")
Back to Journals » International Journal of General Medicine » Volume 15
Phenotypic and Genotypic Signatures of VWF Exon 18 in Eastern Saudi Patients Previously Diagnosed with Type 1 von Willebrand Disease
Authors Alzahrani FM , Al Faris AA, Bashawri LA, Hassan FM , El-Masry OS, Aldossary MA, Al Sultan O, Borgio JF , Alsahli MA , Goodeve A
Received 9 March 2022
Accepted for publication 17 May 2022
Published 2 June 2022 Volume 2022:15 Pages 5385—5394
DOI https://doi.org/10.2147/IJGM.S364818
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Faisal M Alzahrani,1 Asma A Al Faris,1 Layla A Bashawri,1 Fathelrahman Mahdi Hassan,2 Omar S El-Masry,1 Maryam A Aldossary,1 Osama Al Sultan,3 J Francis Borgio,4 Mohammed A Alsahli,5 Anne Goodeve6
1Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia; 2Department of Hematology and Immunohematology, College of Medical Laboratory Science, Sudan University of Science and Technology, Khartoum, Sudan; 3Department of Internal Medicine, King Fahad Hospital of the University, Imam Abdulrahman Bin Faisal University, Khobar, Saudi Arabia; 4Department of Genetic Research, Institute for Research and Medical Consultations (IRMC), Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia; 5Department of Medical Laboratories, College of Applied Medical Sciences, Qassim University, Buraydah, 51452, Saudi Arabia; 6Medical School, University of Sheffield, Sheffield, UK
Correspondence: Faisal M Alzahrani, Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia, Tel +00966566655776, Email [email protected]
Introduction: von Willebrand disease (VWD) is the most prevalent bleeding disease, which is associated with either low levels of von Willebrand factor (VWF) or abnormality in its structure. Three types of the disease have been described; type 1 (VWD1) and 3 (VWD3) are caused by deficiency of VWF and type 2 (VWD2) is caused by production of defective VWF. The aim of the current study was to characterize gene variants of VWF gene; exon 18 in particular, in a cohort of Saudi families as well as healthy control subjects.
Methods: A total of 19 families comprising 60 subjects of type 1 VWD were enrolled in the study. Participants were divided into 22 index cases, 21 affected family members and 17 unaffected family members ranging in age from 6 to 70 years. Blood samples were collected from all participants to measure activated partial thromboplastin time test (APTT), von Willebrand antigen level (VWF:Ag), Factor VIII activity (FVIII:C) and ristocetin cofactor activity (VWF:RCo), platelet count, determining the ABO blood group and for genetic analysis by Sanger sequencing.
Results: The results indicated that VWD1 patients have lower levels of VWF and factor VIII than the non-affected family members and the control subjects. In addition, five gene variants were reported in VWF exon 18; of these, c.2365A>G and c.2385T>C were more common in the control group and might be protective from VWD.
Discussion: In conclusion, VWF levels are influenced by blood group, and there was no association between variants in exon 18 of VWF gene reported in all groups and the disease status; however, blood group analysis and genome-wide genotyping could help to highlight high-risk groups and improve clinical management of VWD.
Keywords: VWF gene, von Willebrand disease, Saudi Arabia
Introduction
von Willebrand disease (VWD) is the most common bleeding disorder in humans.1 The disease is caused by one mutation or more in the von Willebrand factor (VWF) gene that causes a qualitative or quantitative abnormality in the protein product. VWD is generally inherited as an autosomal dominant trait and affects both genders.2 In this regard, three types of the disease have been described; type 1 (VWD1) and 3 (VWD3) are associated with partial or complete deficiency in VWF in order, whilst type 2 (VWD2) is associated with production of defective VWF.3 The prevalence of this disease varies among different ethnic groups.4 In Saudi Arabia, a study5 showed that the prevalence of VWD is higher than other populations, reaching 3.9% of the Saudi population. The gene of VWF comprises 52 exons producing a large glycoprotein. Also, the presence of a highly similar gene known as pseudo VWF with a highly homologous sequence makes genetic analysis of VWF challenging.6 It was recently reviewed that >700 unique gene variants of VWF were compiled in VWF variant database established by a group of researchers at the University of Sheffield in the UK (http://www.vwf.group.shef.ac.uk/).7 Numerous studies have reported several types of mutations in VWD in different populations; majority of these mutations are missense. Also, low levels of VWF are associated with the presence of mutations in the VWF gene. The authors also indicated that the disease severity might be affected by other factors such as ABO blood group type.8,9 Therefore, the aim of the current study was to characterize the phenotypic and genotypic signatures of patients diagnosed with type 1 VW disease and their family members (affected and non-affected) by recording specific phenotypic scores compared to control values and performing DNA Sanger sequencing, a particular focus was given to exon 18 on VWF gene.
Patients and Methods
A total of 21 families comprising 60 subjects of type 1 VWD (11 males and 49 females). Participants were divided into 22 index cases (who were previously diagnosed with type 1 VWD), 21 affected family members (diagnosed with type 1 VWD) and 17 unaffected family members ranging in age from 6 to 70 years. Phenotypic analysis was used to confirm the diagnosis with type 1 VWD. All participants signed an informed consent prior to participation and answered the International Society on Thrombosis and Haemostasis (ISTH) bleeding score questionnaire (ISTH-SSC) to assess the severity of bleeding symptoms. This study was conducted in accordance with the Declaration of Helsinki. The study was reviewed and approved by the institutional review board at Imam Abdulrahman Bin Faisal University (IRB-2017-03-009). Samples were collected from children and adolescents up to 18 years of age upon receiving signed informed consent from a parent and/or legal guardian and from participants for >18 years for participation in the study.
