")
Back to Journals » OncoTargets and Therapy » Volume 12
Pharmacological inhibition of USP7 promotes antitumor immunity and contributes to colon cancer therapy
Authors Fu C, Zhu X, Xu P, Li Y
Received 6 August 2018
Accepted for publication 11 September 2018
Published 15 January 2019 Volume 2019:12 Pages 609—617
DOI https://doi.org/10.2147/OTT.S182806
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Takuya Aoki
Changqing Fu,1 Xiaojue Zhu,2 Peiqi Xu,2 Yonghao Li2
1Clinical Laboratory, Zhangjiagang Fifth People’s Hospital, Suzhou University, Suzhou, Jiangsu 215621, People’s Republic of China; 2Clinical Laboratory, Zhangjiagang First People’s Hospital, Suzhou University, Suzhou, Jiangsu 215600, People’s Republic of China
Background: Effectiveness of clinical therapy such as chemotherapy for solid tumors is limited by acquired drug resistance and side effects. Available antitumor immunity methods showed promising prospect of cancer therapy. However, more drug targets for boosting antitumor immunity still need to be explored and selective and effective compounds are yet to be developed.
Purpose: To study the effect and possible mechanism of compound P5091, a selective USP7 inhibitor, on CT26 xenografts growth in mice.
Materials and methods: CT26 xenografts model was employed to examine the anti-tumor effect of P5091. RT-PCR and ELISA analysis were used to detect the level of IFN-γ, TNF-α and IL-10 in tumor tissue and serum, respectively. IFN-γ expression in CD4+ and CD8+ T cells was analyzed by intracellular stain. The level of FOXP3 in Treg cells was confirmed by intracellular stain and western blotting.
Results: Compound P5091, a selective USP7 inhibitor, was found to inhibit CT26 xenografts growth in mice, which is comparable to the effect of Anti-PD-1 antibody. RT-PCR analysis showed that P5091 treatment decreased IL-10 mRNA level in tumor tissue while elevated mRNA level of IFN-γ and TNF-α. Moreover, ELISA analysis manifested decreased of IL-10 and elevation of IFN-γ and TNF-α in serum from tumor bearing mice. Intracellular stain showed increased IFN-g expression both in CD4+ and CD8+ T cells after P5091 treatment. Furthermore, P5091 treatment caused FOXP3 loss in Treg cells decreased the proportion of Treg cells in tumor bearing mice.
Conclusion: Our study here showed that P5091 may be a candidate for cancer immunotherapy.
Keywords: USP7, Treg, antitumor immunity, colon cancer
Introduction
Colorectal cancer (CRC) is one of the most common forms of cancer worldwide affecting people’s life and health. Surgical management, radiation, chemotherapy, and ongoing developments in immune therapy are now being used as efficient cure tools for CRC treatment. On the one hand, clinical chemotherapeutic drugs, such as 5-FU, cisplatin, Bortezomib, and others showed promising cancer treatment efficiency, but dose-limiting toxicities and the development of resistance limit their long-term utility. On the other hand, the use of therapeutic monoclonal antibodies (mAbs) in oncology, which mainly includes anti-PD-1, anti-PD-L1, and anti-CTLA4,1,2 has gained widespread recognition. Unfortunately, the clinical responses observed following mAb treatment remain largely heterogeneous, their duration is still highly unpredictable, and they have a high and unbearable economic cost.3 Also, these therapies are associated with a diverse spectrum of immune-related adverse events (irAEs) that are typically transient, but occasionally severe, or even fatal, like cytokine storm. Therefore, effective and safe treatments for cancer are still needed to be explored.4
In recent years, significant efforts in understanding and modulating the immune response in cancer have been made. In this context, immunosuppressive cells, including Tregs and myeloid-derived suppressor cells, have come under intense investigation for their proposed roles in suppressing tumor-specific immune responses and establishing an immunosuppressive tumor microenvironment, thus enabling tumor immune evasion. An accumulation of FOXP3+ Tregs within tumor tissue and/or draining lymph nodes has a negative prognostic effect for many solid tumors, including lung cancer, colon cancer, and other malignancies.5–9 Thus, Treg depletion or functional suppression could be a new strategy for cancer immunotherapy.10,11
The ubiquitin proteasome system is a major nonlysosomal pathway by which intracellular protein degradation is mediated via proteasome holoenzyme, ubiquitin ligases, and deubiquitylating (DUB) enzymes. USP7 is a cysteine protease among the 100 DUB members and is known to regulate various physiological processes by deubiquitinating and stabilizing the proteins involved. Studies on USP7 show that it has been closely implicated in tumorigenesis, cancer metastasis, and HIV progression.12–16 Inhibition of USP7 is capable of inducing cell death in ovarian cancers17 and overcoming Bortezomib resistance by inducing apoptosis in multiple myeloma cells.18 Moreover, USP7 stabilizes expression of Tip60 and FOXP3, which are essential for Treg cells.19 Treg cell play central role in regulation of immune responses to self-antigens, allergens, and commensal microbiota as well as immune responses to infectious agents and tumors.20 Transcriptional factor FOXP3 serves as a lineage specification factor of Treg cells while its deficiency is responsible for systemic overactivated immune response,21 indicating that USP7 is a promising target for antitumor therapy.
