")
Back to Journals » Journal of Pain Research » Volume 12
Pharmacokinetics, safety, and efficacy of tapentadol oral solution for treating moderate to severe pain in pediatric patients
Authors Muse D , Tarau E, Lefeber C , Sohns M, Brett M , Goldberg J, Rosenburg R
Received 4 December 2018
Accepted for publication 27 February 2019
Published 31 May 2019 Volume 2019:12 Pages 1777—1790
DOI https://doi.org/10.2147/JPR.S197039
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Katherine Hanlon
Derek Muse,1 Eva Tarau,2 Claudia Lefeber,3 Melanie Sohns,3 Martin Brett,3 Jutta Goldberg,3 Ronald Rosenburg3
1Jean Brown Research, Salt Lake City, UT, USA; 2Grünenthal USA Inc., Overland Park, KS, USA; 3Grünenthal GmbH, Aachen, Germany
Background: This trial is part of the global pediatric clinical development program investigating the administration of the strong analgesic tapentadol in children and adolescents.
Patients and methods: The single site, open-label phase 2 trial evaluated the pharmacokinetic profile of tapentadol and its major metabolite, tapentadol-O-glucuronide, as well as safety and tolerability and efficacy of a single dose of tapentadol oral solution (1 mg/kg) in patients (2 to <18 years) undergoing dental, ear, nose, or throat surgery. Blood sampling and pain intensity measurements were conducted using age-appropriate schedules and rating scales, respectively. Adverse events were monitored throughout the trial.
Results: Sixty-six patients were treated. They were stratified by age: Group 1 (12 to <18 years), n=21; Group 2 (6 to <12 years), n=28; and Groups 3 (3 to <6 years) and 4 (2 to <3 years), n=17. Serum tapentadol concentrations observed in these pediatric patients were within the range observed in adults after administration of a single tapentadol immediate-release dose (50–100 mg), whereas those of the metabolite tapentadol-O-glucuronide were within the same range or lower than in adults who received comparable single doses of tapentadol. Pain intensity improved over time across all age groups. The most common treatment-emergent adverse events were nausea (24.2%), vomiting (16.7%), dizziness (9.1%), and headache (6.1%).
Conclusion: A single dose of tapentadol oral solution (1 mg/kg) administered to pediatric patients (2 to <18 years) resulted in serum tapentadol concentrations within the targeted range shown to be safe and efficacious in adults. Tapentadol demonstrated good tolerability and safety; within the limitations of the trial design, improvements in postsurgical pain intensity were observed across the age groups. Tapentadol may provide a new treatment option in the management of moderate to severe pediatric pain.
Keywords: analgesics, pediatric pain management, moderate to severe pain, opioid receptors, tapentadol
Introduction
The impact of painful experience on children is significant, and short- and long-term effects can occur, including a lowered pain tolerance for months after a pain-producing event1 and the development of chronic pain.2,3 Current pharmacological treatment options for pain include nonsteroidal anti-inflammatory drugs and other nonopioid analgesic medications, as well as weak and strong opioids, corresponding to World Health Organization Step II and Step III.4 For children, opioids remain an important component of pharmacological analgesia but are often used off-label. The general lack of systematically studied and authorized medications for pediatric patients led to the establishment of regulatory frameworks for the development of pediatric medications by both the European Medicines Agency and the Food and Drug Administration.5,6
The strong centrally acting analgesic tapentadol with the two mechanisms of action µ-opioid receptor agonism and noradrenaline reuptake inhibition7,8 is the first pain medication to undergo a formal pediatric clinical trial program. Tapentadol is approved and marketed in several countries as an extended-/prolonged-release formulation for managing chronic pain and as an immediate-release (IR) formulation for managing acute pain in adults. Phase 3 trials in patients with acute postsurgical pain9,10 and pain from end-stage joint disease11 have demonstrated that tapentadol IR provides similar pain relief to oxycodone IR but with an improved tolerability profile, particularly gastrointestinal tolerability, and might thus provide a useful treatment option in pediatric pain management.