Sample Collection and Downstream Analysis
Three blood samples were collected from each participant, 2 samples were collected in sodium citrate tubes and one sample in EDTA vacutainer. The sodium citrate sample was used for activated partial thromboplastin time test (APTT), von Willebrand antigen level (VWF Ag), Factor VIII activity (FVIII:C) and ristocetin cofactor activity (VWF:RCo) to assess the phenotypic profiles. VWF Ag, FVIII:C, and VWF:RCO were measured by STAR Max® - Stago (Diagnostica Stago, France) hemostasis analyzer. VWF Ag was determined by ELISA method using the Asserachrom® VWF Ag kit (#00942, Diagnostica Stago, France). Factor VIII: C was quantified using a chromogenic method by employing the triniCHROM factor VIII: C kit (#T2608, Diagnostica Stago, France). VWF:RCO was also assessed by an automated method using STA-VWF:RCO kit (#01191, Diagnostica Stago, France) according to the manufacturer’s protocol. The EDTA sample was used for measuring the platelet count, determining the ABO blood group and for genetic analysis.
Genotype Analysis
Herein, exon 18 of the VWF gene was selected for genotyping analysis from the VWF variant database (http://www.vwf.group.shef.ac.uk/), which indicated 107 identified gene variants in this exon. The primers (Forward: 5′ TAGGGGACCAAAGGACAGTG 3′ and Reverse: 5′ CCGTGTTTAGCCCTTGTTTC 3′) used to amplify exon 18 were previously reported.8,10 The primer annealing sequences were cross-checked for the presence of single nucleotide variants at www.ensebmble.org.
PCR and Gel Purification of PCR-Product
DNA was extracted from participants’ blood using Relia prepTM blood g DNA mini prep system kit (Promega Corporation, USA) according to the manufacturer’s protocol. The PCR reaction was performed in a final volume of 30 µL containing 15 µL master mix (Taq DNA polymerase enzyme, buffer, deoxy nuclease triphosphate (dNTPs), MgCl2 and loading dye), 2 µL of each primer, 2 µL of the template DNA (100 ng) and 9 µL nuclease free water (PCR reagents were purchased from MOLEQULE-ON®, Auckland, New Zealand). The thermal cycling (Maxi TM thermal cycler by ESCO technologies, USA) was set to an initial denaturation at 94 °C for 7 minutes, denaturation at 94 °C for 1 minute, annealing at 60 °C for 1 minute (35 cycles), extension at 72 °C for 1 minute, and a final extension at 72 °C for 7 minutes. The expected product size of this PCR reaction is 349 bp. Product size was confirmed by 2% agarose gel electrophoresis and compared with DNA ladder (MOLEQULE-ON). The DNA bands were then visualized under a UV-light box then purified the amplified PCR product. Gel purification was performed using MQ PCR/Gel product purification kit (MOLEQULE-ON).
DNA Sequencing
Purified amplicon for exon 18 of VWF gene was sequenced using the chain termination method developed by Frederick Sanger.11 The sequencing was done using the forward and the reverse primers separately by Applied Biosystems ABI 3730xl Capillary DNA analyzer. The results were analyzed by UCSC genome browser https://genome.ucsc.edu/ by the BLAT tool. This tool compares the analyzed sequence with the human von Willebrand Factor reference sequence. The results were also analyzed by special software programs: Vario Box program (Bioinformatics. UA. PT, Portugal), sequence analysis software program (ABI, USA), and Mutation Surveyor V4.0.8 program (soft Genetics, USA). All sequences were compared to the following VWF GenBank reference sequences, Homo sapiens von Willebrand Factor (VWF), mRNA NCBI reference sequence: NM_000552.4 and GenBank accession NC_000012.12 region: 5949015.6123196.
Statistical Analysis
The mean, the median, the range, the 25th and 75th percentiles were calculated and the p value for the comparison of gender and blood groups for each research group was calculated using the Chi-square test. The p value for the comparisons of bleeding scores, number of bleeding symptoms and the phenotypic laboratory results between different study groups was calculated by the Kruskal–Wallis test. The comparison between phenotypic results between groups was done with the Mann–Whitney U-test. Cross Tabulation with correlations and Chi-square test were utilized to calculate correlation coefficient and estimate the significance of association between genotypes and disease phenotypes and laboratory parameters. Statistical analysis was performed using SPSS program (version 22; IBM Corp., USA). The p value was considered statistically significant at p ≤0.05.
Results
The study sample comprised 22 index cases, 21 affected family members and 17 unaffected family members. Table 1 summarizes the demographic characteristics and age groups within study groups. The results for these groups were compared to a normal control group comprising 100 healthy volunteers whose demographic data are also presented. There was a significant difference between female and male representation amongst all three study groups (p < 0.001).
