")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 13
Pharmacological Profile of Viltolarsen for the Treatment of Duchenne Muscular Dystrophy: A Japanese Experience
Received 20 September 2021
Accepted for publication 12 November 2021
Published 16 December 2021 Volume 2021:13 Pages 235—242
DOI https://doi.org/10.2147/CPAA.S288842
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Rohini Roy Roshmi, Toshifumi Yokota
Department of Medical Genetics, University of Alberta, Edmonton, Alberta, Canada
Correspondence: Toshifumi Yokota
Department of Medical Genetics, University of Alberta, Edmonton, Alberta, Canada
Tel +1 7804921102
Fax +1 7804921998
Email [email protected]
Abstract: Duchenne muscular dystrophy (DMD) is a fatal, X-linked recessive disorder characterized by progressive muscle loss and cardiorespiratory complications. Mutations in the DMD gene that eliminate the production of dystrophin protein are the underlying causes of DMD. Viltolarsen is a drug of phosphorodiamidate morpholino oligomer (PMO) chemistry, designed to skip exon 53 of the DMD gene. It aims to produce truncated but partially functional dystrophin in DMD patients and restore muscle function. Based on a preclinical study showing the ability of antisense PMOs targeting the DMD gene to improve muscle function in a large animal model, viltolarsen was developed by Nippon Shinyaku and the National Center of Neurology and Psychiatry in Japan. Following clinical trials conducted in Japan, Canada, and the United States showing significant improvements in muscle function, viltolarsen was approved for medical use in Japan in March 2020 and the United States in August 2020, respectively. Viltolarsen is a mutation-specific drug and will work for 8% of the persons with DMD who carry mutations amenable to exon 53 skipping. This review summarizes the pharmacological profile of viltolarsen, important clinical trials, and challenges, focusing on the contribution of Japanese patients and researchers in its development.
Keywords: phosphorodiamidate morpholino oligomer, exon skipping, dystrophin, efficacy, timeline, safety
Graphical Abstract:
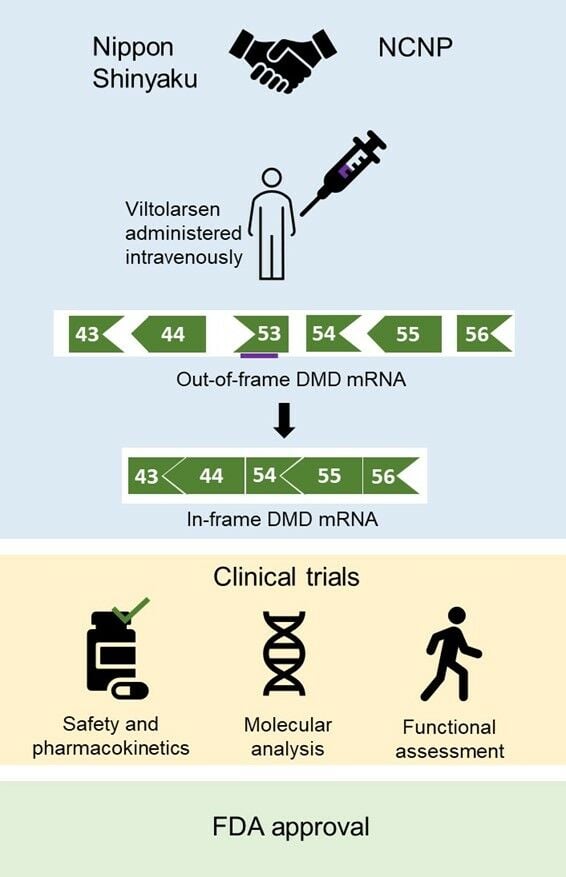
Introduction
Duchenne muscular dystrophy (DMD) is a lethal, X-linked recessive disorder with a prevalence rate of 1 in 3000–6000 boys globally.1 It is caused due to null mutations in the DMD gene.2 The DMD gene codes for dystrophin, a rod-shaped structural protein essential for muscle function.3 Dystrophin links the cytoskeleton of muscle fibres to the extracellular matrix and is essential for the stabilization of sarcolemma. In the absence of dystrophin, muscle fibres are damaged upon contraction and are replaced by fibrotic and adipose tissue.2 As a result of severe muscle damage, patients with DMD start experiencing lower body weakness at the age of 3–5 years. As the disease is characterized by progressive muscle loss, most of them lose ambulation during their early teens and die prematurely during their late twenties, due to cardiorespiratory complications.4,5
The DMD gene is the longest human gene with 79 exons.6 60% of patients with DMD have large deletions in the DMD gene.4,7 Other DMD-causing mutations include exonic duplications, mid-intronic mutations, point mutations, missense mutations, nonsense mutations, etc.8 Due to its long sequence of 2.4 million base pairs, the DMD gene is very susceptible to mutations. Despite being a common genetic disorder, it has been extremely difficult to design a therapy that will work for most patients with DMD as mutations among patients are highly variable. Fortunately, two mutation hotspots, exons 45–55 and exons 2–19, have been identified in the gene and are potential targets for therapeutics.9,10
