine Center of Fudan University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Purpose: To investigate the mitochondria-related mechanism of Gynura segetum (GS)-induced apoptosis and the protective effect of phosphocreatine (PCr), a mitochondrial respiration regulator.
Methods: First, the mechanism was explored in human hepatocyte cell line. The mitochondrial oxidative stress was determined by fluorescence assay. The level of sirtuin 3 (SIRT3), acetylated superoxide dismutase 2 (Ac-SOD2), SOD2, and apoptosis were detected by Western blotting. Mito-TEMPO and cell lines of viral vector-mediated overexpression of SIRT3 and SIRT3H248Y, were used to further verify the mechanism of GS-induced apoptosis. GS-induced liver injury mice models were built by GS through intragastric administration and interfered by PCr through intraperitoneal injection. A total of 30 C57BL/6J mice were assigned to 5 groups and treated with either saline, PCr (100 mg/kg), GS (30 g/kg), or PCr (50 or 100 mg/kg)+GS (30 g/kg). Liver hematoxylin and eosin (HE) staining, immunohistochemical analysis, and blood biochemical evaluation were performed.
Results: GS induced hepatocyte apoptosis and elevated levels of mitochondrial ROS in L-02 cells. The expression of SIRT3 was decreased. Downregulation of SIRT3 was associated with increased levels of Ac-SOD2, which is the inactivated enzymatic form of SOD2. Conversely, when overexpressing SIRT3 in GS-treated cells, SOD2 activity was restored, and mitochondrial ROS levels and hepatocyte apoptosis declined. Upon administration of PCr to GS-treated cells, they exhibited a significant upregulation of SIRT3 and were protected against apoptosis. In animal experiments, serum ALT level and mitochondrial ROS of the mice treated with GS and 50 mg/kg PCr were significantly attenuated compared with only GS treated. The changes in SIRT3 expression were also consistent with the in vitro results. In addition, immunohistochemical analysis of the mouse liver showed that Ac-SOD2 was decreased in the PCr and GS co-treated group compared with GS treated group.
Conclusion: GS caused liver injury by dysregulating mitochondrial ROS generation via a SIRT3-SOD2 pathway. PCr is a potential agent to treat GS-induced liver injury by mitochondrial protection.
Keywords: Gynura segetum, apoptosis, mitochondrial, ROS, SIRT3, phosphocreatine
Introduction
Gynura segetum (GS), called Tusanqi in China, is a herb used in traditional Chinese medicine and is applied externally after trauma and fractures. However, cases have consistently been reported that oral intake of GS leads to liver injury,1 causing hepatic sinusoidal obstruction syndrome (HSOS) which is characterized by hepatomegaly, ascites, and jaundice.2 It has also been reported that 16% of the patients died of liver failure after taking GS.3 However, there are currently few specific and effective therapies available for GS-induced HSOS.
GS contains high levels of pyrrolizidine alkaloids (PAs).4 PAs can induce acute toxicity, chronic toxicity, and genotoxicity by reacting with cellular proteins and DNA after they are metabolized to pyrrolic ester metabolites by the liver cytochrome P450 enzymes.5 This toxic effect can occur in many organs, including the liver, kidneys, lungs, and blood vessels, with liver injury being the most common and severe site of damage.6
Previous studies reported that PAs induce hepatotoxicity by dysregulating the cellular redox balance.7 Notably, the mitochondria are organelles that are closely associated with oxidative stress. PAs induce mitochondrial dysfunction,8,9 including loss of mitochondrial membrane potential and mitochondrial fragmentation, as well as increased release of cytochrome c into the cytoplasm, which suggests that mitochondria may be involved in PA-induced apoptosis and liver injury. However, the underlying mechanism remains unclear.
Superoxide dismutase 2, mitochondrial (SOD2), a primary antioxidant located in the mitochondrial matrix, scavenges excess mitochondrial reactive oxygen species (ROS) to maintain mitochondrial function so the cell can efficiently produce ATP.10 Sirtuin 3 (SIRT3), a member of the mammalian sirtuin family of proteins, exhibits deacetylase activity in mitochondria.11 SIRT3 binds to and deacetylates SOD2, leading to SOD2 enzymatic activation. Finally, deacetylated SOD2 exerts its antioxidant effect to maintain the mitochondrial oxidation/anti-oxidation balance and normal mitochondrial function.12 However, to the best of our knowledge, there have been no studies regarding the role of the SIRT3-SOD2 pathway in regulating mitochondrial dysfunction induced by GS. Therefore, we aim to investigate GS-induced liver toxicity and explore the underlying mechanism controlling mitochondrial dysfunction, caused by dysregulation of the oxidation/anti-oxidation system in GS-induced hepatocyte apoptosis.
Several studies demonstrated that phosphocreatine (PCr) is essential in maintaining membrane stability, regulating mitochondrial respiration to preserve ATP homeostasis, and resisting oxidative damage, thereby attenuating the occurrence of apoptosis.13–15 These suggest that it might act as a mitochondrion protective agent. Therefore, we hypothesized that PCr could act as a protective agent to prevent mitochondrial damage and designed experiments to observe the effect of PCr on GS-induced hepatocyte apoptosis, both in vivo and in vitro.
Materials and methods
GS extract and quantification of pyrrolizidine alkaloids
GS was gifted by the patients who were diagosed with a HSOS after GS taking. To purify the herbal extract for this study, 100 g dried rhizome of GS was soaked in water (200 mL) for 1 hr. The solution was then made up to 300 mL and boiled for 1 hr, as previously described.16 After boiling, the solution was filtered with a 0.22 μm Ministart® filter (Sartorius, Göttingen, Germany). The PAs present in GS were analyzed and quantified by HPLC-UV-MS.17,18 The quantification results are shown in Table S1. For animal experiments, the GS extract was concentrated to 2 g/mL for intragastrical administration to induce a mouse model of HSOS. For in vitro experiments, the GS solution was pre-frozen in liquid nitrogen, then placed in a freeze dryer (Labconco, Missouri, USA) and lyophilized to powder. GS powder was dissolved in Dulbecco’s Modified Eagle Medium (DMEM) (10–013-CV, Corning Inc., Corning, NY, USA), and then filtered with a 0.22 μm Ministart® filter to sterilize the solution.
Cell culture, plasmid construction, and transfection
The human normal liver cells L-02 and HEK293T cells (human embryonic kidney) were kindly provided by Stem Cell Bank, Chinese Academy of Sciences. The plasmid pCDH-SIRT3-EF1-Puro was held by our lab. The plasmid pCDH-SIRT3H248Y-EF1-Puro was constructed by the quick-change method using KOD-Plus (TOYOBO, Osaka, Japan). The lentivirus was packaged in the plasmids. After transfection with 10 µg lentivirus-producing vectors for 6 hrs, 293T cells were cultured with DMEM for 24 hrs. Then, the virus-containing supernatants were collected and filtered. L-02 cells were then infected with high-titration lentivirus and polybrene. Finally, cells were cultured in medium with 2.5 μg/mL puromycin to obtain cell lines that stably expressed SIRT3 and SIRT3H248Y, respectively.
In vitro experimental protocol
Cells were treated with GS at various concentrations (0, 5, 10, and 15 mg/mL) for 48 hrs. For PCr cotreatment, PCr (Laiboten, Harbin, China) was dissolved in DMEM to produce a 2 M stock solution. Then, the cells were pretreated with different concentrations of PCr (0, 2.5, 5, 10, and 20 mM) for 8 hrs, followed by 15 mg/mL GS stimulation for 48 hrs in the presence of PCr. In functional experiments, L-02 cells were treated according to the following groups: (1) Control group: L-02 cells cultured in DMEM; (2) GS group: L-02 cells exposed to 15 mg/mL GS for 48 hrs; (3) GS + PCr group: L-02 cells preincubated with 5 mM PCr for 8 hrs followed by 15 mg/mL GS for 48 hrs in the presence of PCr; (4) PCr group: L-02 cells treated with 5 mM PCr for 56 hrs; (5) Mito-TEMPO group: L-02 cells treated with 50 μM Mito-TEMPO (Sigma, St Louis, MO, USA) for 56 hrs; (6) Mito-TEMPO+GS group: L-02 cells preincubated with 50 μM Mito-TEMPO for 8 hrs followed by 15 mg/mL GS for 48 hrs in the presence of Mito-TEMPO.