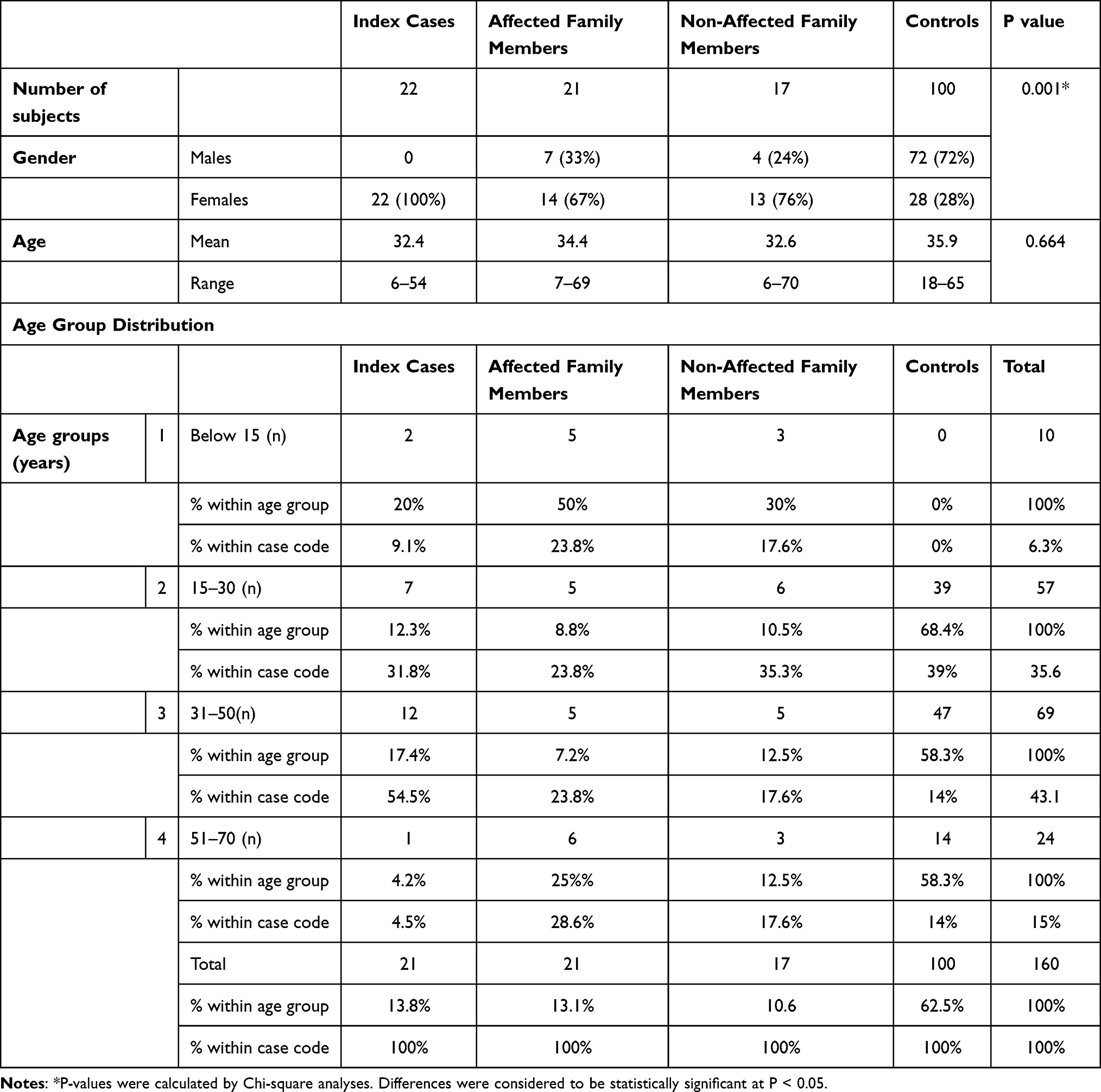 |
Table 1 Demographic Characteristics of Study Groups |
Phenotypic Signatures
Phenotypic characteristics included blood group analysis, bleeding scores and bleeding symptoms. The majority of participants have the O blood group, which was significantly prevalent than other blood groups representing 86.4%, 95.2%, 66.7% of all index, affected and non-affected cases, respectively (p < 0.001). The median bleeding score was significantly higher in the index case group than the affected and the non-affected family member groups (p < 0.001) (Figure 1). Also, the median value for the number of bleeding symptoms recorded in the index group was significantly higher than the other two patient groups (p = 0.003) (Table 2). The frequency of different bleeding symptoms recorded for the three studied groups are summarized in Figure 2. In this regard, the bleeding symptoms reported for the affected family members included epistaxis (30%), cutaneous bleeding (39%), oral cavity bleeding (43%), bleeding after tooth extraction (13%), bleeding from minor wounds (4%), menorrhagia (21%), post-partum hemorrhage (13%), muscle hematomas (8%), and other types of bleeding (8%).