Since the first report of DMD in 1868, advanced medicine has achieved a lot in terms of managing the symptoms of DMD; however, no DMD patient has been cured yet. Many therapies are under research. Since the 1980s, corticosteroids have been prescribed to patients with DMD, along with drugs that intervene with muscle blood flow, antioxidants, and growth hormones.11–13 However, long-term use of corticosteroids can have adverse effects such as weight gain, cataracts, and late puberty.14 More recently, therapies to restore the expression of dystrophin are emerging. Genome editing, gene-replacement, exon skipping, and stop codon read-through approaches are commonly researched for the treatment of DMD.4,15–17
Currently, four phosphorodiamidate morpholino oligomer (PMO) drugs, which result in exon skipping, namely eteplirsen, golodirsen, viltolarsen, and casimersen, have been approved by the Food and Drug Administration (FDA) for the treatment of DMD (Table 1).18–23 However, as these drugs are mutation-specific, they are only applicable to approximately 30% of all patients with DMD. In our previous review paper on viltolarsen, we discussed the pharmacokinetics, pharmacodynamics, and safety of the drug.21 This review paper will recap and update the clinical trials discussed previously, focusing on the pharmacological profile of viltolarsen and the contributions of Japanese researchers and patients in its development.
 |
Table 1 Current FDA Approved PMO Drugs for the Treatment of DMD |
Mechanism of Action
PMOs, like viltolarsen, consist of a short single-stranded DNA analog and a backbone of morpholine ring bonded by phosphorodiamidate linkages.24 They bind to a target sequence, by standard nucleic acid base-pairing, on the out-of-frame dystrophin pre-mRNA and alter splicing, resulting in an in-frame dystrophin mRNA. Consequently, a truncated but functional dystrophin protein is produced. Becker muscular dystrophy (BMD), which is a milder dystrophinopathy, is mostly caused by in-frame mutations in the DMD gene.25 Therefore, the form of dystrophin protein that BMD patients can produce is truncated but it is functional as the progression of BMD is significantly slower than DMD. BMD patients live a life with less severe medical complications and are a major inspiration behind exon skipping therapy.26
Viltolarsen, a PMO that facilitates exon 53 skipping of the DMD gene, was developed based on a pre-clinical study conducted by Yokota et al showing that antisense PMOs for exon skipping restore muscle function accompanied by dystrophin restoration in a dog model of DMD.21,27 Theoretically, exon 53 skipping can treat 8% of all patients with DMD including patients with exons 45–52, 47–52, 48–52, 49–52, 50–52, or exon 52 deletion, among others.28 It is a 21-nucleotide sequence that binds to exon 53 on the out-of-frame dystrophin pre-mRNA and hides exon 53 from the splicing machinery. As a result, exon 53 is skipped, and the open reading frame is restored. The in-frame mRNA is translated to produce a truncated but functional form of dystrophin (Figure 1).