Clinical trials on analgesics are generally associated with numerous challenges, including variability in perceived severity of pain and analgesic responsiveness, and high placebo responses.12 Pediatric analgesic trials are further complicated by the lack of pain models, the need for different pain assessment tools depending on age, and limitations in the ability of parents or guardians to evaluate pain in children and adolescents. Developmental differences in metabolism, excretion, analgesic efficacy, and other factors further confound assessments of efficacy and safety. These challenges are described in detail in a recent review,13 the first article in the thematic series “Tapentadol for moderate to severe acute pain in children and adolescents”. One of the factors to consider for a pediatric trial design is the choice of an appropriate pain model. The treatment of pain after tonsillectomy, a relatively common pediatric surgical procedure associated with significant pain,14 is adequate for pain research in young children (2 to <6 years). Pain related to dental surgery also represents a useful model for analgesic research because the pain is typically of consistent intensity and relatively short duration (24–48 h);15 dental pain models have been extensively used in analgesic trials in adult and pediatric patients.16
The current article presents the results of a pharmacokinetic (PK) trial initiated to derive an appropriate tapentadol dose regimen for different pediatric age groups and is part of the thematic tapentadol pediatric series in this journal. A single dose of tapentadol oral solution (OS) was administered to children and adolescents from 2 to <18 years of age after surgical procedures that are typically associated with moderate to severe postsurgical pain. Evaluations included the PK profile of tapentadol and its major metabolite, tapentadol-O-glucuronide as well as safety and tolerability, and efficacy of the medication.
Patients and methods
This open-label, phase 2 trial was conducted from November 2012 to February 2014 at a single US trial site in accordance with Good Clinical Practice guidelines and the ethical principles laid out in the Declaration of Helsinki. The protocol, amendments, and applicable informed consent forms and assent forms were approved by an institutional review board (Quorum Review, Inc, Seattle, USA). Informed consent was obtained from the parent(s)/legal guardian(s) and/or patient if applicable. The trial is registered with ClinicalTrials.gov (identifier NCT01729728).
The trial included an enrollment period (not to exceed 30 days), the surgical procedure, and the immediate post-recovery period. The treatment and evaluation period lasted for 15 h after dosing with tapentadol OS. A follow-up telephone call took place between day 10 and day 14 following treatment.
Trial population
The trial population comprised patients aged 3 to <18 years who were scheduled for dental surgery or tonsillectomy with or without adenoidectomy and patients aged 2 to <3 years who were scheduled for ear, nose, or throat surgery, including tonsillectomy. Patients were stratified by age generally according to the recommendations of the regulatory authorities. Group 1 included patients aged 12 to <18 years, Group 2 patients aged 6 to <12 years, Group 3 patients aged 3 to <6 years, and Group 4 included patients aged 2 to <3 years.
Patients were eligible to participate with an American Society of Anesthesiologists physical status of I or II and had to be alert, oriented, able to follow commands, able to complete postsurgically required procedures, and able to tolerate oral medication or fluids. To be included in the trial, patients aged 6 to <18 years were required to have a postsurgical pain intensity score of ≥4 on the McGrath Color Analog Scale (CAS)17 or to have a postsurgical pain level requiring opioid treatment (based on the investigator’s clinical judgment relying on usual standard of care). For patients aged 2 to <6 years only the latter assessment was used. Patients were excluded from the trial if they had suicidal tendencies as per the investigator’s impression based on the Columbia-Suicide Severity Rating Scale (C-SSRS;18 performed only in patients ≥12 years of age if the parent or legal guardian provided consent), a history of seizure disorder or brain injury, clinically relevant respiratory disease or abnormal pulmonary function, a history of alcohol or drug abuse, or clinically relevant abnormal findings for laboratory, electrocardiogram (ECG), or vital sign assessments.
