")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Phenotypic characterization of an older adult male with late-onset epilepsy and a novel mutation in ASXL3 shows overlap with the associated Bainbridge-Ropers syndrome
Authors Verhoeven W , Egger J, Räkers E, van Erkelens A, Pfundt R, Willemsen MH
Received 9 October 2017
Accepted for publication 15 January 2018
Published 27 March 2018 Volume 2018:14 Pages 867—870
DOI https://doi.org/10.2147/NDT.S153511
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Willem Verhoeven,1,2 Jos Egger,1,3 Emmy Räkers,4 Arjen van Erkelens,5 Rolph Pfundt,5 Marjolein H Willemsen5
1Vincent van Gogh Institute for Psychiatry, Centre of Excellence for Neuropsychiatry, Venray, the Netherlands; 2Department of Psychiatry, Erasmus University Medical Centre, Rotterdam, the Netherlands; 3Donders Institute for Brain, Cognition and Behaviour, Radboud University, Nijmegen, the Netherlands; 4ASVZ, Centre for People with Intellectual Disabilities, Sliedrecht, the Netherlands; 5Department of Human Genetics, Radboud University Medical Centre, Nijmegen, the Netherlands
Abstract: The additional sex combs like 3 gene is considered to be causative for the rare Bainbridge-Ropers syndrome (BRPS), which is characterized by severe intellectual disability, neonatal hypotonia, nearly absent development of speech and language as well as several facial dysmorphisms. Apart from disruptive autistiform behaviors, sleep disturbances and epileptic phenomena may be present. Here, a 47-year-old severely intellectually disabled male is described in whom exome sequencing disclosed a novel heterozygous frameshift mutation in the ASXL3 gene leading to a premature stopcodon in the last part of the last exon. Mutations in this very end 3' of the gene have not been reported before in BRPS. The phenotypical presentation of the patient including partially therapy-resistant epilepsy starting in later adulthood shows overlap with BRPS, and it was therefore concluded that the phenotype is likely explained by the identified mutation in ASXL3.
Keywords: Bainbridge-Ropers syndrome, ASLX3, frameshift mutation, epilepsy, intellectual disability, array analysis, whole exome sequencing, autism spectrum disorder
Introduction
Bainbridge-Ropers syndrome (BRPS) [OMIM #615485] is a novel syndromal entity that is caused by a mutation in the additional sex combs like 3 (ASLX3) gene located at 18q12.1. In 2013, the syndrome was first described in four patients.1 The ASLX3 gene has a functional role in the process of deubiquitination and is expressed in several organ systems including the central nervous system.2 At present, <30 patients with this syndrome, children and adolescents only, have been published.1,3–6 The phenotype typically includes severe intellectual disability, nearly absent speech and language, hypotonia, and feeding difficulties at young age as well as distinct craniofacial features such as prominent forehead, arched eyebrows, and hypertelorism. Regarding behavior, in addition to aggressive and autistic traits with rocking and hand flapping, periodic agitation and sleep disturbances are frequently described. In a minority of the patients, tonic-clonic seizures or absences occur in childhood.5–7
Here, the first middle-aged male patient with a mutation in ASXL3 and a phenotype reminiscent of BRPS is described. He developed tonic-clonic epilepsy at the end of his fourth decade.
Patients and methods
Case description
The patient is a 47-year-old male with one older sister by 4 years from the same parents, two half-brothers and one half-sister from other fathers. All siblings, including most probably also their mother, were intellectually disabled and developed within a very poor pedagogic family context. Apart from minimal to absent speech and language development, no information is available about his early years. From the age of 6, the patient needed a specialized child day care facility. Two years later, he had to be institutionalized because of severe behavioral problems, particularly continuous extreme screaming, also nocturnal. From his early adolescence on, anxious, aggressive, self-injurious, and compulsive behaviors occurred intermittently contingent upon environmental changes for which various psychotropics were prescribed symptomatically. Social maturity was established at a developmental level of ~5 years. At his mid-thirties, the behavioral pattern was attributed to an autism spectrum disorder for which he was moved to a specialized ward. This resulted in a significant amelioration of his behavioral repertoire. Aged 38, the patient developed erythema migrans due to a borrelia infection that was proven by seroconversion in blood with normal cerebrospinal fluid parameters for which he was adequately treated with antibiotics.