In our study, a selective USP7 inhibitor, P5091, significantly suppressed the growth of CT26, a CRC cell line, as indicated by decreased tumor volume and weight. Tumor cell proliferation was downregulated by P5091 and anti-PD-1 treatment as evidenced by hematoxylin and eosin stain (H&E) and proliferating cell nuclear antigen (PCNA) immunohistochemistry staining. In addition, P5091 treatment suppressed IL-10 and promoted IFN-γ and TNF-α expression, both at the mRNA and protein levels. Further study suggested that P5091 boosted the function of CD4+ and CD8+ T-cells by upregulating IFN-γ expression and inhibited immune suppressor Tregs by reducing FOXP3 expression.
Materials and methods
Reagents
P5091 (P005091) was purchased from Selleck (Shanghai, People’s Republic of China). Dulbecco’s Modified Eagle’s Medium and fetal bovine serum were from Thermo Fisher Scientific (Waltham, MA, USA). Antibodies against FOXP3, PARP, and cleaved-caspase-3 were purchased from Cell Signaling Technology (Danvers, MA, USA) and β-actin was bought from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). The TUNEL assay kit was bought from Vazyme Biotech Co., Ltd (Nanjing, People’s Republic of China). CD8-FITC (FITC-conjugated anti-CD8 antibody), FOXP3-PE (PE-conjugated anti-FOXP3 antibody), and IFN-γ-APC (APC conjugated anti-IFN-γ antibody) were purchase from BioLegend (San Diego, CA, USA). TRIzol reagent and cell culture products were obtained from Takara (Shiga, Japan).
Cell culture
Mouse Colon Carcinoma (CT26) cells were purchased from Keygen Biotech (Nanjing, People’s Republic of China). Cells were maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum and antibiotics (100 U/mL penicillin and 100 U/mL streptomycin) at 37°C in a humidified atmosphere of 5% CO2 (Thermo Fisher Scientific).
Ethics statements
The approval for use of animals in research was performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD, USA) and approved by the Animal Care and Use Committee for Health Sciences of Suzhou University. All animal experiments were carried out according to the Animal Care Guidelines of Suzhou University. The animals were housed under specified-pathogen-free conditions that meet international standards; they were regularly checked by the certified veterinarian responsible for health monitoring, animal welfare supervision, experimental protocols, and procedure revision. At the time of sacrifice, they were euthanized by cervical dislocation. All efforts were made to reduce the number of animals used and to minimize the animals’ suffering.
Animals
Female BALB/c mice, 6–8 weeks old, were purchased from the Experimental Animal Center of Suzhou University (Suzhou, People’s Republic of China). They were maintained with free access to pellet food and water in plastic cages at 21°C±2°C and kept on a 12-hour light/dark cycle.
CT26 xenograft model
Mice received subcutaneous injections of CT26 cells (5×105 cells in 100 μL PBS). Tumor growth was evaluated by measuring the length and width of the tumor mass with calipers every 2 days. P5091 (5, 10 mg/kg, intraperitoneal every day) was administrated from day 3 onward. Intraperitoneal injected anti-PD-1 (5 mg/kg, every 3 days) was used as a positive control. Tumor volumes were measured and calculated using the equation volume = a*b2/2, where “a” is the maximal width and “b” is the maximal orthogonal width. On the 15th day, mice were weighed and euthanized and the tumors were removed and weighed.
RT-PCR
Total RNA from tissue samples was isolated using TRIzol reagent, according to the supplier’s instructions. Total RNA was reverse transcribed into cDNA using Superscript II enzyme and oligo (dT) (Thermo Fisher Scientific). RT-PCR was performed with the Bio-Rad Real-Time PCR Detection System (Bio-Rad, CFX Connect™ SYBR® Green Supermix [Bio-Rad, 1708880, Bio-Rad Laboratories Inc., Hercules, CA, USA]) and threshold cycle numbers were obtained using Bio-Rad CFX Manager Software (Bio-Rad). The program for amplification was 1 cycle of 95°C for 5 minutes followed by 40 cycles of 95°C for 10 seconds, 60°C for 25 seconds, and 95°C for 10 seconds. Actin was used as an internal control. The primer sequences used in this study were as follows: m-β-Actin forward 5′-TGCTGTCCCTGTATGCCTCT-3′; reverse 5′-TTTGATGTCACGCACGATTT-3′.
m-IFN-γ forward 5′-TGAGTATTGCCAAGTTTGAGGTCA-3′; reverse 5′-CGGCAACAGCTGGTGGAC-3′.
m-IL-10 forward 5′-AATAAGGTTTCTCAAGGGGCT-3′; reverse 5′-AGAACCAAGACCCAGACATCAA-3′.
m-TNF-α forward 5′-CGAGTGACAAGCCTGTAGCCC-3′; reverse 5′-GTCTTTGAGATCCATGCCGTTG-3′.