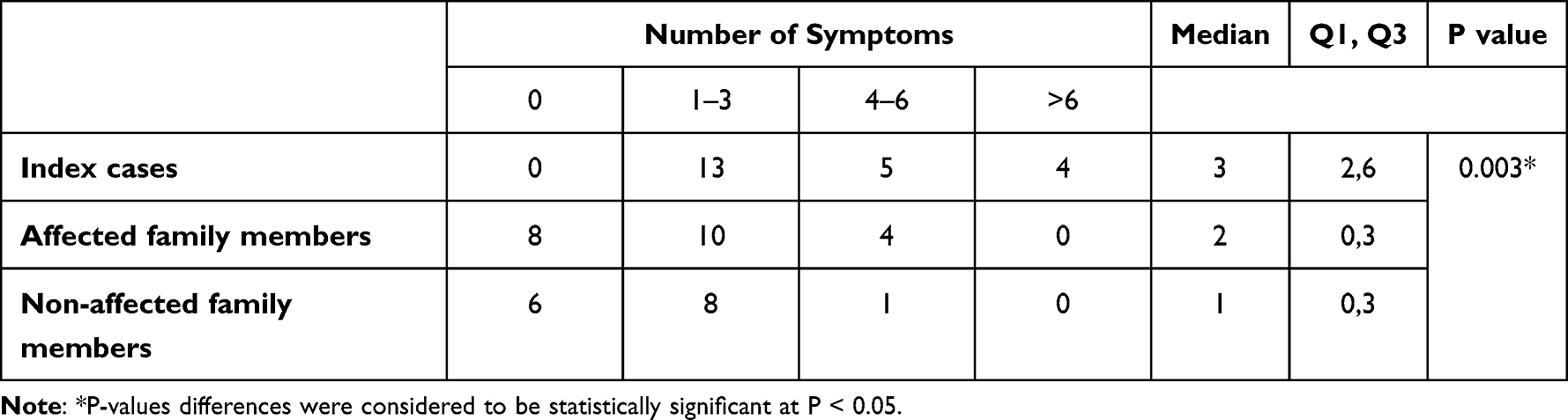 |
Table 2 Number of Bleeding Symptoms Recorded in the Studied Groups |
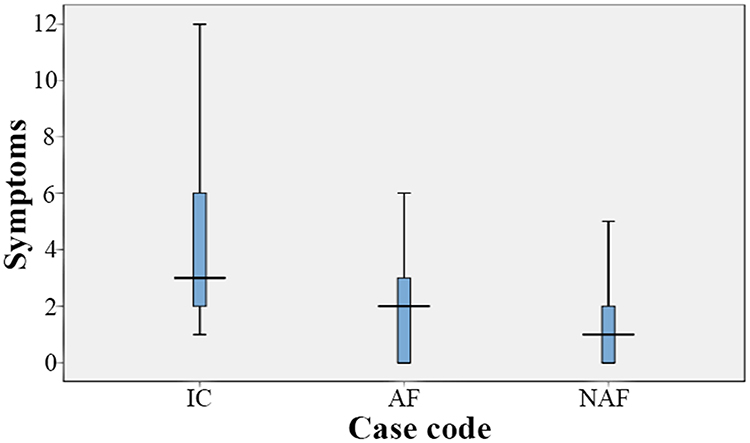 |
Figure 1 Median values and quartiles of bleeding scores in the study groups. Results indicated that the median value of bleeding scores was the highest in the index cases as compared to both the affected and the non-affected family members. Abbreviations: IC, index cases; AFM, affected family members; NAFM, non-affected family members. |
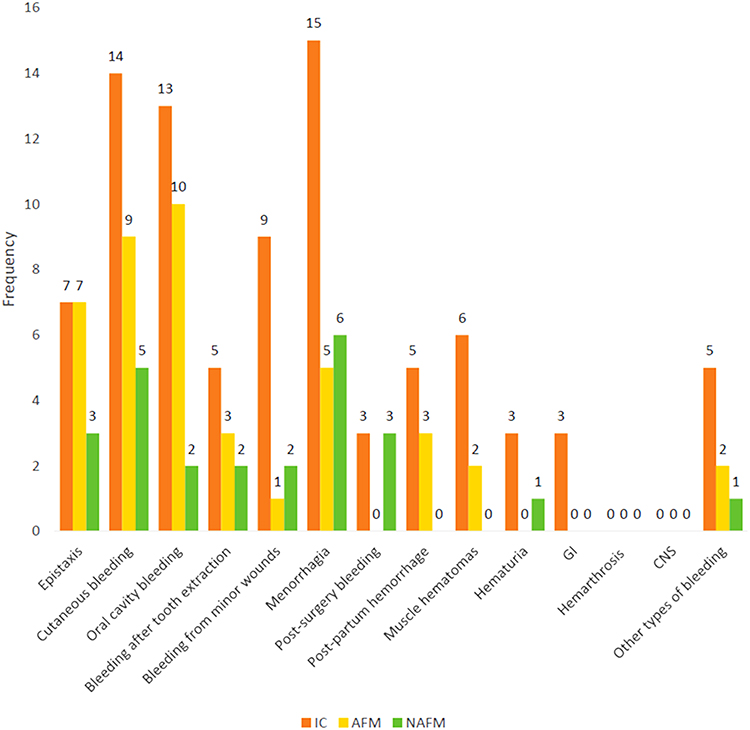 |
Figure 2 Frequency of different bleeding symptoms recorded for the three studied groups. Bleeding symptoms were more frequently reported in the index cases of VWD1 and the affected family members with cutaneous bleeding, oral cavity bleeding and menorrhagia being the most frequent. Abbreviations: GI, gastrointestinal; CNS, central nervous system; IC, index cases (22 females); AFM, affected family members (21 patients, 7 males and 14 females); NAFM, non-affected family members (17 subjects; 4 males and 13 females). |
Comparison between the study groups regarding laboratory investigations that included platelet count, activated partial thromboplastin time (APTT), VWF antigen (VWF:Ag), VWF:RCo, VWF:RCo/VWF:Ag ratio, factor VIII concentration (FVIII:C), bleeding score and number of bleeding symptoms are summarized in Tables 3 and 4. There was no significant difference between studied groups regarding platelet count and APTT. However, individual pair comparisons indicated a significant difference between the studied groups regarding the measured parameters as shown in Table 4.