At the age of 45 years, the patient’s actions became increasingly disruptive with progressive repetitive and compulsive behaviors. In addition, intensified sensitivity to sensory stimuli as well as severe sleep problems developed for which risperidone and lorazepam were prescribed. In this period, generalized epilepsy with three tonic-clonic seizures occurred and treatment with carbamazepine was started (800 mg; 8.7 mg/L). Subsequently, two consecutive 24 h video-EEG registrations disclosed no epileptic configurations. CT and MRI scanning of the brain, the latter under general anesthesia because of his disinhibited behaviors, disclosed no abnormalities. Because of accumulation of severe challenging behaviors and sleep disturbances necessitating nearly permanent individualized care, the patient was referred for expert consultation.
Relevant family context
The patient’s older sister, with about a similar history and later staying at the same institute, was institutionalized from early age on because of severe challenging behaviors. She was also suffering from tonic-clonic seizures, most probably starting in infancy, for which she was initially treated with phenytoine, which was changed to carbamazepine (1,000 mg) in her early twenties. There were no neurological abnormalities or notable dysmorphic features. Development of speech and language was restricted to single words and sounds. She displayed autistic traits with periodic screaming, crying, and anxiety-driven aggressive behaviors. At that time, mean developmental age was 2.5 years. Because of persistent epileptic phenomena, occurring also at night, 10 years later valproic acid was added (1,200 mg), which, however, could not prevent further tonic-clonic seizures. Aged 35 years, she underwent bilateral mastectomy because of carcinoma. One year later, she died during a nocturnal tonic-clonic epileptic status.
The mother of both siblings deceased 4 years later (unknown cause). Their father died many years before. Similar to the early years of the patient and his sister, no additional information is available for both parents.
Results
At examination by a clinical geneticist, a severe intellectually disabled, middle-aged male patient was seen with only mild facial dysmorphisms (Figure 1). Height, weight, and head circumference were 168 cm (−2 SD), 54 kg (BMI: 19), and 53.5 cm (−2.5 SD), respectively. Somatic and neurological examination disclosed no abnormalities. Relevant hematological and biochemical parameters were all normal. His behavior repertoire was evaluated by a neuropsychiatrist and could be best typified by autistic-like repetitive and compulsive behaviors such as rubbing/stroking and licking with increase in unknown situations. Hardly any communication was possible but, based on nonverbal instructions, the patient was able to perform very simple daily activities, for example, bringing his plate to the table and taking his meal.
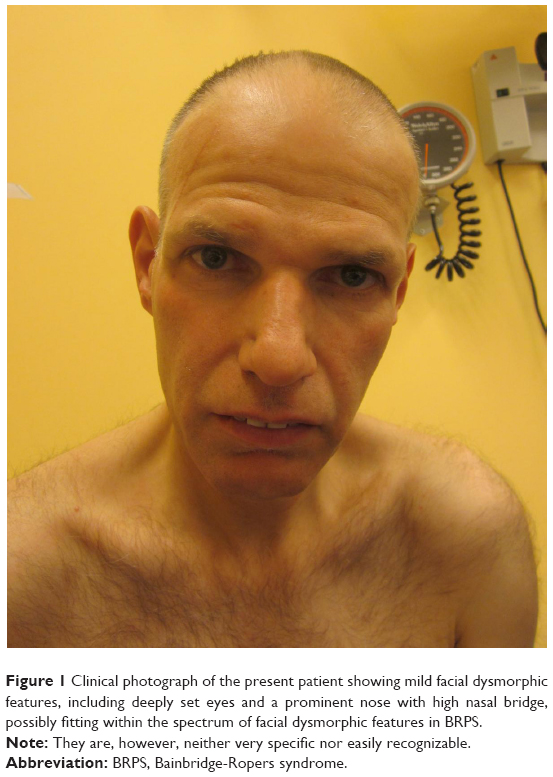 | Figure 1 Clinical photograph of the present patient showing mild facial dysmorphic features, including deeply set eyes and a prominent nose with high nasal bridge, possibly fitting within the spectrum of facial dysmorphic features in BRPS. |
Since etiological investigation was not performed as yet, first, chromosomal analysis using the CytoScan HD array (Applied Biosystems™, Foster City, CA, USA) was done, which disclosed no abnormalities. Fragile-X syndrome was also excluded by DNA diagnostics. Subsequently, whole exome sequencing was performed essentially as described before.8 Target regions were enriched using the Agilent SureSelectXT Human All Exon 50Mb Kit. Whole-exome sequencing was performed on the Illumina HiSeq platform (BGI, Copenhagen, Denmark) followed by analysis of the variants in genes involved in neurodevelopmental diseases. Data were analyzed with Burrows-Wheeler Aligner (read alignment) and Genome Analysis Toolkit (variant calling) software packages. Variants were annotated using an in-house developed pipeline. Prioritization of variants was done by an in-house designed “variant interface” and manual curation. This revealed a heterozygous frameshift mutation in the ASXL3 gene: (Chr18(GRCh37):g.31326509_31326522dup; NM_030632.1:c.6697_6710dup(p.(Ser2238fs)).