Immunohistochemistry
Formalin-fixed paraffin-embedded tissue sections were deparaffinized in xylene, rehydrated through graded ethanol, and then boiled for 10 minutes in citrate buffer (10 mM, pH 6.0) for antigen retrieval. Endogenous peroxidase activity was suppressed by exposure to 3% hydrogen peroxide for 10 minutes. Slides were then blocked with 3% goat serum (Life Technology, 16210-064, Carlsbad, CA, USA) and incubated with diluted PCNA, Granzyme B, or IFN-γ primary antibodies for 2 hours at room temperature at 37°C. They are then incubated with streptavidin-HRP (Shanghai Gene Company, GK500705, Shanghai, People’s Republic of China) for 40 minutes, stained with DAB (Shanghai Gene Company, GK500705) substrate, and counterstained with hematoxylin. Images were acquired by microscopy (Olympus IX51, Olympus Corporation, Tokyo, Japan).
TUNEL assay
Paraffin-embedded tumor sections were stained with TUNEL-FITC (1:50) and then stained with DAPI for 2 minutes. Images were acquired using a fluorescence microscope (Olympus IX51).
Flow cytometry analysis
The murine spleen cells and lymph nodes cells were extracted and stimulated using PMA, ionomycin, and protein transport inhibitor (Thermo Fisher Scientific) for 4 hours. At the end of stimulation, the cells were permeabilized using fixation buffer and 1× permeabilization buffer (Thermo Fisher Scientific). The following antibodies were used for flow cytometry: anti-IFN-γ-APC, anti-FOXP3-FITC, and anti-CD4-PE (Thermo Fisher Scientific). Single-cell suspensions were examined on a FACS Fortessa (BD, San Jose, CA, USA), and the data were analyzed using FlowJo software (FlowJo LLC, Ashland, OR, USA). For sorting, cells were stained with anti-CD4-PE and anti-FOXP3-FITC and double-positive cells were collected.
Cytokine measurement