Prior to administration of tapentadol OS, the use of premedication, intraoperative medication, and opioid analgesics during anesthesia was permitted according to the usual standard of care. During surgery, very short-acting benzodiazepines (ie, half-life [t½] ≤4 h) and local anesthetics were permitted. After the end of anesthesia, nonopioid analgesics were allowed up to 30 mins before dosing with tapentadol OS. Patients were encouraged, but not required, to wait ≥1 h after the intake of tapentadol oral solution before receiving further supplemental nonopioid analgesic medication. Morphine or another opioid could be administered according to medical judgment and usual standard of care if the patient continued to have intolerable pain (based on patient perception) for ≥2 h after the administration of tapentadol OS, despite having received a nonopioid analgesic. Monoamine oxidase inhibitors, strong enzyme-inducing drugs, dextromethorphan, oral neuroleptics, serotonin/noradrenaline reuptake inhibitors, tricyclic antidepressants, anticonvulsants, and antiparkinsonian drugs were prohibited for prespecified time frames prior to and after administration of tapentadol OS.
Dose selection
The OS formulation of tapentadol is available in the two concentrations 20 mg/mL and 4 mg/mL.
Three key assumptions were made when selecting a dose for this pediatric trial:
- in children 2 years of age and older, the underlying pain mechanisms are similar to those in adults
- the metabolism and distribution of tapentadol in pediatric patients older than 2 years of age is similar to that in adults after adjusting for body weight
- the exposure-effect relationship of tapentadol in pediatric patients is similar to that in adults.
Nonlinear mixed-effects modeling (using NONMEM®; Icon Development Solutions, Ellicott City, Maryland, USA) was performed to develop a population PK model with allometric scaling using pooled adult data from phase 1, 2, and 3 trials in the tapentadol acute pain program. Simulations were performed to identify tapentadol doses that would produce total exposures (ie, area under the serum concentration-time curve [AUC]) in pediatric patients that are similar to those reported in adults. The approved adult therapeutic dose range (50–100 mg) was used for comparison.19 Based on the simulation results, the tapentadol dose was set to 1 mg/kg, an average of the predicted dose range (0.7–1.4 mg/kg), which was expected to produce exposures in children that were within the range observed following administration of 50–100 mg of tapentadol in adults. Thus, this dose was anticipated to be efficacious in the pediatric population without compromising the safety of the trial patients.
Assessments
Pharmacokinetics
Blood samples for PK assessments were taken according to age-appropriate schedules (Table 1). The volume of venous blood samples taken was 1 mL per sample in Groups 1–3 and 0.5 mL per sample in Group 4. More frequent sampling was conducted in the adolescent patients (12 to <18 years) to enable noncompartmental PK parameters to be estimated for this group. The sampling schemes for the younger groups were designed to limit the number of blood samples as much as possible in this vulnerable population. Simulations using the body weight allometrically scaled adult population PK model were conducted in order to ensure that the primary population PK parameters in the lower age groups could be sufficiently well characterized with the selected sampling schemes.