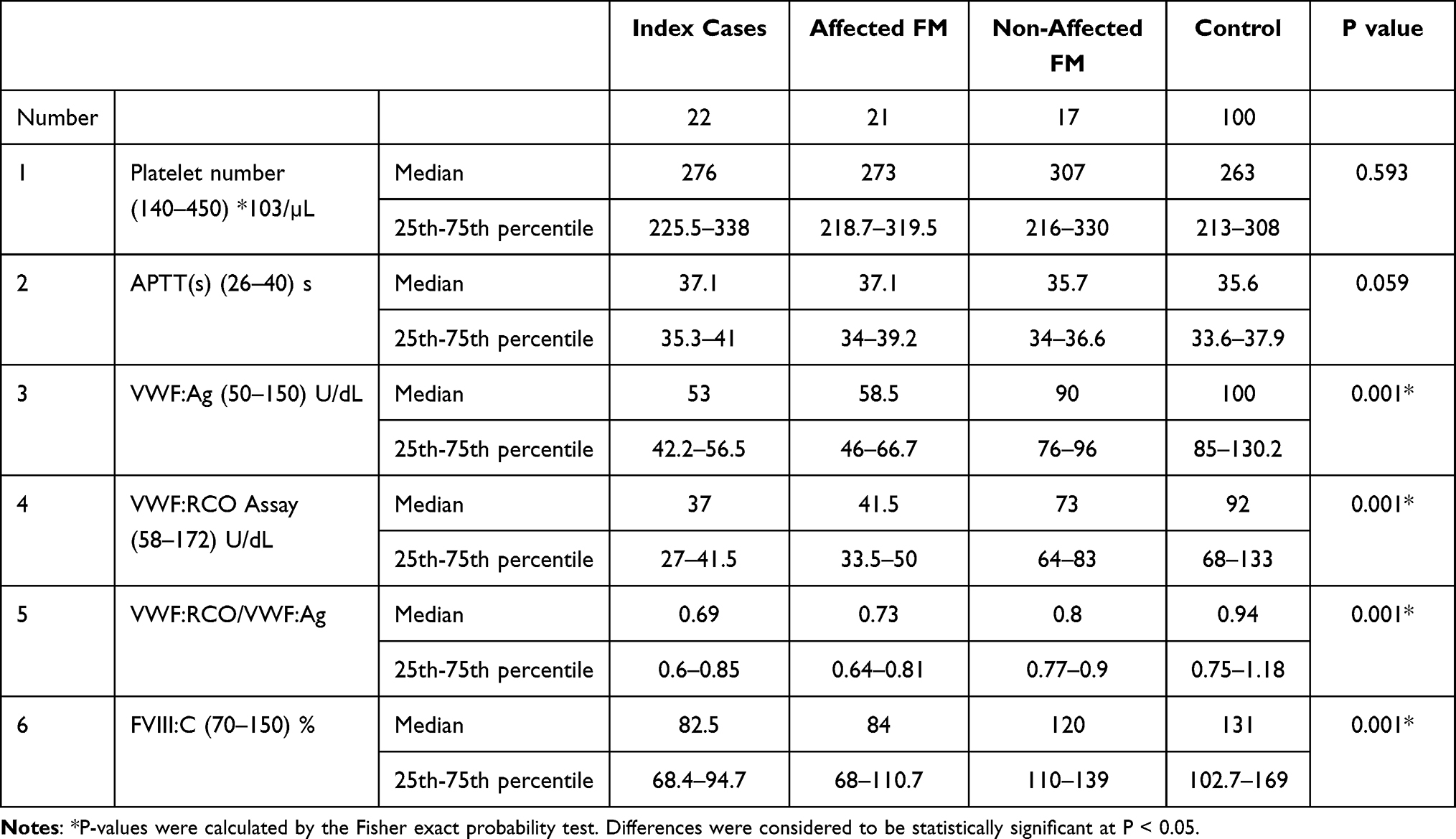 |
Table 3 Values of Laboratory Parameters Measured for All Studied Groups |
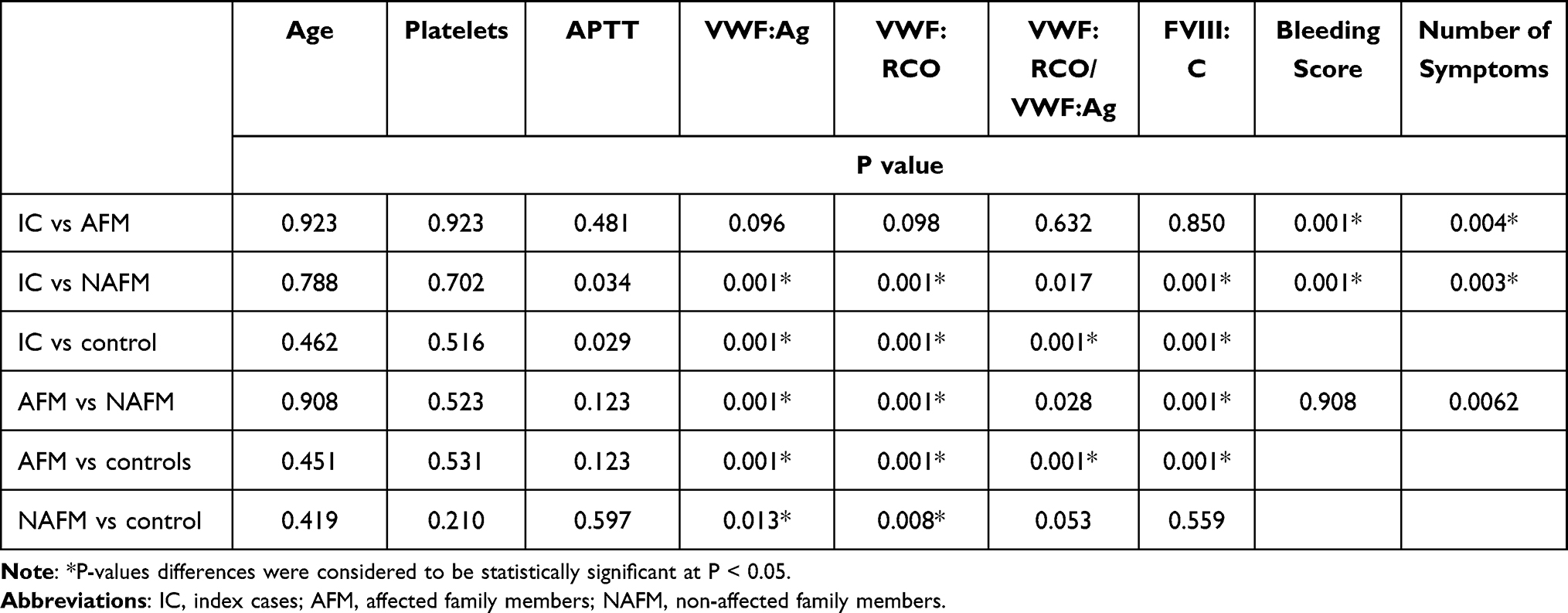 |
Table 4 Statistical Comparison Between the Values of Laboratory Parameters of All Studied Groups |
Genotypic Analysis
The results of Sanger sequencing of VWF exon 18 revealed five variants. Of these, three variants were exonic and two were intronic. Two of the exonic variants were missense changes, and other was synonymous. A summary of these variants and their postulated clinical significance are presented in Table 5. The reported clinical significance of all exonic variants is benign or likely benign in VWD or unspecified otherwise. The distribution of gene variants in exon 18 in study participants are also presented in Table 5. As it could be concluded from Table 5, 8.5% of all affected cases (index cases and affected family members) with O blood group showed gene variants in exon 18 of VWF, furthermore, 60% of non-affected family members had O blood group and inherited with variants in exon 18. Statistical analysis indicated that there was no significant association between c.2365A>G polymorphism and the disease groups (IC and AFM; P = 0.7). Likewise, c.2365A>G genotype was not associated with the control and the non-affected group (P = 0.38). Similarly, c.2385T>C genotype was not significantly associated with neither the disease group nor the non-affected groups (control and non-affected family members) (P = 1 and 0.55, respectively). Of note, these two genotypes were only investigated for correlation with the study groups because they were observed in the healthy control group as well as the disease groups. In this regard, correlation studies in control group indicated that rs1063856 genotype was not statistically correlated to factor VIII levels (P = 0.055). Likewise, the association between the genotype and VWFAg or VWF:RCo was not statistically significant (P = 0.125 and 0.704, respectively). Similarly, rs1063857 was not statistically correlated to the levels of factor VIII in healthy individuals (P = 0.152). Also, neither VWFAg nor VWF:RCo was in a significant correlation with rs1063857 (P = 0.865 and 0.704, respectively).