Cytokines in the serum including IL-10, TNF-α, and IFN-γ (R&D Systems, Inc., Minneapolis, MN, USA) were measured using enzyme-linked immunosorbent assay kits according to the manufacturer’s instructions.
Statistical analysis
All of the experimental data are presented as the mean ± standard error of the mean. The statistical significance of the differences between two groups was determined using Student’s unpaired t-test, and multiple comparisons were analyzed using one-way analysis of variance followed by Dunnett’s post hoc test using GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was taken to indicate statistical significance.
Results
P5091 treatment suppressed colon cancer growth in vivo
To examine the antitumor activity of P5091 in comparison with anti-PD-1 in vivo, a mouse xenograft model of CT26 cells in BALB/C mice was employed. As shown in Figure 1A, P5091 inhibited the growth of CT26 xenografts in a dose-dependent manner, as evidenced by tumor volume and the fact that a high-dose treatment of P5091 had the same effect as anti-PD-1 treatment. The appearance of the tumor after dissection also showed that P5091 and anti-PD-1 treatment could shrink tumor volume (Figure 1B). Moreover, the tumor weight dropped from 1.38 g (control group) to 0.62 g (10 mg/kg P5091), while the anti-PD-1 group decreased to 0.75 g (Figure 1C). Both P5091 and anti-PD-1 treatment exhibited low toxicity, as no significant bodyweight loss was induced in tumor-bearing mice by either treatment (Figure 1D).
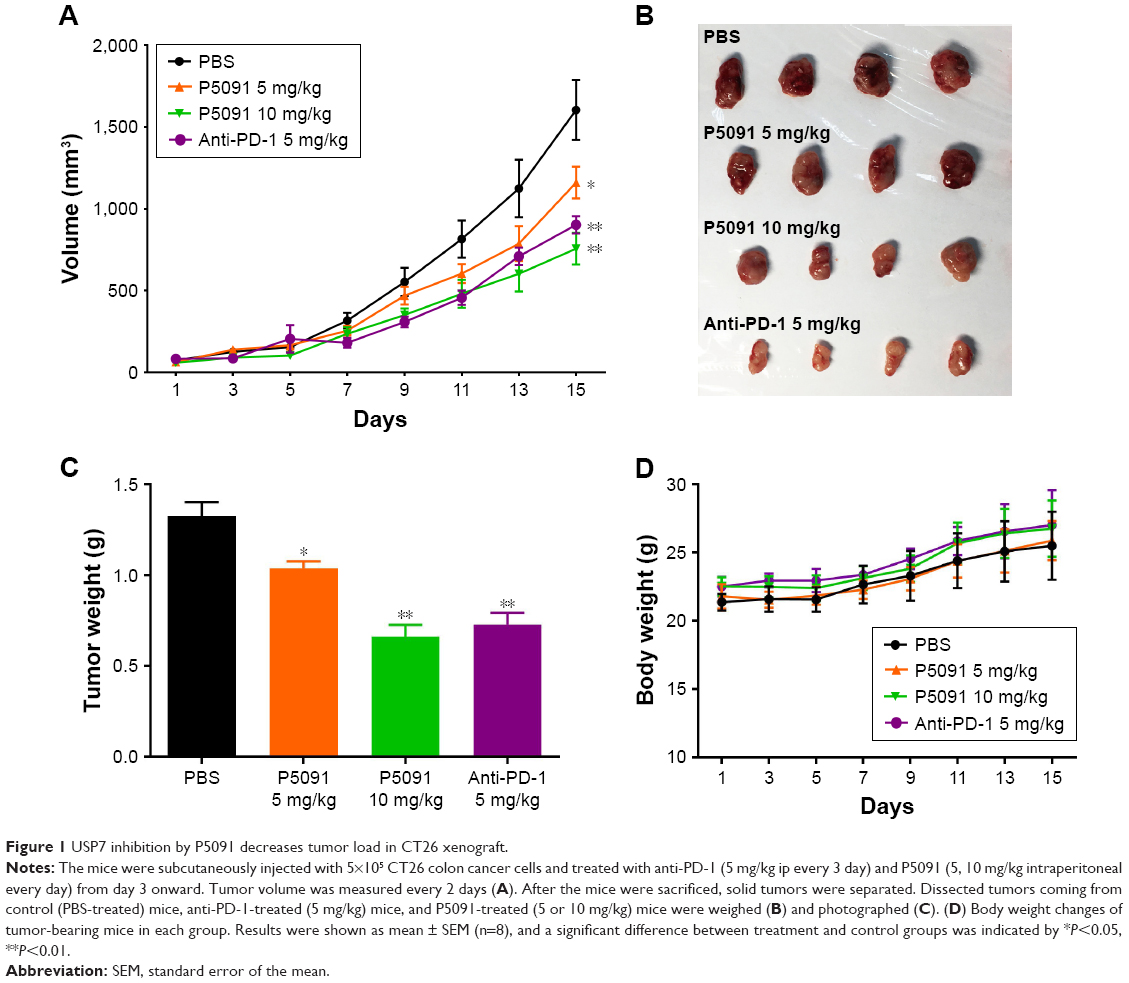 | Figure 1 USP7 inhibition by P5091 decreases tumor load in CT26 xenograft. |
P5091 lead to enhanced proliferation inhibition and apoptosis in tumor tissues