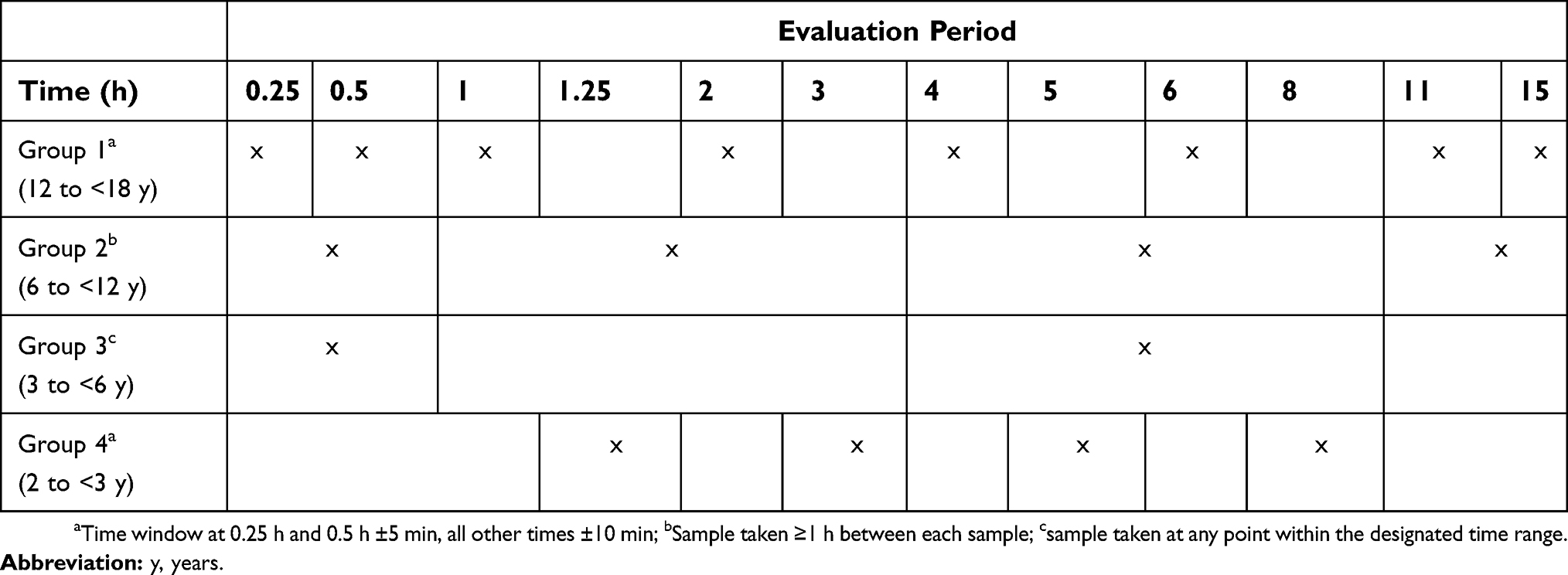 | Table 1 Blood sampling schedules for determination of serum tapentadol and tapentadol-O-glucuronide concentrations |
Serum concentrations of tapentadol and its metabolite, tapentadol-O-glucuronide, were determined using a validated liquid chromatography–tandem mass spectrometry (LC-MS/MS) method. The samples were received in polypropylene vials, deep frozen on dry ice, and properly labeled. All samples were stored in a freezer at ≤−15 °C until analysis. For high-performance liquid chromatography, a gradient with solvent A (0.5% acetic acid) and solvent B (methanol/0.5% acetic acid) was used.
Noncompartmental PK parameters (maximum serum concentration [Cmax], time of Cmax [tmax], terminal phase t1/2, and AUC from time 0 to the time of the last quantifiable concentration [AUC0-t]) for tapentadol and tapentadol-O-glucuronide were estimated for patients aged 12 to <18 years (using the software MODUNA version 2.0.1, a program developed and validated by Grünenthal GmbH). The actual blood sampling times were used for generating individual PK parameters. For patients aged 2 to <12 years, analyses of the PK characteristics are based on sparse sampling requiring modeling and simulation. A population PK analysis was performed using tapentadol and tapentadol-O-glucuronide data from all 4 groups; this analysis is not covered in this publication. A separate manuscript reporting on the population PK modelling and its outcome is currently in preparation for the thematic tapentadol series.