Cell viability
Cell viability was assessed using the Cell Counting Kit-8 (Beyotime Biotechnology, Jiangsu, China). A total of 3×103 L-02 cells were seeded per well in 96-well plates. CCK-8 solution was added at 10 μL per well, and cells were incubated at 37°C for half an hour. Then, the plate was read using a Microplate Reader (ThermoFisher Scientific, Waltham, MA, USA) at 450 nm.
Western blotting
The protein concentration of lysates of L-02 cells and mouse liver tissue were determined using a BCA protein assay kit (Beyotime) and separated by SDS-PAGE. The primary antibodies were against caspase-9 (1:1000, A0281, ABclonal, Woburn, MA, USA), caspase-9 (1:1000, 9508T, Cell Signaling Technology, Danvers, MA, USA), caspase-8 (1:1000, 9746T, Cell Signaling Technology), caspase-3 (1:1000, 9665S, Cell Signaling Technology), cleaved-caspase-3 (1:1000, 9661T, Cell Signaling Technology), SOD2 (1:1000, 13141T, Cell Signaling Technology), SOD2/MnSOD2 (acetyl K68) (1:1000, ab137037, Abcam, Cambridge, MA, USA), SIRT3 (1:500, sc-99143, Santa Cruz, Dallas, TX, USA), and β-Actin (1:2000, Sigma).
Determination of apoptotic cell death
Apoptosis was investigated using the annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection kit (DOJINDO, Kumamoto, Japan) in accord with the manufacturer’s instructions and analyzed on a BD LSR II flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Determination of cellular ATP
Total cellular ATP content was detected using a CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA). In this assay, 4×103 cells were plated per well in an opaque 96-well format. A total of 100 μL Reagent was added per well to react with cells. After equilibration of the plate for approximately 30 mins, the results were measured using an EnVision Multilabel Plate Reader (Perkin Elmer, Waltham, MA, USA).
Measurement of mitochondrial reactive oxygen species (ROS) formation
The generation of mitochondrial superoxide was measured with MitoSOX Red (Invitrogen, Life Technologies, Carlsbad, CA, USA). First, the cells were incubated with MitoSOX Red at a concentration of 5 μM for 10 mins at 37°C and washed three times with Hanks Balanced Salt Solution (HBSS). The images were captured using a Bx51 fluorescence microscope (Olympus, Tokyo, Japan), and the level of fluorescence was detected using BioTek Synergy NEO microplate reader (BioTek, Vermont, USA) at 510/580 nm. For in vivo experiments, freshly isolated, frozen, and non-fixed mouse livers were incubated with MitoSOX Red (5 μM) for 10 mins at 37°C after being sectioned by cryostat at 10 μm. Then, the solution on the slides was removed, and slides were imaged with an inverted confocal microscope FV1200 (Olympus). Finally, the level of fluorescence was quantified using Image-Pro Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD, USA).
Measurement of the mitochondrial transmembrane potential
Tetramethylrhodamine, methyl ester (TMRM) (Invitrogen) was used to measure the mitochondrial transmembrane potential. Notably, the signal is bright in healthy cells with functioning mitochondria. Hoechst 33342 was used to stain nuclei in live L-02 cells. Cells were incubated with 100 nM TMRM for 30 mins at 37°C, after washing 3 times with HBSS, cells were then incubated with Hoechst 33342 for 10 mins at 37°C. The cells were incubated in DMEM until use and images were collected on an inverted confocal FV1200 microscope (Olympus). Finally, the fluorescence intensity was detected using BioTek Synergy NEO microplate reader (BioTek) at 548/574nm.
SOD2 activity
Cu/Zn-SOD and Mn-SOD Assay Kit with WST-8 (Beyotime) was used to determine SOD2 enzymatic activity in accordance with the manufacturer’s instructions. The protein concentrations were measured by BCA assay. The absorption at 450 nm was recorded using a Microplate Reader (Thermo).
Quantitative real-time PCR (qPCR) analysis
Total RNA was extracted from the L-02 cell line using an RNA simple Total RNA Kit (Tiangen, Beijing, China). Then, cDNA was produced using Superscript II reverse transcriptase (TOYOBO). Quantitative PCR amplification was subsequently performed on a CFX96 real-time PCR machine (Bio-Rad, Hercules, CA, USA) using SYBR Green (TOYOBO). GAPDH was used to normalize the relative abundances of the indicated genes. The primer sequences are presented in Table S2.
Mouse model establishment and liver injury assessment
Female C57BL/6J mice (6–8 weeks old) were used in our study. All animals were purchased from VitalRiver Laboratory (Beijing, China) and were kept in an environmentally controlled room with a 12-hr light/dark cycle with ad libitum access to food and water. All animal experimental procedures complied with Guide for Care and Use of Laboratory Animals published by the US National Institutes of Health (8th edition, revised 2011) and were approved by the Animal Care and Use Committee of Zhongshan hospital, Fudan University. After 1 week of adaptive feeding, 30 mice were randomly divided into 5 groups. They were given normal saline, PCr (100 mg/kg/d), GS (30 g/kg/d), PCr (50 mg/kg/d)+GS (30 g/kg/d), or PCr (100 mg/kg/d)+GS (30 g/kg/d), respectively, and were euthanized after 3 weeks of treatment. Liver injury was assessed by the determination of the serum alanine aminotransferase (ALT) activity, aspartate transaminase (AST) activity, and hematoxylin and eosin (H&E) staining of liver sections. Immunohistochemistry was performed against target molecules on paraffin sections utilizing primary antibody against Ac-SOD2 (1:100, ab137037, Abcam) and cleaved-caspase 3 (1:200, 9661T, Cell Signaling Technology).
Statistical analysis
All data were analyzed using GraphPad Prism-7 software (GraphPad Software, La Jolla, CA, USA). Student’s two-tailed t-test and one-way ANOVA were used to determine the statistical significance of differences and values of p<0.05 were considered statistically significant. The results are presented as the mean±SEM. At least three independent replicates of experiments were collected.
Results
GS induced apoptosis in L-02 hepatocytes
To identify the influence of Gynura segetum in the liver, L-02 cells were treated with GS at various concentrations. Notably, GS decreased cell viability in a dose-dependent manner (Figure 1A). When the cells were exposed to 15 mg/mL GS for 48 hrs, the cell viability was reduced to 59.12±5.15% compared with the control group. Next, apoptosis was examined. Cells treated with GS had a decrease in caspase 9, caspase 8, and caspase 3 levels, and an increase in cleaved-caspase 9, cleaved-caspase 8, and cleaved-caspase 3 (Figure 1B). Furthermore, flow cytometry was performed to verify the effect of GS on hepatocyte apoptosis. It was found that L-02 cells treated with 15 mg/mL GS had a significant increase in apoptosis compared with the control group (p<0.001) (Figure 1C and D). Accordingly, these results demonstrated that GS induces apoptosis in a dose-dependent manner.