To further confirm the tumor inhibition effect of P5091, we next tested the proliferation and apoptosis of tumor tissues from each group. A typical pathological characteristic of malignancy, which was shown by H&E staining, was observed in resected specimens of CT26 from the control group. The P5091 group showed massive cancer cell damage, such as condensation of the cytoplasm and pyknosis of nuclei. Interestingly, 10 mg/kg P5091 treatment, the high-dose group, showed more extensive cell damage compared to the anti-PD-1 treatment (Figure 2A). Immunohistochemistry staining of PCNA, a marker for tumor growth, showed that P5091 obviously decreased the protein levels. Meanwhile, PCNA expression in the 10 mg/kg P5091 group was decreased to a level similar to that in the anti-PD-1 group (Figure 2B). TUNEL stain showed that massive apoptotic nuclei in the 10 mg/kg P5091- and anti-PD-1-treated groups (Figure 2C). Further, P5091 treatment also triggered cleavage of PARP and caspase-3 in a dose-dependent manner (Figure 2D). All of these results proved that both P5091 and anti-PD-1 treatment enhanced tumor proliferation inhibition and apoptosis in vivo.
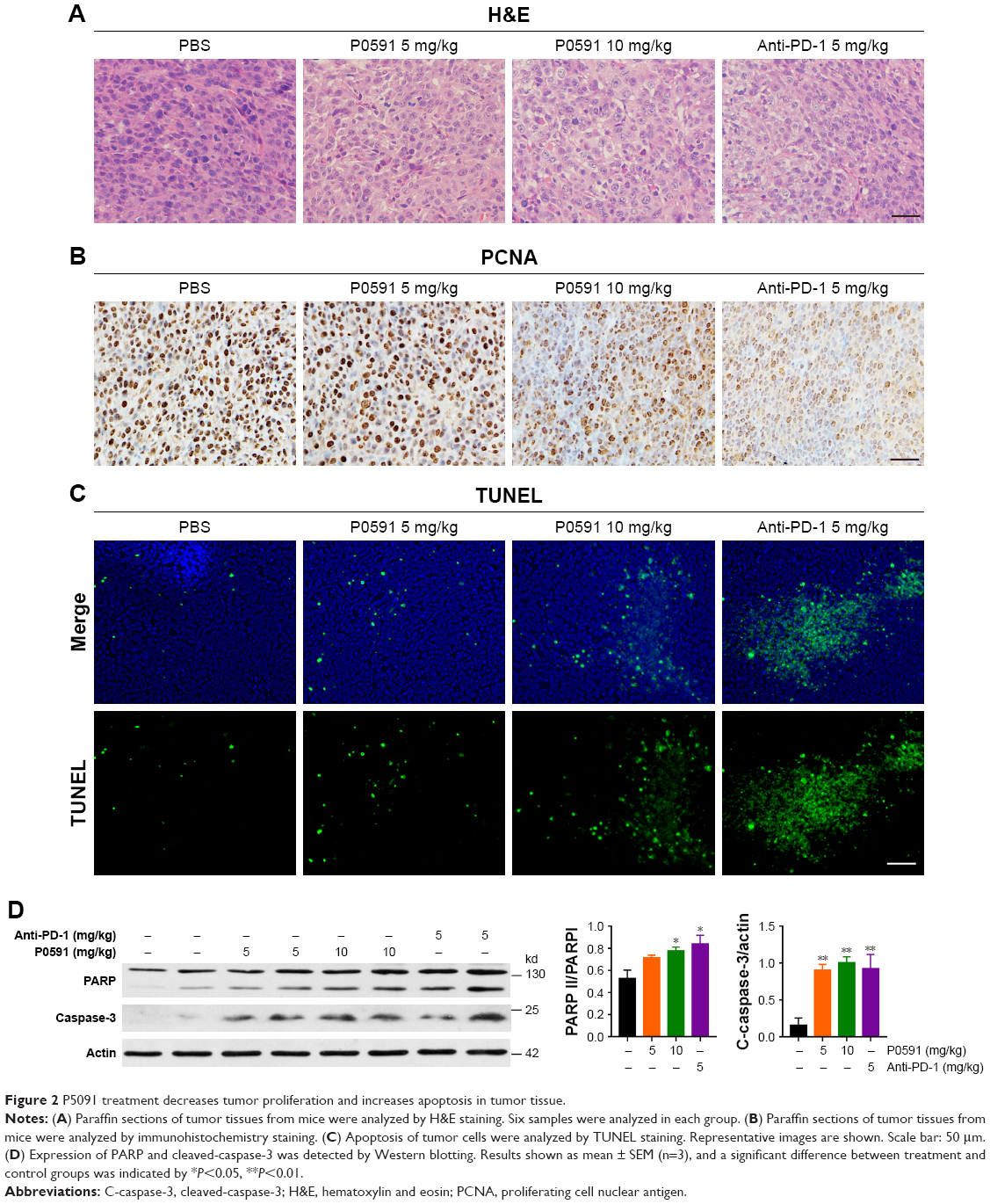 | Figure 2 P5091 treatment decreases tumor proliferation and increases apoptosis in tumor tissue. |
P5091 treatment promoted IFN-γ and TNF-α and suppressed IL-10 expression
To determine the underlying mechanism of how P5091 could suppress colon tumor growth in vivo besides its direct effect on cell cytotoxicity reported before, our study here focused on the effect of P5091 on antitumor immunity. First, we examined the levels of anti-inflammatory cytokine IL-10 and proinflammatory cytokines IFN-γ and TNF-α as IFN-γ and TNF produced by cytotoxic T lymphocytes (CTLs) were crucial for the elimination of established mouse tumors, which could be counterbalanced by IL-10.22,23 As shown in Figure 3A and B, both P5091 and anti-PD-1 treatment boosted the mRNA and protein levels of IFN-γ and TNF-α in tumor tissue and serum, respectively. However, anti-PD-1 treatment had little effect on IL-10 expression, which may due to its effect mainly being centered on T-cells (Figure 3A and B). It has been reported that PD-1 blockade could promote IL-10 release in ovarian cancer, which is thought to be one of the adaptive resistance mechanisms that undermine the efficacy of anti-PD-1 therapy.24