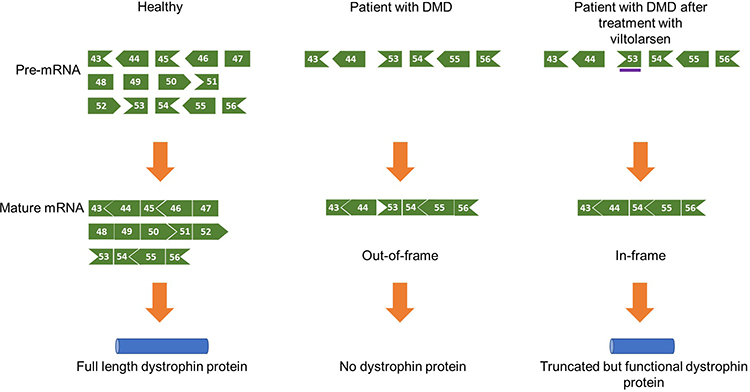 |
Figure 1 Mechanism of action of viltolarsen. Viltolarsen (purple bar) binds to exon 53. As a result, exon 53 is skipped from the mature mRNA, and a truncated but functional dystrophin protein is produced. For example, exons 45–52 deletion in the DMD gene causes an out-of-frame null mutation. Viltolarsen can skip exon 53 and restore the reading frame of the DMD gene, which allows the production of functional dystrophin protein. Adapted from Roshmi, R.R., Yokota, T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today (Barc). 2019, 55(10): 627-639. Copyright © 2019 Clarivate or its licensors. All rights reserved. doi: 10.1358/dot.2019.55.10.3045038.21 Abbreviations: DMD, Duchenne muscular dystrophin; mRNA, messenger RNA. |
The shortened dystrophin is functional as the most essential elements of the protein’s structure stay intact. Dystrophin, a 427 kDa protein, consists of four functional domains: an actin-binding N-terminal (NT) domain, a central rod domain with 24 spectrin repeats, a cysteine-rich domain, and a C-terminal (CT) domain that binds the dystrophin-associated glycoprotein complex (DAGC) in the sarcolemma.25 Exon 53 skipping compromises some of the spectrin repeats in the protein’s central rod domain, like the in-frame mutations seen in BMD patients, who have less severe muscle loss over their lifetime. This indicates that a dystrophin protein with a compromised central domain can still carry out its function of providing muscle strength.26
Golodirsen, a PMO drug designed by Sarepta Therapeutics, is also designed to skip exons 53 of the DMD gene.29 Its mechanism of action is similar to viltolarsen and it applies to the same patient population. No study comparing the two drugs’ efficacy and safety is available now.
Safety Profile
Viltolarsen can be safely administered intravenously to children and adults at a recommended dose of 80 mg/kg of bodyweight weekly.30 It is recommended to monitor kidney function during the treatment because renal toxicity was observed in animals treated with viltolarsen.30 However, kidney function was not compromised in any patient during clinical trials.31–33
In a Phase I clinical trial (Table 2) conducted in Japan, mild treatment-associated adverse drug reactions including proteinuria, eczema, high diastolic pressure, and low ejection fraction were observed.33 Later, proteinuria was eliminated as an adverse effect because proteins were not detected in urine spot samples. None of the participants quit the study due to adverse events or were left with unsolved adverse events at the end of the study.