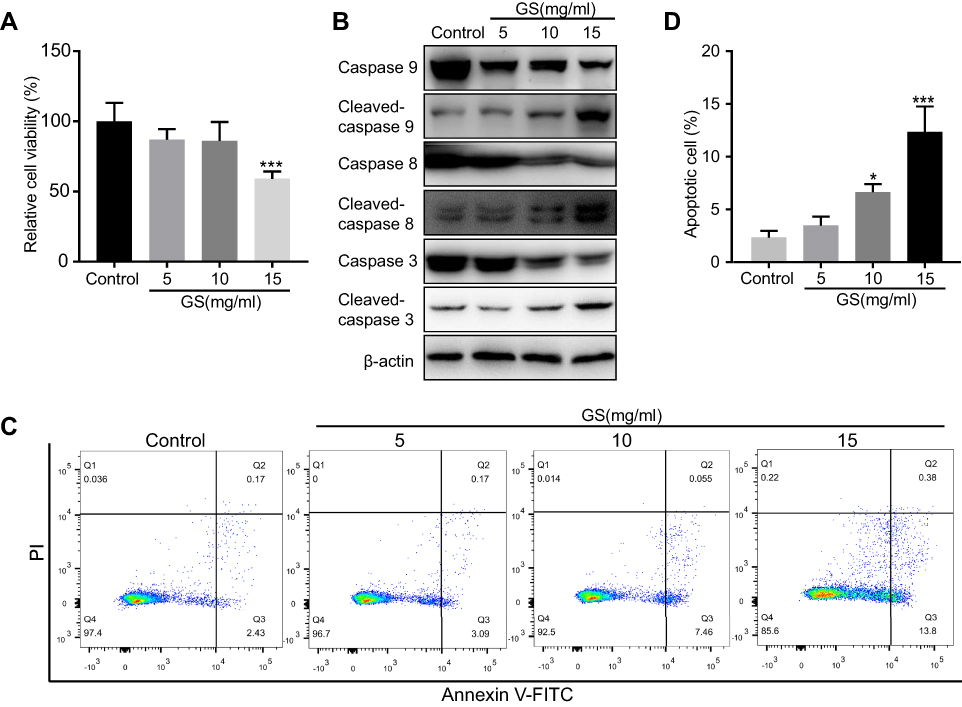 |
Figure 1 Effect of Gynura segetum on apoptosis in the L-02 human hepatocyte cell line. (A) L-02 cells were exposed to different concentrations of GS for 48 hrs, then cell viability determined by CCK8. (B) The expression of apoptosis-related proteins was detected by Western blotting under different concentrations of GS. (C and D) Flow cytometry analysis of apoptosis in L-02 cells treated with 5, 10, and 15 mg/mL GS for 48 hrs. The bar chart shows the proportion of apoptotic cells from three independent assays. The apoptotic rate was calculated as the second quadrant plus the fourth quadrant. Values are the mean±SEM. *p<0.05, ***p<0.001. |
Mitochondrial reactive oxygen species mediates GS-induced apoptosis
It has been reported that ROS are involved in apoptosis in many different diseases19 and that the major component of GS, PAs, induces mitochondrial dysfunction.9 We hypothesized that GS-induced apoptosis was related to the disturbance of mitochondrial stability and especially mitochondrial ROS. A total of 15 mg/mL GS was adopted to induce marked apoptosis in the next experiments based on the previous results. Notably, intracellular adenosine triphosphate (ATP) levels, mainly generated from mitochondria, were decreased 0.55-fold in the GS group when compared with the control group (Figure 2A). Importantly, the decrease in ATP reflects the dysfunction of mitochondria to some extent. To further explore the changes in mitochondrial ROS, MitoSOX Red, a fluorogenic dye for highly selective detection of superoxide in the mitochondria, was used to observe mitochondrial superoxide levels. L-02 cells treated with GS had higher levels of mitochondrial ROS accumulation compared with the control group, as shown by increased MitoSOX Red staining (Figure 2B and C). Thus, mitochondrial oxidative stress was observed in the GS group. In addition, the brightness of TMRM fluorescence was significantly decreased in the GS group, indicating a collapse of mitochondrial membrane potential and mitochondrial dysfunction (Figure 2D and E). Furthermore, the mitochondria-targeted antioxidant Mito-TEMPO, which inhibits mitochondria-derived ROS formation, rescued cells from apoptosis induced by GS (Figure 2F). Taken together, these data suggested that GS-induced apoptosis correlated with mitochondrial dysfunction and that mitochondrial ROS mediated GS-induced cell apoptosis.
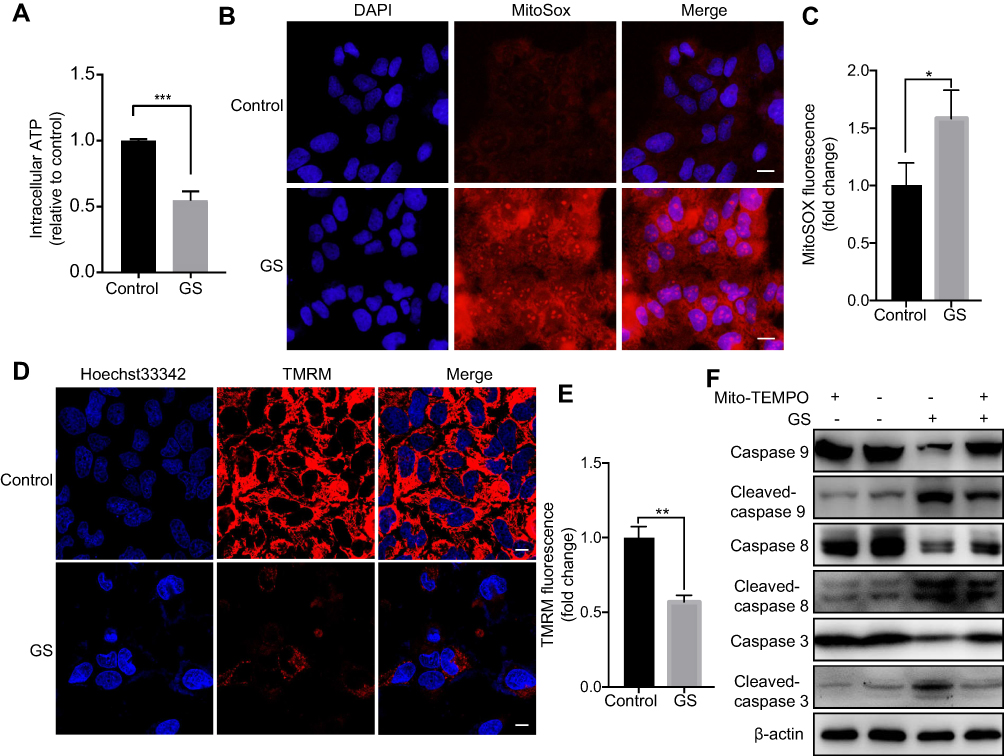 |
Figure 2 Gynura segetum induced apoptosis by increasing mitochondrial ROS. L-02 cells were treated with or without 15 mg/mL GS. (A) The bar chart shows intracellular ATP levels decreased in the GS group compared with the control. (B) Representative fluorescence images showing mitochondrial ROS (red) and nuclei (blue), stained with MitoSOX Red and DAPI, respectively. The scale bar is 20 μm. (C) The MitoSOX Red fluorescence was detected by microplate reader in 3 independent assays. (D) Mitochondrial membrane potential stained with TMRM was observed by confocal microscope in live L-02 cells. Scale Bar, 20 μm. (E) TMRM fluorescence was detected by microplate reader in 3 independent assays. (F) Cells were preincubated with Mito-TEMPO (50 μM) for 8 hrs and then treated with GS and Mito-TEMPO, the level of apoptosis was determined. Values are the mean±SEM. *p<0.05, **p<0.01, and ***p<0.001. |
GS induced apoptosis via a SIRT3-SOD2-mitochondrial reactive oxygen species pathway
Since GS induced increased mitochondrial ROS in cells, we hypothesized that it might be related to an imbalance of the mitochondrial oxidation/anti-oxidation system. SOD2, which localizes to mitochondria and helps to eliminate mitochondrial ROS, plays a protective, anti-apoptotic role against oxidative stress.10,20 We found there was no change in the levels of total SOD2 mRNA and protein expression under normal condition (Figure 3A and B). However, GS treatment significantly decreased SOD2 enzymatic activity compared with the control (Figure 3C). Acetyl-SOD2 is the inactivated form of SOD2. When investigating Ac-SOD2 levels, we found that they were increased 2.7-fold in the GS group compared with the control (Figure 3A). SIRT3 has also been implicated in cell metabolisms, and it possesses NAD+-dependent deacetylase activity, including the regulation of SOD2 deacetylation.21 Importantly, GS-administration significantly decreased SIRT3 mRNA and protein levels (Figure 3D and E). Therefore, the inhibition of SIRT3 might result in the accumulation of Ac-SOD2 in GS-stimulated L-02 cells. Ac-SOD2 accumulation may inhibit mitochondria ROS elimination and thus led to apoptosis.