It concerns a duplication of 14 base pairs leading to a frameshift and premature stop in the 3′-part of the last exon of the ASXL3 gene. This will probably lead to the presence of a truncated protein, rather than nonsense-mediated decay and haploinsufficiency. Other reported mutations in patients with BRPS are most likely leading to haploinsufficiency, although the vast majority of the mutations that have been described so far are located in the last two exons of the ASXL3 gene (The Human Gene Mutation Database).9 Pathogenic mutations in this very end 3′ of the gene have not been reported before. Also, the mutation has been reported neither in the previous literature nor in the Exome Aggregation Consortium (ExAC; http://exac.broadinstitue.org) or Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org). It was classified as possibly pathogenic since the phenotype of the patient, although not highly specific but including intellectual disability, the above-described speech/language impairment, sleep disturbances, behavioral phenomena, and epilepsy, could fit within the spectrum of BRPS. Thus, the ASXL3 mutation was concluded to be a likely explanation. In addition, it is possible that mutations in ASXL3 with another effect than haploinsufficiency may lead to phenotypes with intellectual disabilities that are not typically fitting into the phenotype of BRPS.
To further specify the phenotypic presentation, extensive neuropsychological assessment was performed showing a discrepant profile with a developmental age ranging from ≤1 year (social and communicative skills; Vineland Screener,10 SRZ-P,11) to maximally 2.5 years (visuomotor functioning; Beery VMI12). Due to major impairments in communication, formal testing of attention, memory, and executive functioning could not be performed. Instead, relevant domains were objectivated by proxy-ratings of attention/executive functioning (DEX13 and EFI14: severely disturbed) and psychopathology (PIMRA15: anxiety bound symptoms).
Since no appropriate plasma concentration of risperidone could be achieved due to enzymatic induction by the previously mentioned antiepileptic carbamazepine, the latter was replaced by valproic acid. Also, zopiclone was added to lorazepam, which to some extent improved his sleep pattern. Concerning epilepsy, since the start of antiepileptic drugs, tonic-clonic seizures did no longer occur although regular daytime absences were still noticed. At present, the patient is treated with 1,200 mg valproic acid (60 mg/L), 5 mg risperidone (2.8/29 μg/L), 3.5 mg lorazepam (28 μg/L), and 7.5 mg zopiclone.
Discussion
In this paper, the first middle-aged male patient is described in whom whole exome sequencing demonstrated a never reported novel frameshift mutation in the ASXL3 gene (c.6697_6710dup). Although never reported before and located in the very 3′ end of the gene, this mutation was considered to be likely pathogenic since the phenotype shows overlap with the phenotypic spectrum of BRPS, including severe intellectual disability, autistic-like behavioral problems, and epilepsy that is reported in a minority of the patients diagnosed with BRPS. The facial phenotype is not typically recognizable as BRPS but the phenotype in other patients with this syndrome is variable and not highly specific.6 There are, however, some similarities, for example, in the shape of the nose that can also be recognized in the present patient (Figure 1). It might be that the mutation in the very 3′ end of the ASXL3 gene leads to a less “typical” presentation of BRPS which, however, is not an easily recognizable syndrome per se.6 Furthermore, since individuals with BRPS, in particular adults, are relatively rare, the clinical spectrum may be actually much broader than reported up to now. In contrast to his deceased sister, his epilepsy started not at an early age but in his fourth decade and is partially therapy resistant since absences still persist. However, given the developmental issues, presentation, and course of disease including severe epilepsy, his deceased sister could have had the same genetic disorder. In both, social interaction could be characterized as predominantly anxiety-driven disruptive autistiform behaviors. Since intellectual disability was reported in the mother as well, the mutation in ASXL3 might be inherited from her. Perhaps, she was more mildly affected due to the presence of the mutation in mosaic state. However, half-siblings with the same father were also reported to have intellectual disability. Since segregation analysis was not possible and detailed clinical data are lacking, it remains unclear whether this mutation is inherited and if so, from whom.