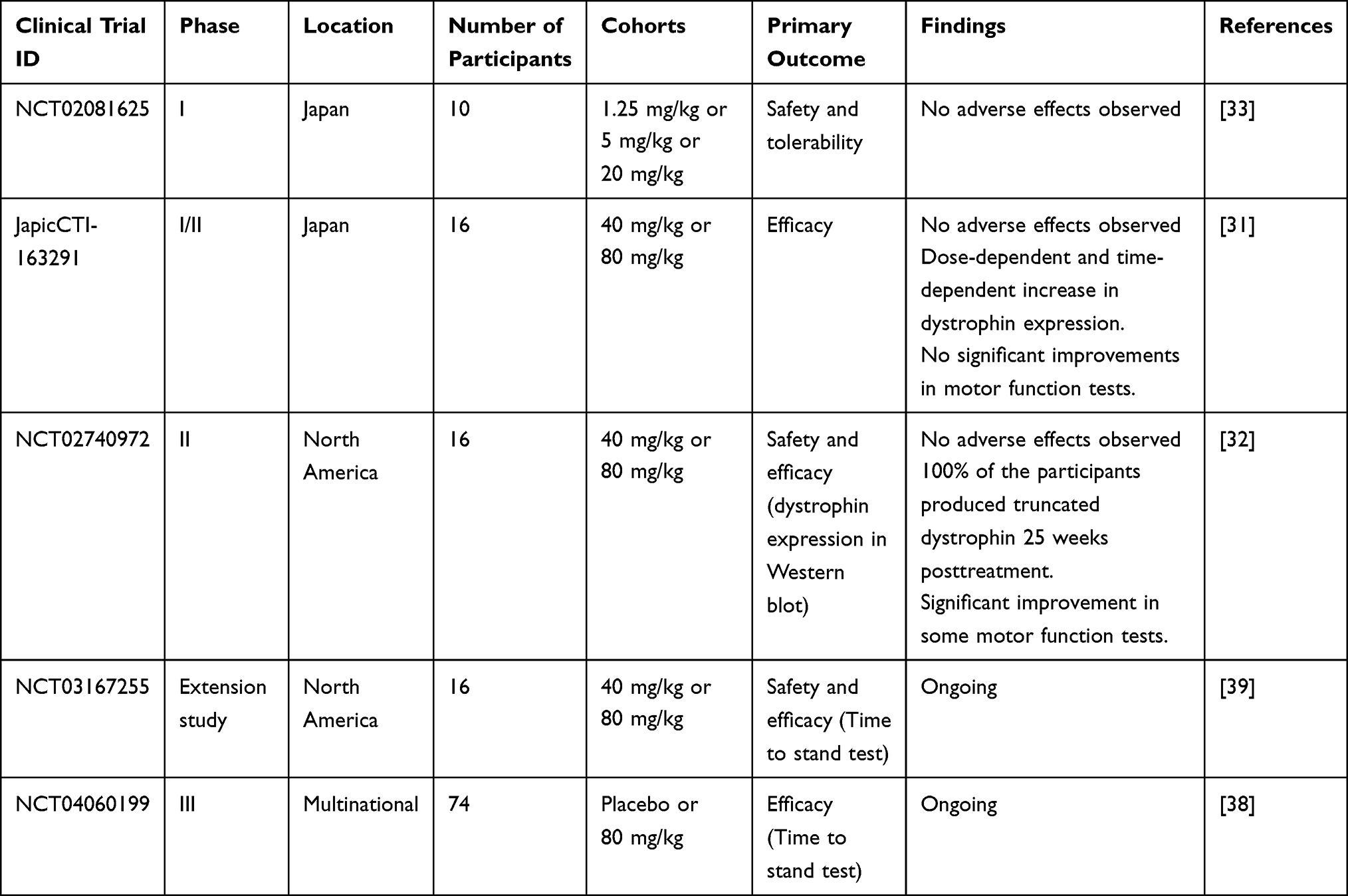 |
Table 2 Summary of Clinical Trials Discussed in the Review Paper |
In a phase I/II clinical trial (Table 2) conducted in Japan, nasopharyngitis and upper respiratory tract infection (URTI) was commonly observed in participants who received 80 mg/kg of viltolarsen weekly.31 All adverse events except one were mild. A severe case of URTI was reported and the participants had to be hospitalized. However, it was later determined to be independent of the treatment. None of the other participants quit the study due to adverse events. In another Phase II clinical trial conducted in North America (Table 2), 94% of the participants suffered mild adverse events.32 However, they were resolved without changing the dose of viltolarsen or the withdrawal of a participant from the study.
As of now, there is no safety data available on viltolarsen for patients with kidney problems, pregnant women, or lactating mothers.30
Pharmacokinetics
The maximum concentration in blood plasma (Cmax) and half-life of viltolarsen were observed to be dose-dependent in both phase I and phase I/II clinical trials conducted in Japan (Table 2).31,33 In the phase I clinical trial conducted by Komaki et al, patients with DMD were intravenously administered 1.25 mg/kg, 5 mg/kg, or 20 mg/kg viltolarsen weekly. Cmax was achieved in less than an hour for the 5 mg/kg and 20 mg/kg cohorts. Within 24 hours, 70–80% of the unchanged drug was removed from the body in the urine. The results were consistent in the phase I/II clinical trial.31–33
Efficacy of Viltolarsen
In the phase I clinical trial conducted by Komaki et al, a dose-dependent increase in exon 53 skipping was observed.33 One patient NS-07, who received 20 mg/kg viltolarsen, showed a 47.5% increase (from pre-treatment 0.3% to posttreatment 47.8%) in exon 53 skipping, compared to only 1% increase in the rest of the 20 mg/kg cohort. NS-07 weighed more than the other participants in the study, hence the impressive results could be attributed to the elevated amount of viltolarsen participant NS-07 received. In a Western blot analysis, only participant NS-07 showed dystrophin expression. The main motive of this study was to evaluate the safety of viltolarsen. The recommended dosage of viltolarsen is 80 mg/kg which is four times more than the highest dosage tested in this study. Hence, the low efficacy can be attributed to the low dosage of viltolarsen administered.