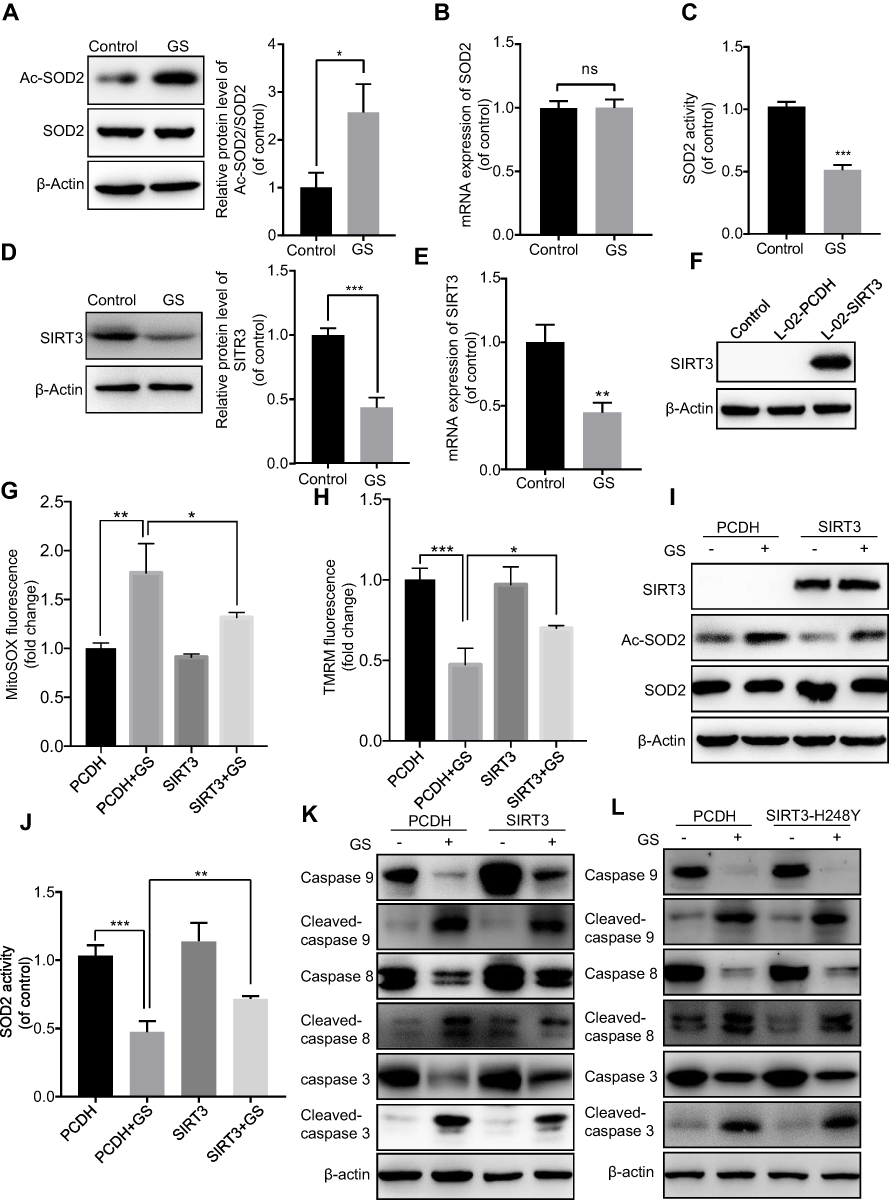 |
Figure 3 Sirtuin 3 mediated Gynura segetum induced apoptosis. L-02 cells were treated with or without 15 mg/mL GS. (A) Representative immunoblot bands of SOD2 and Ac-SOD2 protein, and quantitative analysis of Ac-SOD2 protein expression. (B) SOD2 mRNA level in control and GS treated cells assessed by reverse transcription-PCR. (C) SOD2 activity is shown as a percentage of the control group. (D) Representative immunoblot and quantitative analysis of SIRT3 protein. (E) The mRNA level of SIRT3. (F) Western blot analysis confirmed the overexpression of SIRT3 protein in L-02 cells through lentiviral infection. (G) Fluorescence intensity analysis of mitochondrial ROS. (H) Fluorescence intensity analysis of mitochondrial membrane potential. (I) Immunoblot detection of SIRT3, AC-SOD2, and SOD2 protein. (J) SOD2 activity is shown as a percentage of the control group. (K) Immunoblot detection of apoptosis-related protein in cells overexpressing SIRT3. (L) Immunoblot detection of apoptosis-related protein in cells overexpressing SIRT3H248Y. Values are the mean±SEM. *p<0.05, **p<0.01, and ***p<0.001. |
To further verify the role of SIRT3 in GS-induced apoptosis, we stably overexpressed SIRT3 in the L-02 cell line using a lentivirus (Figure 3F). When overexpressing SIRT3, the mitochondrial ROS was decreased (Figure 3G), and mitochondrial membrane potential was rescued after GS administration (Figure 3H). The protein level of Ac-SOD2 was decreased, and the SOD2 activity was increased accordingly after GS administration in overexpressing SIRT3 cell line (Figure 3I, J and Figure S2A). Regarding apoptosis, there was restoration of caspase 9, 8, and 3 levels and a decline in cleaved-caspase 9, 8, and 3 (Figure 3K and Figure S2B), indicating that overexpression of SIRT3 attenuates apoptosis induced by GS in hepatocytes. Next, the crucial role of the deacetylase activity of SIRT3 in GS-induced cell apoptosis was validated. SIRT3H248Y, which lacks deacetylase activity, was overexpressed in L-02 cells. Western blotting showed that overexpression of the mutant SIRT3H248Y did not rescue GS-induced apoptosis when compared with the wildtype (Figure 3L and Figure S2C). Taken together, these results demonstrated that GS induces apoptosis through a SIRT3-SOD2-mitochondrial ROS pathway.
Phosphocreatine ameliorates GS-induced apoptosis via a SIRT3-SOD2 pathway in L-02 cells
Many studies have found that PCr can protect cells from apoptosis induced via the mitochondrial pathway.14,15,22 The predominant role of PCr in the human body is to maintain ATP for muscular activity. Considering this ability of PCr to restore ATP levels, the suggested therapeutic usages include conditions related to energy exhaustion or energy demands aggravation, such as in ischemic stroke and cardiac bypass surgery. We therefore explored its potential utility in GS-induced apoptosis. As shown in Figure 4A and B, PCr rescued GS-induced apoptosis in L-02 cells at 5 mM, and there was no effect on cell viability when cells were treated with PCr alone in the range of 0 to 20 mM. Next, we tested whether PCr inhibited mitochondrial oxidative stress. The results of MitoSOX Red fluorescence staining showed that mitochondrial ROS were decreased in the PCr co-treated group compared with the GS-treated group, indicating mitochondrial ROS formation was reduced by PCr (Figure 4C and D). Consistent with these results, the collapse of mitochondrial transmembrane potential was also rescued in GS co-treated with PCr cells (Figure 4E and F).