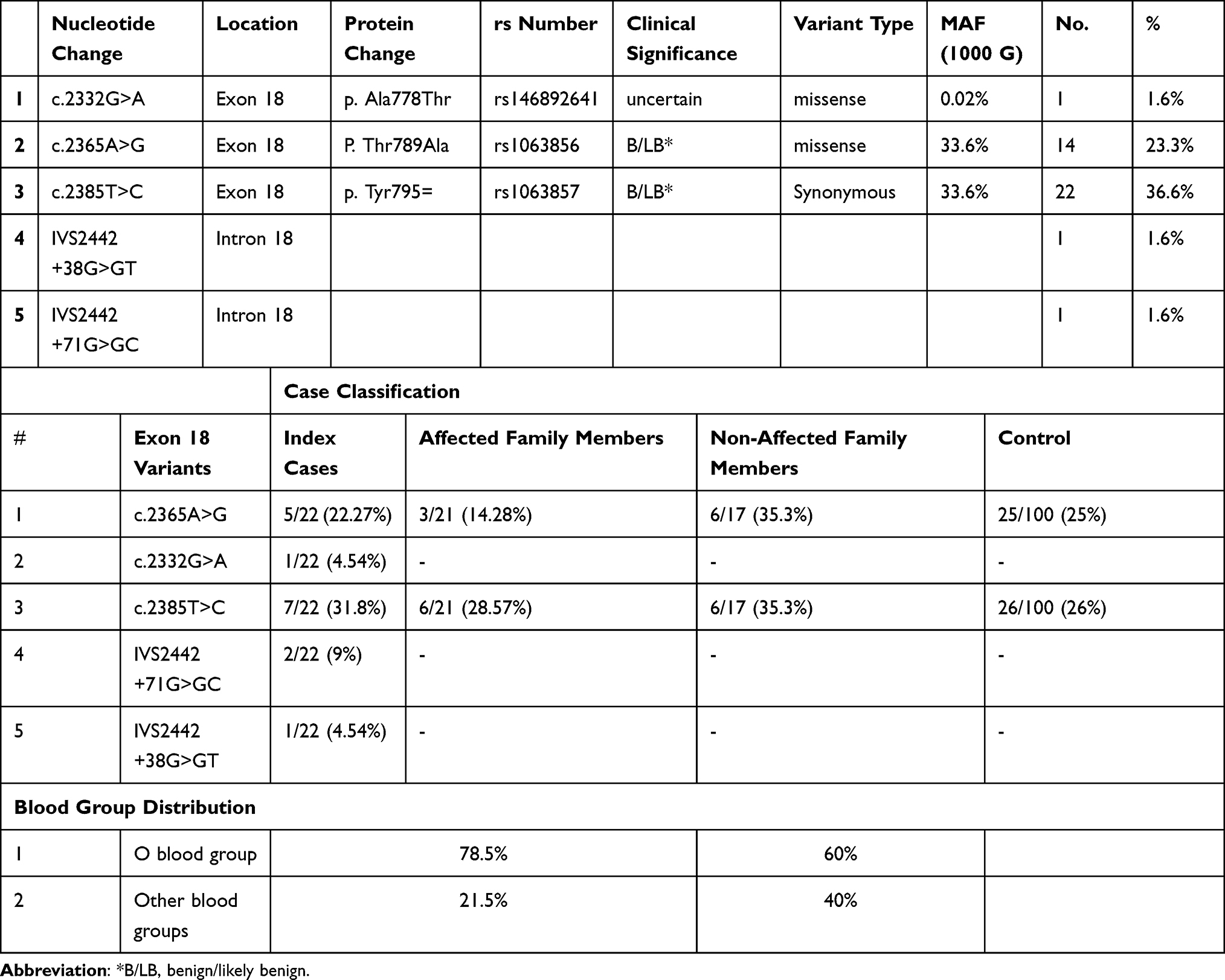 |
Table 5 Gene Variants of VWF Exon 18 and Their Distribution in the Studied Groups |
Discussion
von Willebrand factor (VWF) performs two vital functions in the coagulation cascade being involved in migration of platelets to the site of injuries as well as carrying factor VIII protecting it from degradation until reaching the site of vascular injury. Therefore, maintaining normal levels of this important factor is crucial for coagulation hemostasis.12 In this regard, it was reported that the levels of VWF are affected by a lot of biological factors including blood grouping and genetic variants in VWF gene.13 In the current study, we have employed Sanger sequencing to analyze gene variants of VWF exon 18 and correlate these variants with disease phenotypes in a group of families. For example, it was reported that the single nucleotide variants rs1063856 (c.2365A>G) and rs1063857 (c.2385T>C) are significantly associated with interpersonal variation in VWF plasma levels.14–16 Both variants were reported in the current study in 13.3% and 21.6% of index cases and family members who have VWD, respectively. On the other hand, both variants were reported in 35.3% of non-affected participants. Also, it was reported that VWD is more frequent in individuals having 2365A and 2385T alleles.17 This was in agreement with another study that was done on Swedish VWD patients and found that the T allele of the rs1063857 (c.2385T>C) polymorphism is correlated with the disease.18
In this respect, it was also reported that rs1063856 (c.2365A>G) and rs1063857 (c.2385T>C) are more common in healthy individuals and their occurrence is associated with higher levels of factor VIII and VWF, which seems to be in concordance with our results.19