In the phase I/II clinical trial, a dose-dependent and time-dependent increase in dystrophin expression was observed.31 The average exon skipping levels in the 80 mg/kg cohort (42.4%) were 20.6% higher than that of the 40 mg/kg cohort (21.8%). Additionally, participants in both cohort groups showed greater exon skipping levels in week 24 compared to week 12 of treatment. Western blot results showed an overall 2.78% increase in dystrophin levels in the 80 mg/kg cohort at the end of the study. Contrary to the hopeful results seen in the molecular analysis, participants did not do better in the motor function tests compared to the baseline. However, the decline in performance was less pronounced in participants who received higher 80 mg/kg viltolarsen weekly.
In a phase II clinical trial conducted by Clemens et al, Western blot results exhibited that 100% of the participants produced truncated dystrophin after 25 weeks of treatment.32 On average, 5.7% and 5.9% of normal dystrophin levels were restored in the 40 mg/kg and 80 mg/kg cohorts, respectively. Other methods of measuring efficiency including RT-PCR, mass spectrometry, and immunofluorescence, demonstrated results consistent with the Western blot data of individual participants. The participants also showed significant improvements in some motor function tests including velocity in the time to run/walk 10 m test, 6-minute walk test (6MWT), and time to stand from supine (TTSTAND). For all motor function assessments, viltolarsen-treated participants were compared to natural history controls from The Cooperative International Neuromuscular Research- Duchenne Natural History Study (CINRG-DNHS).34 All participants from this phase II clinical trial are eligible to participate in a 192-week extended study where the efficacy, safety, and motor function will be evaluated further (Table 2).
Recent studies show that dystrophin levels as low as 3% can result in a mild BMD phenotype,35 hence it is hopeful even if the therapy results in little dystrophin production. In the phase II clinical study by Clemens et al, 88% of the participants showed >3% increase in dystrophin expression.32
Timeline to Approval
In 2009, Nippon Shinyaku (NS) and the National Center of Neurology and Psychiatry in Japan (NCNP) started a collaboration to develop viltolarsen for patients with DMD with mutations amenable to exon 53 skipping.22 The phase I study to determine the safety of the drug was initiated in June 2013 and continued until September 2014 in Japan.33,36 Not long after in 2015, viltolarsen received the SAKIGAKE and Orphan Drug designations from the Ministry of Health, Labour and Welfare (MHLW) of Japan. A phase I/II clinical trial (April 2016 to November 2017) and a phase II clinical trial (December 2016 to April 2018) were conducted in Japan and North America respectively to determine the efficacy of viltolarsen.31,32,37 After evaluating results from clinical trials, viltolarsen was granted Fast Track designation, Orphan Drug Status, and Rare Pediatric disease designation in the USA in 2017. In 2019, NS Pharma submitted the New Drug Application (NDA) for viltolarsen. Shortly, on 25th March 2020, viltolarsen was approved in Japan and on 12th August 2020, it was approved by the FDA for treating eligible patients with DMD.35 Currently, there are two ongoing clinical trials to further evaluate the efficacy and safety of viltolarsen: an extension of the phase II clinical trial in North America (July 2017 to October 2021) and RACER53 (April 2020 to December 2024).38,39
Contributions of Japanese Patients and Researchers