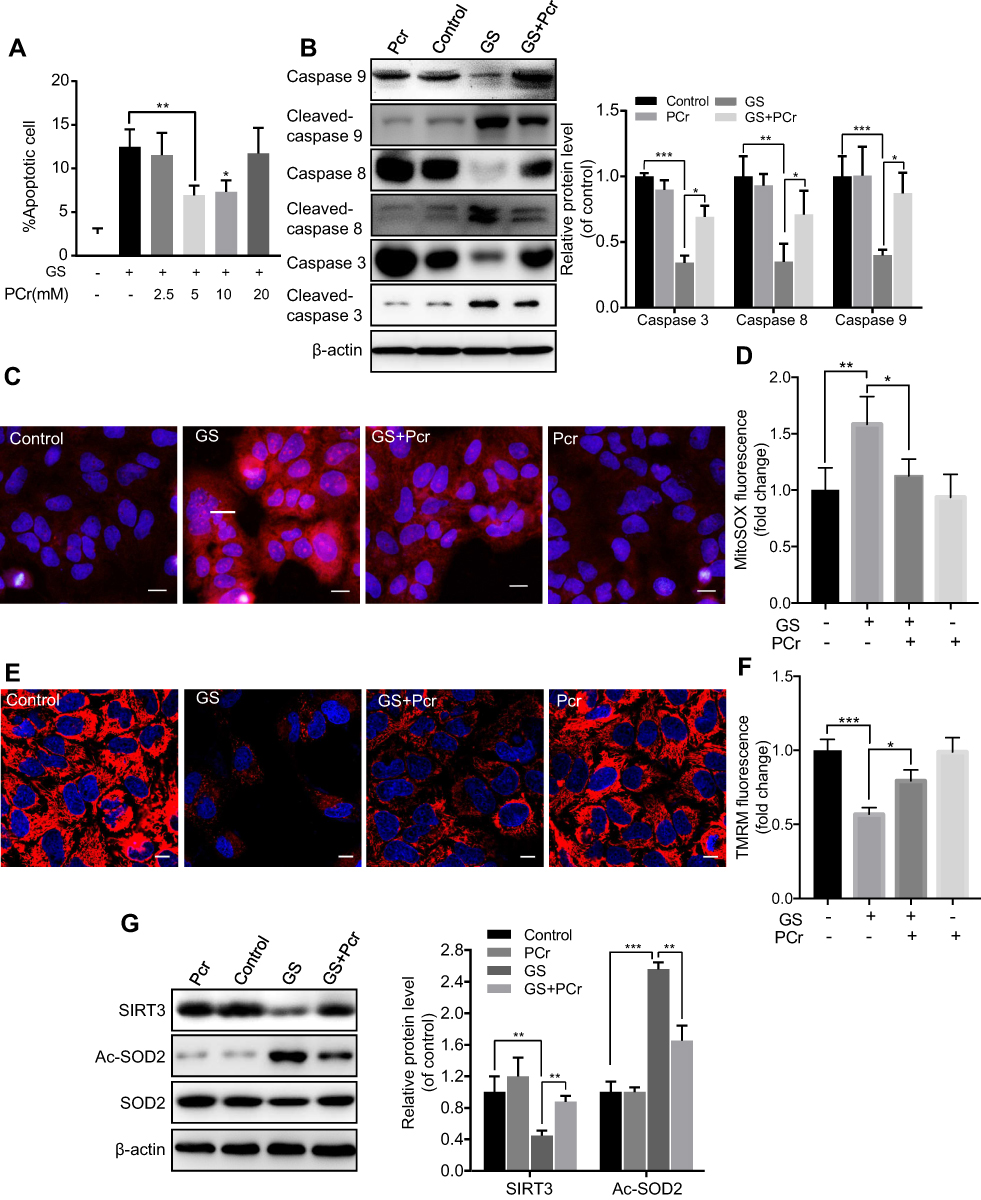 |
Figure 4 Phosphocreatine protected against Gynura segetum-induced apoptosis involving a SIRT3-SOD2 pathway in L-02 cells. (A) Quantitative analysis of apoptosis in L-02 cells treated with 15 mg/mL GS and different concentrations of PCr. (B) Representative Western blot bands of apoptosis-related protein and quantitative analysis of caspase 9, 8, and 3 protein expression. (C) Representative fluorescence images of mitochondrial ROS. (D) Fluorescence intensity analysis of mitochondrial ROS. (E) Representative fluorescence images of mitochondrial membrane potential. (F) Fluorescence intensity analysis of mitochondrial membrane potential. (G) Representative Western blot bands and quantitative analysis of SIRT3 and Ac-SOD2 protein expression in L-02 cells. Values are the mean±SEM. *p<0.05, **p<0.01, and ***p<0.001. |
We have thus demonstrated that GS induced apoptosis through the SIRT3-SOD2-mitochondrial ROS pathway and that PCr could rescue cells from apoptosis induced by GS. We then considered the effect of PCr on this pathway. Therefore, we detected the protein levels of SIRT3 and Ac-SOD2 in the PCr co-treated group. The administration of PCr increased SIRT3 expression and caused a consequent reduction in Ac-SOD2 in GS-treated L-02 cells (Figure 4G). Overall, these findings suggested that PCr ameliorated GS-induced apoptosis by increasing SIRT3 expression and Ac-SOD2 accumulation in L-02 cells.
Phosphocreatine protects against GS-induced liver injury by a SIRT3-SOD2 pathway in mice
We established a GS-induced liver injury model in mice via intragastric GS administration. Considering that hepatomegaly is a typical clinical symptom of patients with liver injury, we recorded the ratio of liver weight to body weight of mice. Importantly, the ratio of liver to body weight in the GS group was significantly higher than the control group (4.27±0.31% vs 5.70±0.16%, p=0.004) (Figure 5A). We then performed H&E staining of liver tissue to observe the liver injury caused by GS. Notably, GS induced apoptosis, necrosis, vacuolar degeneration, marked hepatic sinus dilatation, and hemorrhage (Figure 5D). The damage caused by GS mainly occurred around the central vein (CV) and spread to the portal vein (PV). Thus, we have demonstrated that intragastric GS administration induced liver injury in mice similar in many aspects to human patients with HSOS, specifically hepatomegaly and the H&E staining pattern of liver injury. Thus, this model was adopted to further validate the in vitro results.
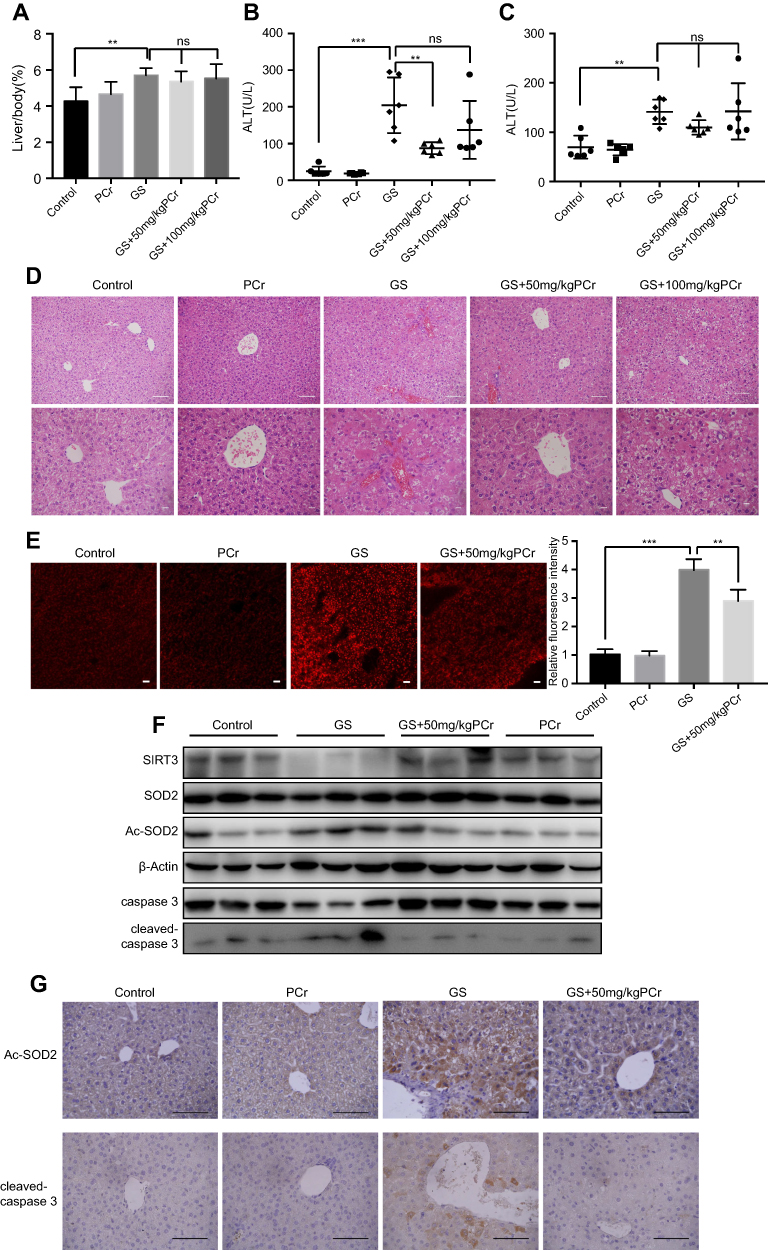 |
Figure 5 Phosphocreatine attenuated Gynura segetum-induced liver injury in mice. (A) The liver/body weight ratios were calculated for 5 groups (n=6). (B and C) Serum ALT and AST levels were analyzed. (D) Changes in the histopathology of mouse livers by H& E staining. First row, scale bar, 50 μm. Second row, scale bar, 20 μm. (E) Mitochondrial ROS stained with MitoSOX Red in mouse liver tissues were observed by confocal microscope, with the bar chart showing quantification of MitoSOX fluorescence. Scale bar is 100 μm. (F) Representative Western blot bands of SIRT3, Ac-SOD2, SOD2, caspase 3, and cleaved-caspase 3 protein expression in mouse liver. (G) Representative images of liver sections for immunostaining analysis using Ac-SOD2 antibody and cleaved-caspase3 antibody respectively. Scale bar, 100 μm. Values are the mean±SEM. **p<0.01, ***p<0.001. |
To determine whether PCr can protect against GS-induced liver injury in vivo, mice were co-treated with PCr (50 or 100 mg/kg) by intraperitoneal injection. The additional administration of PCr (50 or 100 mg/kg) had no significant effect on the ratio of liver to body weight compared with the GS-treated mice (Figure 5A). H&E staining, however, showed significant attenuation of liver injury in the 50 mg/kg PCr co-treated group (Figure 5D), indicating that low dose of PCr could protect against GS-induced liver injury in mice. Furthermore, we tested serum levels of ALT and AST, both of which are well-established indicators of liver injury. There was a marked elevation of ALT and AST in mice administered GS (Figure 5B and C). Treatment with 50 mg/kg PCr significantly decreased ALT levels as compared with the GS group (p=0.0035) (Figure 5B). Altogether, these results demonstrated that 50 mg/kg PCr could alleviate GS-induced liver injury in vivo. Therefore, 50 mg/kg PCr group was adopted to the following experiments. As described earlier, PCr attenuated GS-induced mitochondrial ROS elevation in vitro. To explore the changes in mitochondrial ROS in vivo, we measured mitochondrial ROS levels in mouse liver tissues by using 50 mg/kg PCr. We observed that mitochondrial ROS were elevated by GS, but were significantly lower in the PCr co-treated mice versus the GS group (Figure 5E). Next, we examined the expression of SIRT3 in the mouse liver and found that PCr restored the GS-mediated SIRT3 reduction in protein levels in mice (Figure 5F). Meanwhile, the upregulation of acetylated-SOD2 induced by GS was also attenuated by PCr, with the total amount of SOD2 remaining constant (Figure 5F). Immunohistology analysis of the mouse liver also validated the changes in Ac-SOD2 protein level in GS-induced liver injury and PCr co-treated group (Figure 5G).