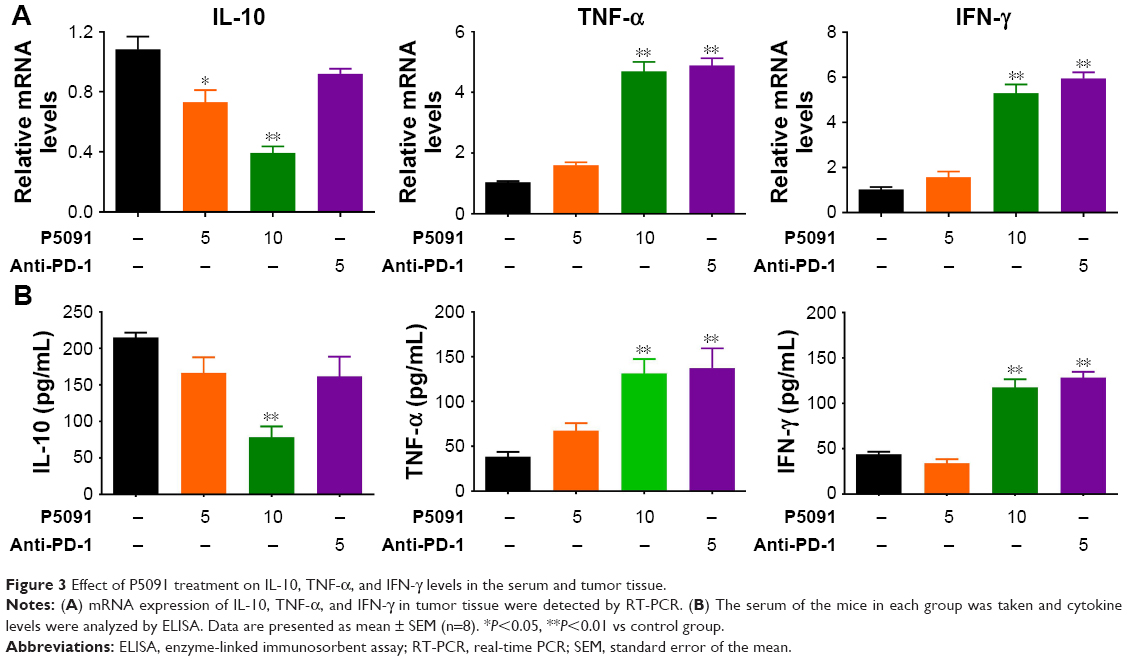 | Figure 3 Effect of P5091 treatment on IL-10, TNF-α, and IFN-γ levels in the serum and tumor tissue. |
P5091 treatment promoted the CD4+ and CD8+ T-cell-mediated antitumor immune response
IFN-γ secreted by CD4+ and CD8+ T-cells carries out its antitumor immunity by direct cytotoxicity to tumor cells, triggering either Fas-mediated targeted cell death or perforin-induced cytolysis. Taking this into account, we tested the cytotoxicity of CD4+ and CD8+ T-cells by IFN-γ staining. The intracellular stain showed a significant increase of IFN-γ expression in both CD4+ and CD8+ T-cells in the P5091- and anti-PD-1-treated groups (Figure 4A and B). These results showed that P5091 and anti-PD-1 treatment boosted the antitumor immune response.
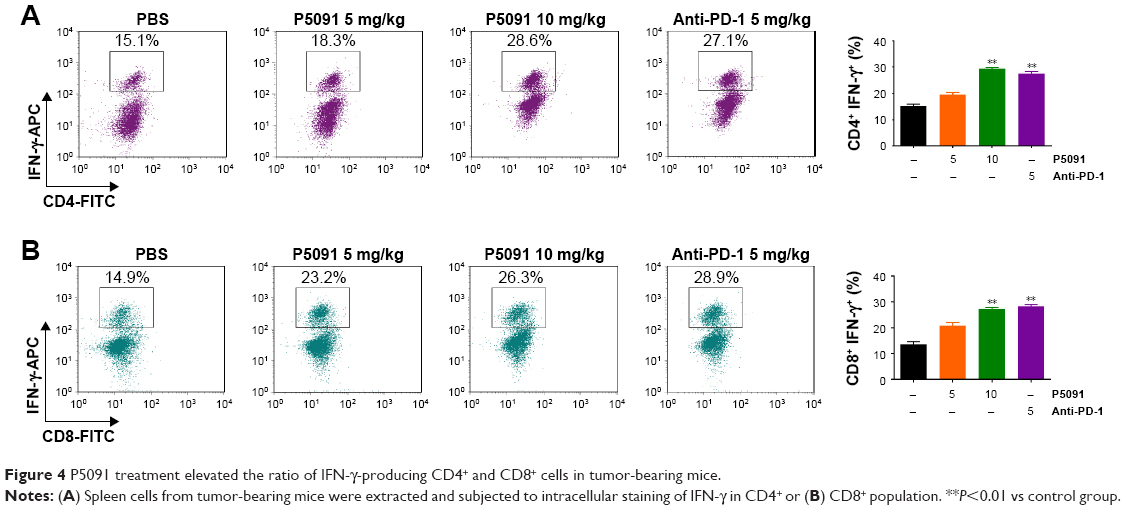 | Figure 4 P5091 treatment elevated the ratio of IFN-γ-producing CD4+ and CD8+ cells in tumor-bearing mice. |
P5091 treatment inhibited FOXP3+ Treg function in the tumor microenvironment
FOXP3+ Treg cells are known to suppress protective host immune responses to a wide variety of solid tumors. We next tried to find out the effect of P5091 on the function of FOXP3+ Treg cells. The intracellular stain showed a significant decrease in FOXP3 expression in CD4+ T-cells in the P5091-treated group, while no change was observed in anti-PD-1 group (Figure 5A and B). Western blot also showed the same tendency of FOXP3 expression in CD4+ T-cells (Figure 5C and D). Taken together, P5091 treatment not only boosted the antitumor immune response but also suppressed the protective host immune response in the tumor microenvironment.