Japanese researchers have made significant contributions to the field of exon skipping therapy.41 Currently four PMO drugs have been approved by the FDA for the treatment of DMD; these PMO drugs operate by the principles of exon skipping in the DMD gene, which was first shown by Takeshima et al in 1995.41 A 16-year-old Japanese DMD patient had a 52 bp deletion within exon 19 of the DMD gene.40 cDNA from the patient did not have exon 19, which led researchers to investigate an exonic splicing enhancer site. Hence, it was proposed that antisense oligonucleotides (AOs) can be targeted towards splicing enhancers to inhibit the splicing of exon 19. Following up on these findings, the exon skipping strategy was developed. A 31-year-old Japanese patient harboured a nonsense mutation in exon 27.42 The patient was expected to have DMD; however, he had a BMD phenotype. Further assessment of the patient’s cDNA showed that exon 27 was naturally skipped. It was concluded that in-frame mRNA has the ability to produce functional dystrophin. In 2009, Yokota et al demonstrated the first successful PMO-mediated exon skipping accompanied by bodywide restoration of dystrophin protein in a severe animal model of DMD, called canine X-linked muscular dystrophy in Japan (CXMDj), which was established at the NCNP. This study directly led to the collaboration between Nippon Shinyaku and the NCNP, and development of viltolarsen.27
A national registry of Japanese DMD/BMD patients called Remudy has made a considerable contribution to the enrollment of DMD patients. Remudy was developed in collaboration with Translational Research in Europe–Assessment and Treatment of Neuromuscular Diseases (TREAT-NMD) in 2009.43 DMD/BMD patients’ clinical and molecular genetics characteristics are reported and stored at Remudy. Clinical symptoms, diagnostic test results such as muscle biopsy, genetic test results, and epidemiological information of patients can be accessed through Remudy. These data can help find eligible patients for clinical trials and keep patients informed about upcoming trials. Only consenting patients with confirmed genetic status are registered in the database. As genetic testing is not covered by public health insurance in Japan, Remudy arranges to sequence their DMD gene free of cost. As of August 2021, 2030 patients with dystrophinopathies have requested to register with Remudy.44
Conclusion
Viltolarsen is the second exon 53 skipping drug approved by the FDA. It has shown promising results in clinical trials, but it is only applicable for 8% of patients with DMD worldwide. Additionally, a one-year supply of viltolarsen for a child weighing 25 kilogram is expected to be $587,000, which further makes accessibility difficult for patients who make an average income.45 This cost is further expected to increase in the future when the child grows up, due to increased body weight.
Overall, treatment with viltolarsen resulted in dystrophin expression in muscle cells in the clinical trial and improvements in motor function tests. However, the number of participants in the clinical trials was low and the duration of the study was too short to make robust conclusions about improvements in muscle function. Two clinical trials are still running to access the long-term efficacy of viltolarsen. One of them is RACER53 which plans to recruit 74 participants who will receive viltolarsen for a course of 48 weeks and the efficacy of viltolarsen will be determined by their performance in motor function tests (Table 2).38 In addition, viltolarsen may not be as efficient in treating the heart because PMO uptake by cardiac tissue is usually poor.46 It is of major concern as death from cardiac complications are common in patients with DMD.
For decades it has been difficult to design therapies that cater to all patients with DMD due to the variability in mutations among them. So far, 31% of patients with DMD can be theoretically treated with the PMO drugs approved by the FDA. The approval of viltolarsen is yet another small victory for persons with DMD, their families, and researchers.
Acknowledgments
This work was supported by Muscular Dystrophy Canada, the Friends of Garrett Cumming Research Fund, the HM Toupin Neurological Science Research Fund, Canadian Institutes of Health Research (CIHR), Alberta Innovates: Health Solutions (AIHS), Jesse’s Journey, and the Women and Children’s Health Research Institute (WCHRI).
Disclosure
TY is a co-founder and shareholder of OligomicsTx Inc., which aims to commercialize antisense technology. The authors report no other conflicts of interest in this work.