Western blots and immunohistological analysis showed that PCr could alleviate GS-induced liver cells apoptosis (Figure 5F and G). Thus, we have successfully established a GS-induced liver injury mouse model and found that PCr protects against GS-induced liver injury, via the SIRT3-SOD2 pathway in mice.
Discussion
GS is widely used in China and more than 600 cases of liver injury after GS administration have been reported in recent years.2,23,24 However, there are currently few effective drugs to cure this disease, resulting in a high mortality rate. Previous studies have found hepatotoxic PAs cause liver cell damage and mitochondrial dysfunction.9,25 PAs cause hepatotoxicity through the mitochondria-mediated apoptotic pathway.26 However, few studies have been undertaken to uncover the mechanism of PA-induced hepatotoxicity on mitochondria. In our study, we focused on the specific mechanism underlying GS-induced hepatotoxicity and revealed that GS induced apoptosis through SIRT3-SOD2-mediated mitochondrial oxidative stress.
First, GS was found to cause hepatocyte cytotoxicity in a dose-dependent manner. Previous studies have shown that PAs can induce apoptosis.8,27 Our result was therefore consistent with previous studies, and we also found that GS can induce apoptosis in a dose-dependent manner. GS-induced hepatotoxicity is attributed to oxidative stress.7,28 Being central to energy metabolism, mitochondria are the main organelles producing ROS, and high levels of ROS are harmful to cells.29,30 ROS also play an important role in apoptosis.31 Therefore, we hypothesized that the increase in mitochondrial ROS is a key mechanism for the induction of apoptosis in GS.
Here, we found GS can increase mitochondrial ROS levels. TMRM signal, which accumulates in active mitochondria with intact membrane potentials, was dimmed or disappeared in GS-treated cells. These findings suggest that GS causes depolarization and collapse of the mitochondrial membrane potential. Furthermore, after administration of Mito-TEMPO, a specific scavenger of mitochondrial superoxide, cells were partially rescued from GS-induced apoptosis. These results suggest that GS induces apoptosis by increasing mitochondrial ROS. Applying antioxidants or a protective agent targeting mitochondrial may be a potential way to prevent GS-induced cytotoxicity. In addition, since sinusoidal structure and endothelial cells are also the targets in GS-induced liver injury based on previous studies,32 it would be interesting to investigate GS-induced oxidative stress in the endothelial cells.
We have confirmed that increased mitochondrial ROS can mediate apoptosis in GS-treated cells, but it was unclear how GS specifically leads to mitochondrial ROS elevation. Steady-state regulation of mitochondrial ROS is affected by the tricarboxylic acid cycle, electron transport chain, oxidative phosphorylation, and various enzymes involved in mitochondrial free radical scavenging systems, among which SOD2, the most important antioxidant in mitochondria, is involved in maintaining mitochondrial oxidation/antioxidant system balance.11,33 Overexpression of SOD2 can rescue cells from oxidation-induced apoptosis.34 Here, we found that GS did not alter the expression of overall SOD2 but affected its activity, with GS increasing the levels of Ac-SOD2, the form of SOD2 with no antioxidant activity, in L-02 cells. The increased levels of Ac-SOD2 were further verified by immunohistology in liver tissues of patients with liver injury caused by administration of GS (Figure S1), supporting our hypothesis that GS induces hepatic apoptosis through disrupting mitochondrial ROS levels.
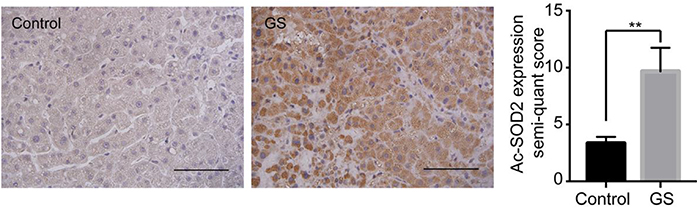 |
Figure S1 Representative images of liver sections from patients for immunostaining analysis using an Ac-SOD2 antibody. Scale bar, 100 μm. **p<0.01. |
Ac-SOD2 relies on the deacetylation function of SIRT3 to become active SOD2.35 SIRT3, localized to the mitochondria, maintains mitochondrial homeostasis by deacetylating its downstream target molecules, including SOD2.36 After GS treatment, the expression of SIRT3 was reduced both at the protein and mRNA level.
To further verify that the reduction in SIRT3 is correlated with GS-induced apoptosis, we applied GS to cell lines with overexpressing SIRT3. The higher level of SIRT3 significantly attenuated GS-induced apoptosis, while SIRT3H248Y, having lost its deacetylation function, could not decrease apoptosis. This indicates that SIRT3 inhibits GS-induced apoptosis, potentially through upregulating SOD2’s antioxidation activity with subsequent clearing of mitochondrial-derived ROS. Because SIRT3 inhibition and SOD2 acetylation play an etiological role in GS-induced hepatotoxicity, they might serve as therapeutic targets to rescue GS-induced apoptosis. In addition, our experimental results demonstrated that GS-induced apoptosis could not be prevented by overexpressing SIRT3 completely, which indicates the reduction of SIRT3 is just one of the pathways of GS-induced apoptosis. GS could also induce apoptosis through another mechanism such as formation of pyrrole-ATP5B adduct.37
Moreover, PCr can inhibit the excessive production of ROS in cells and restore mitochondrial membrane potential,15,22 and PCr has been shown to reduce apoptosis.13–15 Therefore, we tested whether PCr can protect against GS-induced apoptosis. We found that 5mM PCr attenuated GS-induced apoptosis significantly in L-02 cells. PCr partially reduced mitochondrial ROS and restored mitochondrial membrane potential caused by GS. In addition, we found that PCr rescued SIRT3 protein level in GS co-treated cells. The expression level of Ac-SOD2 was reduced, and SOD2 activity was partially restored. These findings suggested that PCr inhibited mitochondrial ROS production and rescued cells from apoptosis through the SIRT3-SOD2 pathway. However, these findings require further verification. As for the potential mechanism by which PCr regulates SIRT3 expression, we supposed that PCr might facilitate the expression of SIRT3 by activating the PI3K/AKT-PGC1α signaling pathway.38
Then, we verified this hypothesis in vivo. We treated mice with a dose of 30 g/kg/d GS and 50 or 100 mg/kg/d PCr, where the dose of GS was consistent with a previously published paper on GS-induced liver injury.16 After three weeks of administration, serum ALT and AST levels, and H&E staining of liver tissues showed GS induced liver injury and that GS consistently induced apoptosis. Notably, 50 mg/kg PCr significantly attenuated the increased level of ALT caused by GS, while 100 mg/kg PCr did not show a significant rescue effect. Furthermore, histological analysis also showed that 50 mg/kg PCr markedly reduced the areas of hepatocyte injury with little effect on hepatic sinus hemorrhage. These data proved that low dose of PCr attenuates GS-induced hepatotoxicity, which was consistent with the results in L-02 cells. This may provide the basis for clinical investigations. As 50 mg/kg PCr showing alleviation for GS-induced liver injury, the 50 mg/kg PCr group was adopted for the next experiments. We further detected mitochondrial ROS in the liver of mice and found that the high level of mitochondrial ROS induced by GS was also attenuated by PCr. Thus, the changes in SOD2, Ac-SOD2, and SIRT3 induced by GS in mice were reversed by PCr, which was consistent with in vitro results. The proposed model of how PCr attenuates GS-induced liver injury was shown in Figure 6. Therefore, PCr attenuates GS-induced liver injury by acting on mitochondria to rescue mitochondrial function. Similarly, PCr may possibly be applied to other types of drug-induced liver injury that damage mitochondria, such as acetaminophen. In addition, PCr was used by intraperitoneal injection in our study, instead of regular intravenous injection. Further studies are needed to investigate the detailed pharmacokinetics of PCr in the treatment of GS-induced liver injury. Considering that creatine can synthesize PCr in the liver, oral administration of creatine may have similar protective effects. However, this hypothesis needs to be explored.