Conclusion
Extensive genetic analysis including whole exome sequencing is mandatory in patients with unexplained developmental delay and intellectual disability, also in adulthood and older age. The latter not only to detect the genetic etiology of syndromic intellectual disability, but especially also to further elucidate course into adulthood and prognosis of newly discovered disorders like Bainbridge-Ropers syndrome.
Acknowledgments
Written informed consent was obtained from the legal representative (manager of the institute; since no family members are alive) for publication of this paper on the patient and his family and to include a picture of the face of the patient. The patient was referred by the Centre of Consultation and Expertise, region West. The authors are indebted to Mrs Anneke Baselier, consultant psychologist, and to the staff members of the ASVZ-institute for intellectual disabilities for their careful assessment of the behavior status of the patient. Thanks are extended to Mrs Linde van Dongen, psychologist from the Centre of Excellence for Neuropsychiatry of the Vincent van Gogh Institute for Psychiatry, who performed neuropsychological testing.
Disclosure
The authors report no conflicts of interest in this work.
References
Bainbridge MN, Hu H, Muzny DM, et al. De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome Med. 2013;5(2):11. | ||
Srivastava A, Ritesh KC, Tsan YC, et al. De novo dominant ASXL3 mutations alter H2A deubiquination and transcription in Bainbridge-Ropers syndrome. Hum Mol Genet. 2016;25(3):597–608. | ||
Dinwiddle DL, Soden SE, Saunders CJ, et al. De novo frameshift mutation in ASXL3 in a patient with global developmental delay, microcephaly, and craniofacial anomalies. BMC Med Genomics. 2016;6:32. | ||
Hori I, Miya F, Ohashi K, et al. Novel splicing in the ASXL3 gene causing Bainbridge Ropers syndrome. Am J Med GenetA. 2016;170(7):1863–1867. | ||
Kuechler A, Czeschik JC, Graf E, et al. Bainbridge-Ropers syndrome caused by loss-of-function variants in ASXL3: a recognizable condition. Eur J Hum Genet. 2017;25(2):183–191. | ||
Balasubramanian M, Willoughby J, Fry AE, et al. Delineating the phenotypic spectrum of Bainbridge-Ropers syndrome: 12 new patients with de novo heterozygous, loss-of-function mutations in ASXL3 and review of the published literature. J Med Genet. 2017;54(8):537–543. | ||
Contreras-Capetillo SN, Vilchis-Zapata ZH, Ribbón-Conde J, Pinto-Escalante D. Global developmental delay and postnatal microcephaly: Bainbridge-Ropers syndrome with a new mutation in ASXL3. Neurología. Epub 2017 Apr 18. | ||
Neveling K, Feenstra I, Gilissen C, et al. Post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. HumMutat. 2013;34(12):1721–1726. | ||
Stenson PD, Mort M, Ball EV, et al. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136(6):665–677. | ||
Scholte E, Van Duijn E, Dijkxhoorn Y. Vineland Screener 0–6 Years: Manual of the Dutch Adaptation. Leiden, The Netherlands: PITS; 2008. | ||
Kraijer DW, Kema GN. Sociale Redzaamheidsschaal Voor Verstandelijk Gehandicapten Van Hoger Niveau (SRZ-P). Amsterdam, The Netherlands: Pearson; 2004. | ||
Beery KE, Buktenica NA, Beery NA. The Beery-Buktenica Developmental Test of Visual-Motor Integration (Beery VMI). 5th ed. San Antonio, Texas: Pearson; 2004. | ||
Wilson B, Alderman N, Burgess P, Emslie H, Evans J. Behavioural Assessment of the Dysexecutive Syndrome (BADS). London, UK: Pearson; 1997. | ||
Janssen GT, De Mey HR, Egger JI. Executive functioning in college students: evaluation of the Dutch executive function index (EFI-NL).Int J Neurosci. 2009;119(6):792–805. | ||
Matson JL, Kazdin AE, Senatore V. Psychometric properties of the psychopathology instrument for mentally retarded adults. Appl Res Ment Retard. 1984;5(1):81–89. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.