The results of the current study indicated that VWF:Ag, VWF:RCo, VWF:RCo/VWF:Ag and FVIII:C values were significantly higher in the control and non-affected family members than in the index cases and the affected family members (Tables 4 and 5). This could explain the higher bleeding score in VWD patients (IC cases). In this context, it was reported that there is an association between ABO blood group and the level of both factor VIII and VWF, where people who have blood group O have lower levels of both factors.20 This seems to be in concordance with the results of the current study that showed that the majority of the index cases and the affected subjects who had variation in exon 18 of VWF have blood group O and they have lower levels of VWF and factor VIII as compared to the healthy control group or the non-affected family members (Tables 5). This was also in agreement with another recent study,21 which reported lower levels of VWF in O blood group than non-O blood group individuals. Likewise, O blood group was found to be associated with type 1 VWD in Swedish patients.18 This confirms the finding reported in the current study showing the prevalence of O blood group among Saudi patients of VWD. In this context, as far as blood grouping is a concern in VWD, another group22 recommended the investigation of blood group amongst VWD laboratory panels as this might contribute to reduction of panel requests for VWD patients. VWF gene was observed with highest number of novel HGMD heterozygous variants among the blood and bleeding disorder genes reported from the Saudi population.23,24 Further analysis of other exons in VWF gene in the study subjects can reveal the molecular basis of VWD.
The study could conclude that VWF levels are influenced by blood group and presence of certain gene variants indicating that both routine hematology testing and genetic analysis are important to decide on clinical management and classification of patients.24–26 The results of the current study as well as other studies identified a few gene variants of VWF exon 18 that could be protective from VWD; therefore, individual’s genotyping is advocate to screen for high-risk group that allows early diagnosis and therapeutic intervention.
Ethical Approval
This study was conducted in accordance with the Declaration of Helsinki. This study was reviewed and approved by the institutional review board at Imam Abdulrahman Bin Faisal University (IRB-2017-03-009).