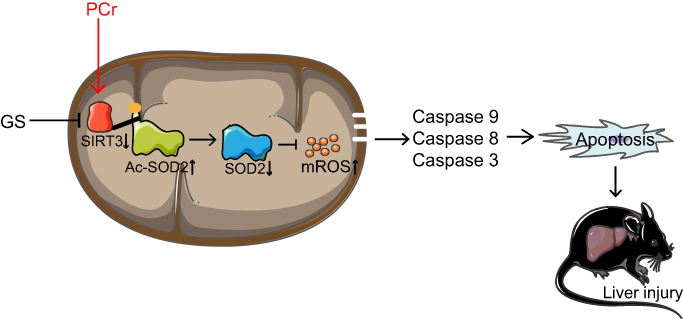 |
Figure 6 Proposed model of how phosphocreatine attenuates Gynura segetum-induced liver injury created via a SIRT3-SOD2 pathway. |
Conclusion
In summary, we made the novel discovery that GS induces apoptosis through excessive accumulation of mitochondrial ROS. Additionally, the SIRT3-SOD2 signaling pathway plays a vital role in modulating GS-induced mitochondrial ROS formation. More importantly, our study demonstrated for the first time that PCr protects against GS-induced liver injury via the SIRT3-SOD2-mitochondrial ROS pathway. Thus, PCr may be of therapeutic value for GS-induced liver injury in the future.
Abbreviation list
GS, Gynura segetum; PAs, pyrrolizidine alkaloids; ROS, reactive oxygen species; SIRT3, sirtuin 3; SOD2, superoxide dismutase 2; Ac-SOD2, acetyl-SOD2; PCr, phosphocreatine; ATP, adenosine triphosphate; ALT, alanine aminotransferase; AST, aspartate aminotransaminase.
Acknowledgments
We thank Ronggui Hu (Professor at Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences) for providing research instruction and technical support. This study was funded by the Shanghai Committee of Science and Technology, People’s Republic of China (Grant 12401907400) and Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (KEF152006).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gao H, Ruan JQ, Chen J et al. Blood pyrrole-protein adducts as a diagnostic and prognostic index in pyrrolizidine alkaloid-hepatic sinusoidal obstruction syndrome. Drug Des Devel Ther. 2015;9:4861–4868. doi:10.2147/DDDT.S87858
2. Wang X, Qi X, Guo X Tusanqi-related sinusoidal obstruction syndrome in China. Medicine. 2015;94(23):e942 . doi:10.1097/MD.0000000000000874
3. Wang Y, Qiao D, Li Y, Xu F Risk factors for hepatic veno-occlusive disease caused by Gynura segetum: a retrospective study. BMC Gastroenterol. 2018;18(1):156. doi:10.1186/s12876-018-0863-2
4. Lin G, Wang JY, Li N et al. Hepatic sinusoidal obstruction syndrome associated with consumption of Gynura segetum. J Hepatol. 2011;54(4):666–673. doi:10.1016/j.jhep.2010.07.031
5. Fu PP, Xia Q, Lin G, Chou MW Pyrrolizidine alkaloids—genotoxicity, metabolism enzymes, metabolic activation, and mechanisms. Drug Metab Rev. 2004;36(1):1–55. doi:10.1081/DMR-120028426
6. Zhao Y, Xia Q, Yin JJ, Lin G, Fu PP Photoirradiation of dehydropyrrolizidine alkaloids—formation of reactive oxygen species and induction of lipid peroxidation. Toxicol Lett. 2011;205(3):302–309. doi:10.1016/j.toxlet.2011.06.020
7. Ji L, Liu T, Wang Z Pyrrolizidine alkaloid clivorine induced oxidative injury on primary cultured rat hepatocytes. Hum Exp Toxicol. 2010;29(4):303–309. doi:10.1177/0960327110361757
8. Yang X, Wang H, Ni H et al. Inhibition of Drp1 protects against senecionine-induced mitochondria-mediated apoptosis in primary hepatocytes and in mice. Redox Biol. 2017;12:264–273. doi:10.1016/j.redox.2017.02.020
9. Lu Y, Ma J, Song Z et al. The role of formation of pyrrole–ATP synthase subunit beta adduct in pyrrolizidine alkaloid-induced hepatotoxicity. Arch Toxicol. 2018;92(11):3403–3414. doi:10.1007/s00204-018-2309-6
10. Kinugasa H, Whelan KA, Tanaka K et al. Mitochondrial SOD2 regulates epithelial–mesenchymal transition and cell populations defined by differential CD44 expression. Oncogene. 2015;34(41):5229–5239. doi:10.1038/onc.2014.449
11. Chen Y, Qing W, Sun M et al. Melatonin protects hepatocytes against bile acid-induced mitochondrial oxidative stress via the AMPK-SIRT3-SOD2 pathway. Free Radical Res. 2015;49(10):1275–1284. doi:10.3109/10715762.2015.1067806
12. Gao J, Feng Z, Wang X et al. SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell Death Differ. 2018;25(2):229–240. doi:10.1038/cdd.2017.144
13. Tang L, Xia Z, Zhao B et al. Phosphocreatine preconditioning attenuates apoptosis in ischemia-reperfusion injury of rat brain. J Biomed Biotechnol. 2011;2011:1–4
14. Sun Z, Lan X, Ahsan A et al. Phosphocreatine protects against LPS-induced human umbilical vein endothelial cell apoptosis by regulating mitochondrial oxidative phosphorylation. Apoptosis. 2016;21(3):283–297. doi:10.1007/s10495-015-1210-5
15. Ahsan A, Han G, Pan J et al. Phosphocreatine protects endothelial cells from oxidized low-density lipoprotein-induced apoptosis by modulating the PI3K/Akt/eNOS pathway. Apoptosis. 2015;20(12):1563–1576. doi:10.1007/s10495-015-1175-4
16. Zhu H, Chu Y, Huo J, Chen Z, Yang L Effect of prednisone on transforming growth factor-β1, connective tissue growth factor, nuclear factor-κBp65 and tumor necrosis factor-α expression in a murine model of hepatic sinusoidal obstruction syndrome induced by Gynura segetum. Hepatol Res. 2011;41(8):795–803. doi:10.1111/j.1872-034X.2011.00830.x
17. Ruan J, Li N, Xia Q et al. Characteristic ion clusters as determinants for the identification of pyrrolizidine alkaloid N-oxides in pyrrolizidine alkaloid-containing natural products using HPLC-MS analysis. J Mass Spectrom. 2012;47(3):331–337. doi:10.1002/jms.2969
18. Lin G, Zhou KY, Zhao XG, Wang ZT, But PP Determination of hepatotoxic pyrrolizidine alkaloids by on-line high performance liquid chromatography mass spectrometry with an electrospray interface. Rapid Commun Mass Spectrom. 1998;12
19. Wong CH, Iskandar KB, Yadav SK et al. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS One. 2010;5(4):e9996. doi:10.1371/journal.pone.0009996
20. Becuwe P, Ennen M, Klotz R, Barbieux C, Grandemange S Manganese superoxide dismutase in breast cancer: from molecular mechanisms of gene regulation to biological and clinical significance. Free Radical Bio Med. 2014;77:139–151. doi:10.1016/j.freeradbiomed.2014.08.026
21. Dikalova AE, Itani HA, Nazarewicz RR et al. Sirt3 Impairment and SOD2 hyperacetylation in vascular oxidative stress and hypertensionnovelty and significance. Circ Res. 2017;121(5):564–574. doi:10.1161/CIRCRESAHA.117.310933
22. Tokarska-Schlattner M, Epand RF, Meiler F et al. Phosphocreatine interacts with phospholipids, affects membrane properties and exerts membrane-protective effects. PLoS One. 2012;7(8):e43178. doi:10.1371/journal.pone.0043178
23. Kan X, Ye J, Rong X et al. Diagnostic performance of contrast-enhanced CT in pyrrolizidine alkaloids-induced hepatic sinusoidal obstructive syndrome. Sci Rep-Uk. 2016;6(1)
24. Zhou H, Wang YJ, Lou H, Xu X, Zhang M Hepatic sinusoidal obstruction syndrome caused by herbal medicine: CT and MRI features. Korean J Radiol. 2014;15(2):218. doi:10.3348/kjr.2014.15.2.218
25. Waizenegger J, Braeuning A, Templin M, Lampen A, Hessel-Pras S Structure-dependent induction of apoptosis by hepatotoxic pyrrolizidine alkaloids in the human hepatoma cell line HepaRG: single versus repeated exposure. Food Chem Toxicol. 2018;114:215–226. doi:10.1016/j.fct.2018.02.036
26. Wei Y, Weng D, Li F et al. Involvement of JNK regulation in oxidative stress-mediated murine liver injury by microcystin-LR. Apoptosis. 2008;13(8):1031–1042. doi:10.1007/s10495-008-0237-2
27. Ji LL, Zhang M, Sheng YC, Wang ZT Pyrrolizidine alkaloid clivorine induces apoptosis in human normal liver L-02 cells and reduces the expression of p53 protein. Toxicol In Vitro. 2005;19(1):41–46. doi:10.1016/j.tiv.2004.06.003
28. Liang QN, Liu TY, Ji LL, Min Y, Xia YY Pyrrolizidine alkaloid clivorine-induced oxidative stress injury in human normal liver L-02 cells. Drug Discov Ther. 2009;3
29. Pervaiz S, Clément M Hydrogen peroxide-induced apoptosis: oxidative or reductive stress? Methods Enzymol. 2002;352:150–159.