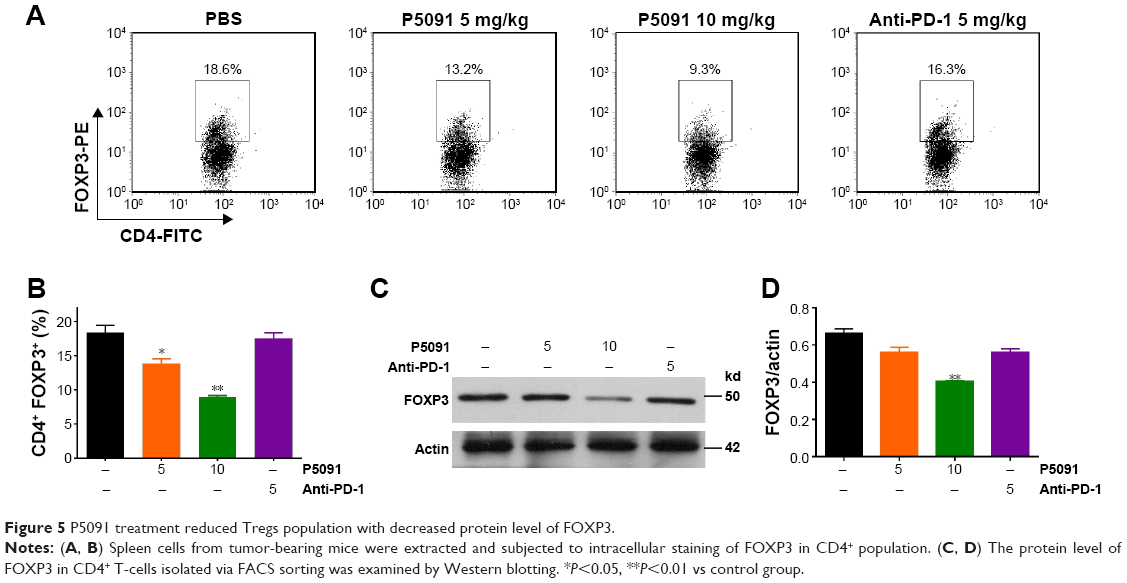 | Figure 5 P5091 treatment reduced Tregs population with decreased protein level of FOXP3. |
Discussion
The deubiquitinating enzyme USP7 plays a critical role in its substrate proteins’ involvement in tumors, DNA repair, immune response, viral recognition, and so on.25,26 Deregulation of USP7 has been implicated in the pathogenesis of many human diseases, including cancer. USP7 promotes cell proliferation through the stabilization of Ki-67 protein in non-small-cell lung cancer cells.27 USP7 inhibitors hold promise as a new strategy for the treatment of cancer. It is reported that USP7 inhibitors sensitize various cancer cells to PARP-inhibitor drugs.28,29 In addition, small-molecule compounds that inhibit USP7 deubiquitinating activity induce cancer cell line apoptosis via the p53 pathway.30,31 P5091 is a reported selective USP7 inhibitor and it inhibited Wnt-β-catenin pathway and contributes to the apoptosis of colon cancer cells.32 Our study here aimed to examine the effect of P5091 on antitumor immunity and its possible role in cancer immunotherapy.
In our study, we found that P5091 promoted colon cancer cell apoptosis and death in CT26 xenografted mice and that the effect was similar when compared to an anti-PD-1-treated group. In the mechanism study, we confirmed the direct effect of P5091 on both the CT26 and HCT116 cancer cell line (data not shown). It has been reported that USP7 inhibition by P5091 for CRC inhibition is p53-independent.32 Both p53 and PTEN show no mutation in CT26.33 Also, the antitumor immunity in the microenvironment was specifically examined. A previous study reported by others showed that USP7 knockdown by RNA interference impaired FOXP3+ Treg function and promoted antitumor immunity.19 The results from in vivo experiments showed boosted expression of IFN-γ in both CD4+ and CD8+ T-cells, which reflected an increase in cytotoxicity and the function of T-cells by P5091 treatment. Furthermore, P5091 also inhibited FOXP3 expression in CD4+ T-cells, indicating an impaired immune tolerance in the tumor microenvironment. Based on the findings by others and our study, we think that P5091 can both directly kill the cancer and boost the antitumor immunity for cancer growth inhibition (one stone, two birds).
Currently, therapeutic mAbs such as anti-PD-1 for boosting immunity are widely used for the treatment of cancer in clinical trials. Moreover, there is a tendency to combine chemotherapeutic drugs and mAb treatment for cancer patients, as the former always lead to impaired immunity. However, deficiencies of mAbs, such as inadequate pharmacokinetics and tissue accessibility, high economic cost, and side effects including cytokine storms, limit their use to some extent.
Thus, more effective and economic compounds for immunity promotion imperatively need to be explored. Moreover, adaptive resistance mechanisms such as IL-10 release after anti-PD-1 treatment could also cripple its efficacy.24 Our research showed that Tregs can be restrained by USP7 inhibition, resulting in the decrease of IL-10, which suggest that USP7 inhibitors may be a new option for colon cancer combination therapy such as combination with anti-PD-1. Taken together, the study here proposes that USP7 may be a new target for immune regulation in cancer treatment.
Disclosure
The authors report no conflicts of interest in this work.