References
1. Mendell JR, Shilling C, Leslie ND, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. 2012;71(3):304–313. doi:10.1002/ana.23528
2. Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. doi:10.1016/0092-8674(87)90579-4
3. Zubrzycka-Gaarn EE, Bulman DE, Karpati G, et al. The Duchenne muscular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature. 1988;333(6172):466–469. doi:10.1038/333466a0
4. Manzur AY, Kinali M, Muntoni F. Update on the management of Duchenne muscular dystrophy. Arch Dis Child. 2008;93(11):986–990. doi:10.1136/adc.2007.118141
5. Mah J. Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr Dis Treat. 2016;12:1795–1807. doi:10.2147/NDT.S93873
6. Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509–517. doi:10.1016/0092-8674(87)90504-6
7. van Deutekom JCT, van Ommen G-JB. Advances in Duchenne muscular dystrophy gene therapy. Nat Rev Genet. 2003;4(10):774–783. doi:10.1038/nrg1180
8. Juan-Mateu J, Gonzalez-Quereda L, Rodriguez MJ, et al. DMD mutations in 576 dystrophinopathy families: a step forward in genotype-phenotype correlations. Ervasti JM, ed. PLoS One. 2015;10(8):e0135189. doi:10.1371/journal.pone.0135189
9. Oudet C, Hanauer A, Clemens P, Caskey T, Mandel J-L. Two hot spots of recombination in the DMD gene correlate with the deletion prone regions. Hum Mol Genet. 1992;1(8):599–603. doi:10.1093/hmg/1.8.599
10. Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2(12):731–740. doi:10.1016/S1474-4422(03)00585-4
11. Werneck LC, Lorenzoni PJ, Ducci RD-P, Fustes OH, Kay CSK, Scola RH. Duchenne muscular dystrophy: an historical treatment review. Arq Neuropsiquiatr. 2019;77(8):579–589. doi:10.1590/0004-282x20190088
12. Rutter MM, Collins J, Rose SR, et al. Growth hormone treatment in boys with Duchenne muscular dystrophy and glucocorticoid-induced growth failure. Neuromuscul Disord. 2012;22(12):1046–1056. doi:10.1016/j.nmd.2012.07.009
13. Drachman DB, Toyka KV, Myer E. Prednisone in Duchenne muscular dystrophy. Lancet. 1974;304(7894):1409–1412. doi:10.1016/S0140-6736(74)90071-3
14. Wood CL, Straub V, Guglieri M, Bushby K, Cheetham T. Short stature and pubertal delay in Duchenne muscular dystrophy. Arch Dis Child. 2016;101(1):101–106. doi:10.1136/archdischild-2015-308654
15. Echevarría L, Aupy P, Goyenvalle A. Exon-skipping advances for Duchenne muscular dystrophy. Hum Mol Genet. 2018;27(R2):R163–R172. doi:10.1093/hmg/ddy171
16. Sheikh O, Yokota T. Developing DMD therapeutics: a review of the effectiveness of small molecules, stop-codon readthrough, dystrophin gene replacement, and exon-skipping therapies. Expert Opin Investig Drugs. 2021;30(2):167–176. doi:10.1080/13543784.2021.1868434
17. Verhaart IEC, Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol. 2019;15(7):373–386. doi:10.1038/s41582-019-0203-3
18. Aartsma-Rus A, Corey DR. The 10th oligonucleotide therapy approved: golodirsen for Duchenne muscular dystrophy. Nucleic Acid Ther. 2020;30(2):67–70. doi:10.1089/nat.2020.0845
19. Casimersen (Amondys 45) for Duchenne muscular dystrophy. Med Lett Drugs Ther. 2021;63(1627):e104–e105. Available from: http://www.ncbi.nlm.nih.gov/pubmed/34181634. Accessed November 15, 2021.
20. Shirley M. Casimersen: first approval. Drugs. 2021;81(7):875–879. doi:10.1007/s40265-021-01512-2
21. Roshmi RR, Yokota T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today. 2019;55(10):627. doi:10.1358/dot.2019.55.10.3045038
22. Dhillon S. Viltolarsen: first approval. Drugs. 2020;80(10):1027–1031. doi:10.1007/s40265-020-01339-3
23. Lim KR, Maruyama R, Yokota T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des Devel Ther. 2017;11:533–545. doi:10.2147/DDDT.S97635
24. Nan Y, Zhang Y-J. Antisense phosphorodiamidate morpholino oligomers as novel antiviral compounds. Front Microbiol. 2018;9. doi:10.3389/fmicb.2018.00750
25. Gao QQ, McNally EM. The dystrophin complex: structure, function, and implications for therapy. In: Comprehensive Physiology. Wiley; 2015:1223–1239. doi:10.1002/cphy.c140048