Acknowledgment
This project is funded by the National Science, Technology and Innovation Program (NSTIP)-King Abdul-Aziz City for Science and Technology (KACST), Kingdom of Saudi Arabia, Award Number (11-MED1417- 46). We would like to acknowledge the KACST for funding through Imam Abdulrahman Bin Faisal University that supported our project. Also I would like to thank King Fahad University Hospital for providing the facilities and some equipment needed to produce and complete this strategic project.
Disclosure
All authors confirm that they have no conflicts of interest to declare in this work.
References
1. Swami A, Kaur V. Von Willebrand disease: a concise review and update for the practicing physician. Clin Applied Thrombosis. 2017;23(8):900–910.
2. Leebeek FWG, Eikenboom JCJ. Von Willebrand’s disease. N Engl J Med. 2016;375(21):2067–2080.
3. Sharma R, Flood VH. Advances in the diagnosis and treatment of Von Willebrand disease. Blood. 2017;130(22):2386–2391.
4. Montgomery RR, Flood VH. What have we learned from large population studies of Von Willebrand disease? Hematology. 2016;2016(1):670–677.
5. Abu-Douleh E, Al-Numair N, Albanyan A, Alsuliman A, Bayoumi N, Owaidah T. Prevalence of Von Willebrand disease among university students in Riyadh, Saudi Arabia. J Applied Hematol. 2018;9:136.
6. Manderstedt E, Lind-Hallden C, Lethagen S, Hallden C. Genetic Variation in the Von Willebrand Factor Gene in Swedish Von Willebrand Disease Patients. Companion j Thrombosis Haemostasis. 2018;2(1):e39–e48.
7. Manderstedt E, Lind-Halldén C, Lethagen S, Genetic HC. Variation in the Von Willebrand factor gene in Swedish von Willebrand disease patients. Companion j Thrombosis Haemostasis. 2018;2(1):e39–e48.
8. Goodeve A, Eikenboom J, Castaman G, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 Von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 Von Willebrand Disease (MCMDM-1VWD). Blood. 2007;109(1):112–121.
9. James PD, Notley C, Hegadorn C, et al. The mutational spectrum of type 1 Von Willebrand disease: results from a Canadian cohort study. Blood. 2007;109(1):145–154.
10. Alyami NH. Characterization of Mutations Identified in Patients Historically Diagnosed with Type 1 Von Willebrand Disease. University of Sheffield; 2014.
11. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463–5467.
12. Mufti AH, Ogiwara K, Swystun LL, et al. The common VWF single nucleotide variants c.2365A>G and c.2385T>C modify VWF biosynthesis and clearance. Blood Adv. 2018;2(13):1585–1594.
13. Orstavik KH, Kornstad L, Reisner H, Berg K. Possible effect of secretor locus on plasma concentration of factor VIII and Von Willebrand factor. Blood. 1989;73(4):990–993.
14. van Loon JE, Kavousi M, Leebeek FW, et al. Von Willebrand factor plasma levels, genetic variations and coronary heart disease in an older population. J Thrombosis Haemostasis. 2012;10(7):1262–1269.
15. Zabaneh D, Gaunt TR, Kumari M, et al. Genetic variants associated with Von Willebrand factor levels in healthy men and women identified using the HumanCVD BeadChip. Ann Hum Genet. 2011;75(4):456–467.
16. Zhou Z, Yu F, Buchanan A, et al. Possible race and gender divergence in association of genetic variations with plasma Von Willebrand factor: a study of ARIC and 1000 genome cohorts. PLoS One. 2014;9(1):e84810.
17. Shahbazi S, Alavi S, Mahdian R. Classification of exon 18 linked variants of VWF gene in Von Willebrand disease. Int J Mol Epidemiol Genet. 2012;3(1):77–83.
18. Manderstedt E, Lind-Halldén C, Lethagen S, Genetic HC. Variation in the Von Willebrand Factor Gene in Swedish Von Willebrand Disease Patients. Companion j Thrombosis Haemostasis. 2018;02(01):e39–e48.
19. Flood VH, Johnsen JM, Kochelek C, et al. Common VWF sequence variants associated with higher VWF and FVIII are less frequent in subjects diagnosed with type 1 VWD. Res Practice Thrombosis Haemostasis. 2018;2(2):390–398.
20. Liu X, Chen X, Yang J, Guo R. Association of ABO blood groups with Von Willebrand factor, factor VIII and ADAMTS-13 in patients with lung cancer. Oncol Lett. 2017;14(3):3787–3794.
21. Murray GP, Post SR, Post GR. ABO blood group is a determinant of Von Willebrand factor protein levels in human pulmonary endothelial cells. J Clin Pathol. 2020;73(6):347.
22. Archer NM, Forbes PW, Brugnara C. Knowledge of Blood Group Decreases Von Willebrand Factor Panel Testing in Children. HemaSphere. 2017;1(1):548.
23. Borgio JF. Heterogeneity in biomarkers, mitogenome and genetic disorders of Arab population with special emphasis on large-scale whole-exome sequencing. Arch Med Sci. 2021;1:58. 10.5114/aoms/145370.
24. Baz B, Abouelhoda M, Owaidah T, et al. Molecular classification of blood and bleeding disorder genes. NPJ Genom Med. 2021;6:62. doi:10.1038/s41525-021-00228-2
25. James PD, Connell NT, Ameer B, et al. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of Von Willebrand disease. Blood Adv. 2021;5(1):280–300.
26. Nichols WL, Hultin MB, James AH, et al. Von Willebrand disease (VWD): evidence‐based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA) 1. Haemophilia. 2008;14(2):171–232.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.