30. Pervaiz S, Clement M Superoxide anion: oncogenic reactive oxygen species? Int J Biochem Cell Biol. 2007; 39 (7–8): 1297–1304. doi:10.1016/j.biocel.2007.04.007
31. Izeradjene K, Douglas L, Tillman DM, Delaney AB, Houghton JA Reactive oxygen species regulate caspase activation in tumor necrosis factor–related apoptosis-inducing ligand–resistant human colon carcinoma cell lines. Cancer Res. 2005;65(16):7436–7445. doi:10.1158/0008-5472.CAN-04-2628
32. Lin G, Wang JY, Li N, Li M, Gao H Hepatic sinusoidal obstruction syndrome associated with consumption of Gynura segetum. J Hepatol. 2011;54(4):666–673. doi:10.1016/j.jhep.2010.07.031
33. Pi H, Xu S, Reiter RJ et al. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy. 2015;11(7):1037–1051. doi:10.1080/15548627.2015.1052208
34. Shi X, Bai Y, Zhang G et al. Effects of over-expression of SOD2 in bone marrow-derived mesenchymal stem cells on traumatic brain injury. Cell Tissue Res. 2018;372(1):67–75. doi:10.1007/s00441-017-2716-7
35. Chen Y, Zhang J, Lin Y et al. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011;12(6):534–541. doi:10.1038/embor.2011.65
36. Wang Q, Li L, Li CY et al. SIRT3 protects cells from hypoxia via PGC-1alpha- and MnSOD-dependent pathways. Neuroscience. 2015;286:109–121. doi:10.1016/j.neuroscience.2014.11.045
37. Lu Y, Ma J, Song Z et al. The role of formation of pyrrole–ATP synthase subunit beta adduct in pyrrolizidine alkaloid-induced hepatotoxicity. Arch Toxicol. 2018;92(11):3403–3414. doi:10.1007/s00204-018-2309-6
38. Song C, Zhao J, Fu B et al. Melatonin-mediated upregulation of Sirt3 attenuates sodium fluoride-induced hepatotoxicity by activating the MT1-PI3K/AKT-PGC-1alpha signaling pathway. Free Radic Biol Med. 2017;112:616–630. doi:10.1016/j.freeradbiomed.2017.09.005
Supplementary materials
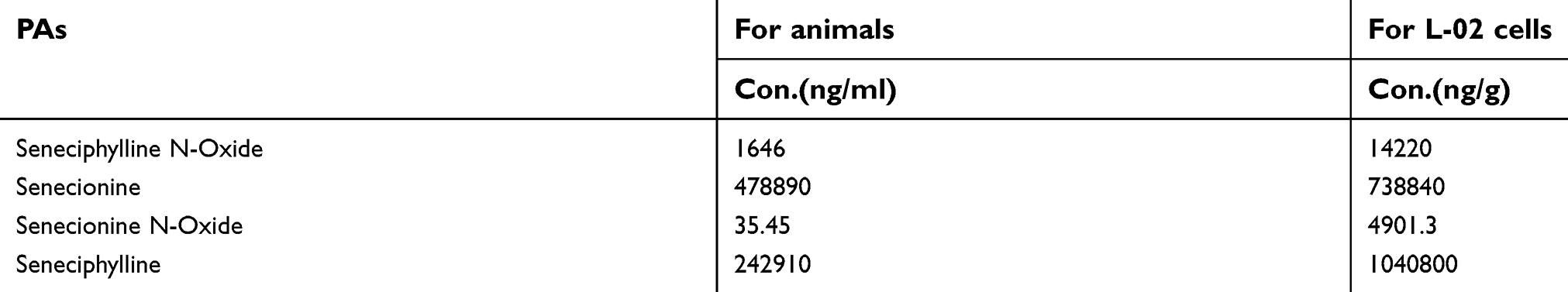 |
Table S1 The quantification of Gynura segetum |
 |
Table S2 qPCR primers used in the study |
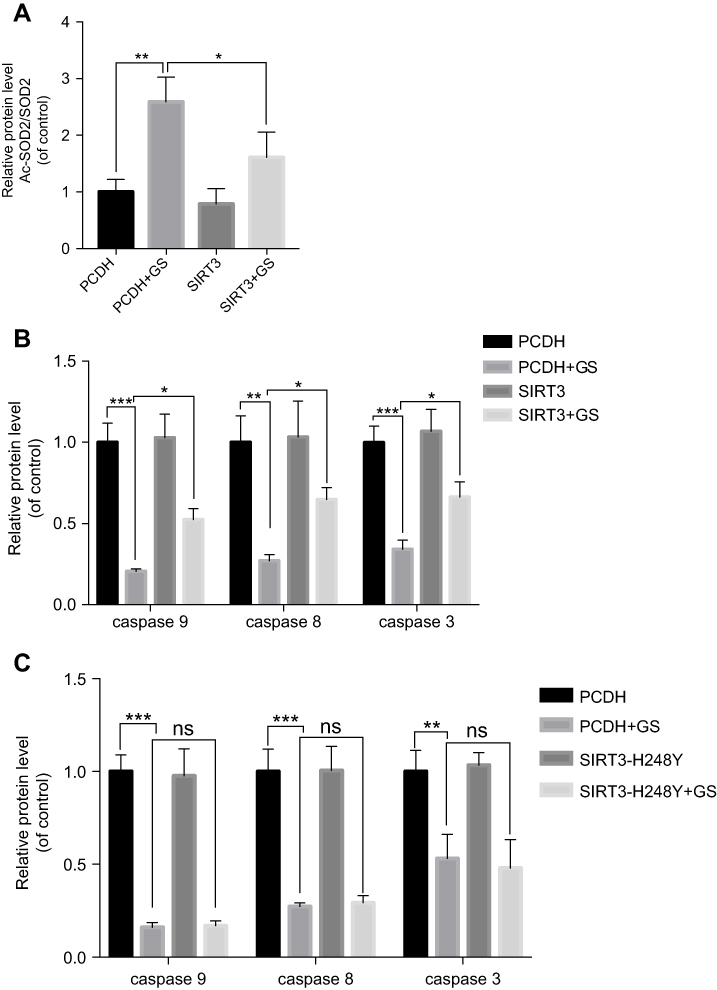 |
Figure S2 Quantitative analysis of Western blot bands. (A) Quantitative analysis of Ac-SOD2 in cells overexpressing SIRT3. (B) Quantitative analysis of apoptosis-related protein in cells overexpressing SIRT3. (C) Quantitative analysis of apoptosis-related protein in cells overexpressing SIRT3H248Y. Values are the mean±SEM. *p<0.05, **p<0.01, and ***p<0.001. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.