26. Le Rumeur E. Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies. Bosn J Basic Med Sci. 2015;15(3). doi:10.17305/bjbms.2015.636
27. Yokota T, Lu Q, Partridge T, et al. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol. 2009;65(6):667–676. doi:10.1002/ana.21627
28. Watanabe N, Nagata T, Satou Y, et al. NS-065/NCNP-01: an antisense oligonucleotide for potential treatment of exon 53 skipping in Duchenne muscular dystrophy. Mol Ther Nucleic Acids. 2018;13:442–449. doi:10.1016/j.omtn.2018.09.017
29. Heo Y-A. Golodirsen: first approval. Drugs. 2020;80(3):329–333. doi:10.1007/s40265-020-01267-2
30. Viltolarsen. Am J Health Pharm. 2020;77(24):2032–2033. doi:10.1093/ajhp/zxaa324
31. Komaki H, Takeshima Y, Matsumura T, et al. Viltolarsen in Japanese Duchenne muscular dystrophy patients: a Phase 1/2 study. Ann Clin Transl Neurol. 2020;7(12):2393–2408. doi:10.1002/acn3.51235
32. Clemens PR, Rao VK, Connolly AM, et al. Safety, tolerability, and efficacy of viltolarsen in boys with Duchenne muscular dystrophy amenable to exon 53 skipping. JAMA Neurol. 2020;77(8):982. doi:10.1001/jamaneurol.2020.1264
33. Komaki H, Nagata T, Saito T, et al. Systemic administration of the antisense oligonucleotide NS-065/NCNP-01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Sci Transl Med. 2018;10(437). doi:10.1126/scitranslmed.aan0713
34. The Cooperative International Neuromuscular Research Group (CINRG). Available from: https://cinrgresearch.org/duchenne-natural-history/.
35. NS PHARMA’S VILTEPSOTM (viltolarsen) injection now FDA-approved in the U.S. for the treatment of Duchenne muscular dystrophy in patients amenable to exon 53 skipping therapy. NS Pharma News Release; 2020. Available from: https://www.nspharma.com/pdfs/Viltepso_Approval_Press_Release.pdf.
36. ClinicalTrials.gov Exploratory Study of NS-065/NCNP-01 in DMD. Available from: https://clinicaltrials.gov/ct2/show/NCT02081625.
37. ClinicalTrials.gov safety and dose finding study of NS-065/NCNP-01 in boys with Duchenne Muscular Dystrophy (DMD). Available from: https://clinicaltrials.gov/ct2/show/NCT02740972.
38. ClinicalTrials.gov Study to assess the efficacy and safety of viltolarsen in ambulant boys with DMD (RACER53). Available from: https://clinicaltrials.gov/ct2/show/NCT04060199.
39. ClinicalTrials.gov Extension Study of NS-065/NCNP-01 in boys with Duchenne Muscular Dystrophy (DMD). Available from: https://clinicaltrials.gov/ct2/show/NCT03167255.
40. Matsuo M, Takeshima Y, Nishio H. Contributions of Japanese patients to development of antisense therapy for DMD. Brain Dev. 2016;38(1):4–9. doi:10.1016/j.braindev.2015.05.014
41. Takeshima Y, Nishio H, Sakamoto H, Nakamura H, Matsuo M. Modulation of in vitro splicing of the upstream intron by modifying an intra-exon sequence which is deleted from the dystrophin gene in dystrophin Kobe. J Clin Invest. 1995;95(2):515–520. doi:10.1172/JCI117693
42. Shiga N, Takeshima Y, Sakamoto H, et al. Disruption of the splicing enhancer sequence within exon 27 of the dystrophin gene by a nonsense mutation induces partial skipping of the exon and is responsible for Becker muscular dystrophy. J Clin Invest. 1997;100(9):2204–2210. doi:10.1172/JCI119757
43. Nakamura H, Kimura E, Mori-Yoshimura M, et al. Characteristics of Japanese Duchenne and Becker muscular dystrophy patients in a novel Japanese national registry of muscular dystrophy (Remudy). Orphanet J Rare Dis. 2013;8(1):60. doi:10.1186/1750-1172-8-60
44. Remudy. Available from: http://www.remudy.jp/news/2021/09/005992.html.
45. Viltolarsen (viltepso) for Duchenne muscular dystrophy. Med Lett Drugs Ther. 2020;62(1609):167.
46. Meyers TA, Townsend D. Cardiac pathophysiology and the future of cardiac therapies in Duchenne muscular dystrophy. Int J Mol Sci. 2019;20(17):4098. doi:10.3390/ijms20174098
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.