Efficacy
Pain intensity was evaluated prior to dosing and at approx. 15 min, 30 min, 1, 2, 4, 6, 11, and 15 h after dosing, and at discharge. Pain intensity was evaluated using age-appropriate rating scales. Patients in Group 1 used the self-reporting scales 100-mm visual analog scale (VAS; from 0=no pain to 100=worst imaginable pain) and McGrath CAS (from 0=no pain to 10=worst imaginable pain). In Group 2, self-reported pain intensity was evaluated using the McGrath CAS followed by the 6-point Faces Pain Scale-revised (FPS-R).20,21 The FPS-R shows six faces with scores of 0, 2, 4, 6, 8, 10 where 0=no pain and 10=very much pain. In Group 3, the observational face, legs, activity, cry, and consolability (FLACC) assessment (scored from 0 to 10 with 0=no pain)22 was used, followed by the self-reported FPS-R. In Group 4, only the FLACC assessment was used to evaluate pain intensity. The dose, time, and route of each intake of supplemental analgesic medication were also recorded during the treatment phase as a measure of analgesic efficacy.
Safety/tolerability
Safety data included hematology, clinical chemistry, and urinalysis values; 12-lead ECG data; respiratory rate, systolic and diastolic blood pressure, heart rate, and oxygen saturation by pulse oximetry; and C-SSRS. Pregnancy tests were carried out predose (for exclusion) and at discharge. Tolerability was assessed through adverse event (AE) monitoring.
Statistical analyses
The sample size was specified to limit the exposure in pediatric patients while providing sufficient data to explore a population PK model. To this end, 56 evaluable patients were planned. For Group 1 (aged 12 to <18 years), 16 to 19 patients were planned for enrollment, and for Group 2 (aged 6 to <12 years), 21 to 24 patients were planned for enrollment (total of 40 patients across both groups). For Group 3 (aged 3 to <6 years), 12 patients (including ≥4 aged 3 years) were planned for enrollment, and for Group 4 (aged 2 to <3 years), 4 patients were planned for enrollment. To ensure patient safety, recruitment followed a staggered approach by age, starting with recruitment of adolescents and older children in Groups 1 and 2, followed by recruitment of younger patients in Group 3 and finally the youngest patients in Group 4.
To ensure that sufficient data points were available to adequately characterize noncompartmental PK parameters in the adolescent group and primary population PK parameters in the younger patients, patients with an insufficient number of blood samples were considered non-evaluable for PK analyses and were replaced. Non-evaluable patients were defined as those in Group 1 with <6 blood samples that could be analyzed for serum concentrations of tapentadol and tapentadol-O-glucuronide; those in Group 2 with <4 such blood samples, those in Group 3 with <2 such blood samples, and those in Group 4 with <3 such blood samples. Patients who vomited within 3 h of administration of tapentadol OS and those who did not take the complete amount of tapentadol OS were also considered to be non-evaluable and were replaced. Replacement patients received the same sequence of treatments and completed the same procedures and assessments as the initially entered patients.
The data obtained from all 4 groups were combined for statistical analyses when appropriate. Groups 3 and 4 were combined in one group for a larger sample size, except for analysis of the FPS-R (only used by Group 3) and presentation of serum concentration-time data (due to differences in the sampling schemes). Descriptive statistics were used to summarize the standard descriptive PK parameters for Group 1 and serum concentrations of tapentadol and tapentadol-O-glucuronide per assessment point (all groups).
All patients who received tapentadol OS were included in efficacy and safety/tolerability analyses. Descriptive statistics were used to summarize pain intensity scores at predefined time points. The sum of pain intensity differences over 4 h (SPID4) was calculated as the weighted sum of the pain intensity differences (PID) within ≤4 h after dosing:
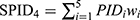
with 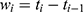 and
and 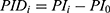 ,
, 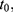 …
…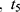 pre-defined assessment time points (pre-dose, 0.25, 0.5, 1, 2 and 4 hrs after dosing) and
pre-defined assessment time points (pre-dose, 0.25, 0.5, 1, 2 and 4 hrs after dosing) and 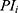 pain intensity score measured at time point
pain intensity score measured at time point 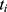 .
.