References
Sliwkowski MX, Mellman I. Antibody therapeutics in cancer. Science. 2013;341(6151):1192–1198. | ||
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | ||
Park JA, Cheung NV. Limitations and opportunities for immune checkpoint inhibitors in pediatric malignancies. Cancer Treat Rev. 2017;58:22–33. | ||
Milling L, Zhang Y, Irvine DJ. Delivering safer immunotherapies for cancer. Adv Drug Deliv Rev. 2017;114:79–101. | ||
deLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res. 2012;18(11):3022–3029. | ||
Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep. 2015;5:15179. | ||
Tao H, Mimura Y, Aoe K, et al. Prognostic potential of FOXP3 expression in non-small cell lung cancer cells combined with tumor-infiltrating regulatory T cells. Lung Cancer. 2012;75(1):95–101. | ||
Salama P, Phillips M, Grieu F, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol. 2009;27(2):186–192. | ||
Shou J, Zhang Z, Lai Y, Chen Z, Huang J. Worse outcome in breast cancer with higher tumor-infiltrating FOXP3+ Tregs: a systematic review and meta-analysis. BMC Cancer. 2016;16:687. | ||
Finotello F, Trajanoski Z. New strategies for cancer immunotherapy: targeting regulatory T cells. Genome Med. 2017;9(1):10. | ||
Crunkhorn S. Cancer immunotherapy: Targeting regulatory T cells. Nat Rev Drug Discov. 2017;16(11):754. | ||
Vishnoi M, Boral D, Liu H, et al. Targeting USP7 identifies a metastasis-competent state within bone marrow-resident melanoma CTCs. Cancer Res. 2018;78(18):5349–5362. | ||
Yeasmin Khusbu F, Chen FZ, Chen HC. Targeting ubiquitin specific protease 7 in cancer: A deubiquitinase with great prospects. Cell Biochem Funct. 2018;36(5):244–254. | ||
Biswas K, Philip S, Yadav A, et al. BRE/BRCC45 regulates CDC25A stability by recruiting USP7 in response to DNA damage. Nat Commun. 2018;9(1):537. | ||
Bhattacharya S, Chakraborty D, Basu M, Ghosh MK. Emerging insights into HAUSP (USP7) in physiology, cancer and other diseases. Signal Transduct Target Ther. 2018;3:17. | ||
Ali A, Raja R, Farooqui SR, Ahmad S, Banerjea AC. USP7 deubiquitinase controls HIV-1 production by stabilizing Tat protein. Biochem J. 2017;474(10):1653–1668. | ||
Wang M, Zhang Y, Wang T, et al. The USP7 Inhibitor P5091 Induces Cell Death in Ovarian Cancers with Different P53 Status. Cell Physiol Biochem. 2017;43(5):1755–1766. | ||
Chauhan D, Tian Z, Nicholson B, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22(3):345–358. | ||
Wang L, Kumar S, Dahiya S, et al. Ubiquitin-specific Protease-7 Inhibition Impairs Tip60-dependent Foxp3+ T-regulatory Cell Function and Promotes Antitumor Immunity. EBioMedicine. 2016;13:99–112. | ||
Rudensky AY. Regulatory T cells and Foxp3. Immunol Rev. 2011;241(1):260–268. | ||
Kim J, Lahl K, Hori S, et al. Cutting edge: depletion of Foxp3+ cells leads to induction of autoimmunity by specific ablation of regulatory T cells in genetically targeted mice. J Immunol. 2009;183(12):7631–7634. | ||
Zhang B, Karrison T, Rowley DA, Schreiber H. IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J Clin Invest. 2008;118(4):1398–1404. | ||
Lee S, Margolin K. Cytokines in cancer immunotherapy. Cancers (Basel). 2011;3(4):3856–3893. | ||
Lamichhane P, Karyampudi L, Shreeder B, et al. IL10 Release upon PD-1 Blockade Sustains Immunosuppression in Ovarian Cancer. Cancer Res. 2017;77(23):6667–6678. | ||
Zlatanou A, Sabbioneda S, Miller ES, et al. USP7 is essential for maintaining Rad18 stability and DNA damage tolerance. Oncogene. 2016;35(8):965–976. | ||
Nicholson B, Suresh Kumar KG. The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem Biophys. 2011;60(1–2):61–68. | ||
Zhang C, Lu J, Zhang QW, et al. USP7 promotes cell proliferation through the stabilization of Ki-67 protein in non-small cell lung cancer cells. Int J Biochem Cell Biol. 2016;79:209–221. | ||
Malapelle U, Morra F, Ilardi G, et al. USP7 inhibitors, downregulating CCDC6, sensitize lung neuroendocrine cancer cells to PARP-inhibitor drugs. Lung Cancer. 2017;107:41–49. | ||
Morra F, Merolla F, Napolitano V, et al. The combined effect of USP7 inhibitors and PARP inhibitors in hormone-sensitive and castration-resistant prostate cancer cells. Oncotarget. 2017;8(19):31815–31829. | ||
Colland F, Formstecher E, Jacq X, et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther. 2009;8(8):2286–2295. | ||
Fan YH, Cheng J, Vasudevan SA, et al. USP7 inhibitor P22077 inhibits neuroblastoma growth via inducing p53-mediated apoptosis. Cell Death Dis. 2013;4:e867. | ||
An T, Gong Y, Li X, et al. USP7 inhibitor P5091 inhibits Wnt signaling and colorectal tumor growth. Biochem Pharmacol. 2017;131:29–39. | ||
Castle JC, Loewer M, Boegel S, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014;15:190. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.