The intake of supplemental analgesic medication between the first intake of trial drug and the start of the discharge visit was analyzed descriptively, including an analysis of the time to first intake of supplemental analgesic (Kaplan-Meier analysis). Patients who did not take any supplemental analgesics until the discharge visit were censored for the Kaplan-Meier analysis at the time point of discharge. For all AEs, the incidence, type, intensity, treatment, onset, duration, relationship to trial drug, and outcome were recorded. Treatment-emergent AEs (TEAEs) were defined as AEs that started after the first intake of the trial drug or within its therapeutic reach (48 h after intake). Pre-treatment AEs that worsened after trial drug administration were recorded as a new AE. Adverse events were encoded using the Medical Dictionary for Regulatory Activities (MedDRA), version 16.1.
Results
Patients
Of the 86 patients who enrolled in the trial, 66 were allocated to and received treatment (Group 1: n=21; Group 2: n=28; Groups 3+4: n=17; Figure 1). Of the 20 patients who were enrolled but not allocated to trial treatment, 16 were enrollment failures (did not fulfill ≥1 of the inclusion criteria or fulfilled ≥1 of the exclusion criteria), one patient did not enroll due to an AE, and the remaining 3 did not enroll for other (unspecified) reasons. A total of 12.1% (8/66) of treated patients were discontinued from the trial. Six of these patients met the protocol-defined discontinuation criterion of vomiting within the first 3 h after tapentadol administration, and the other 2 patients were discontinued due to protocol deviations (inadequate blood samples).
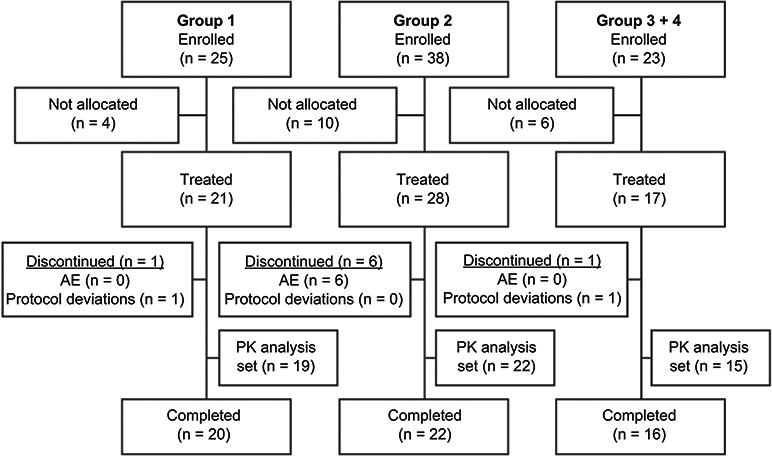 | Figure 1 Patient flow chart. Notes: Group 1, 12 to <18 years, group 2, 6 to <12 years, group 3, 3 to <6 years, and group 4, 2 to <3 years.Abbreviations: AE, adverse event; PK, pharmacokinetic. |
Fifteen patients in Group 1 completed the C-SSRS. Only one of those, a 17-year old female provided information related to lifetime nonspecific active suicidal thoughts at trial enrollment and reported the ability to easily control those thoughts. This patient completed the trial without any report of psychiatric AEs. No other patient displayed suicidal ideation or behavior.
Patient baseline and demographic characteristics are summarized in Table 2. For the overall trial population, the mean ± standard deviation (SD) age was 9.3±4.9 years, and 51.5% (34/66) of patients were female. The majority of patients in Group 1 (16/21 [76.2%]) had dental surgery, the other 5 patients underwent tonsillectomy (23.8%). All patients in the younger age groups (<12 years) had a tonsillectomy. Overall, 68.2% (45/66) of patients had a documented prior disease and 90.9% (60/66) of patients had a documented concomitant disease, some of which were related to the reasons for surgery (eg, tonsillitis and tonsillar hypertrophy). Prior medication use was reported for all patients, and concomitant medication use was reported for 80.3% (53/66) of patients. Most prior/concomitant medications (>80%) were used for the conditions for which patients underwent surgery or for control of postsurgical pain.
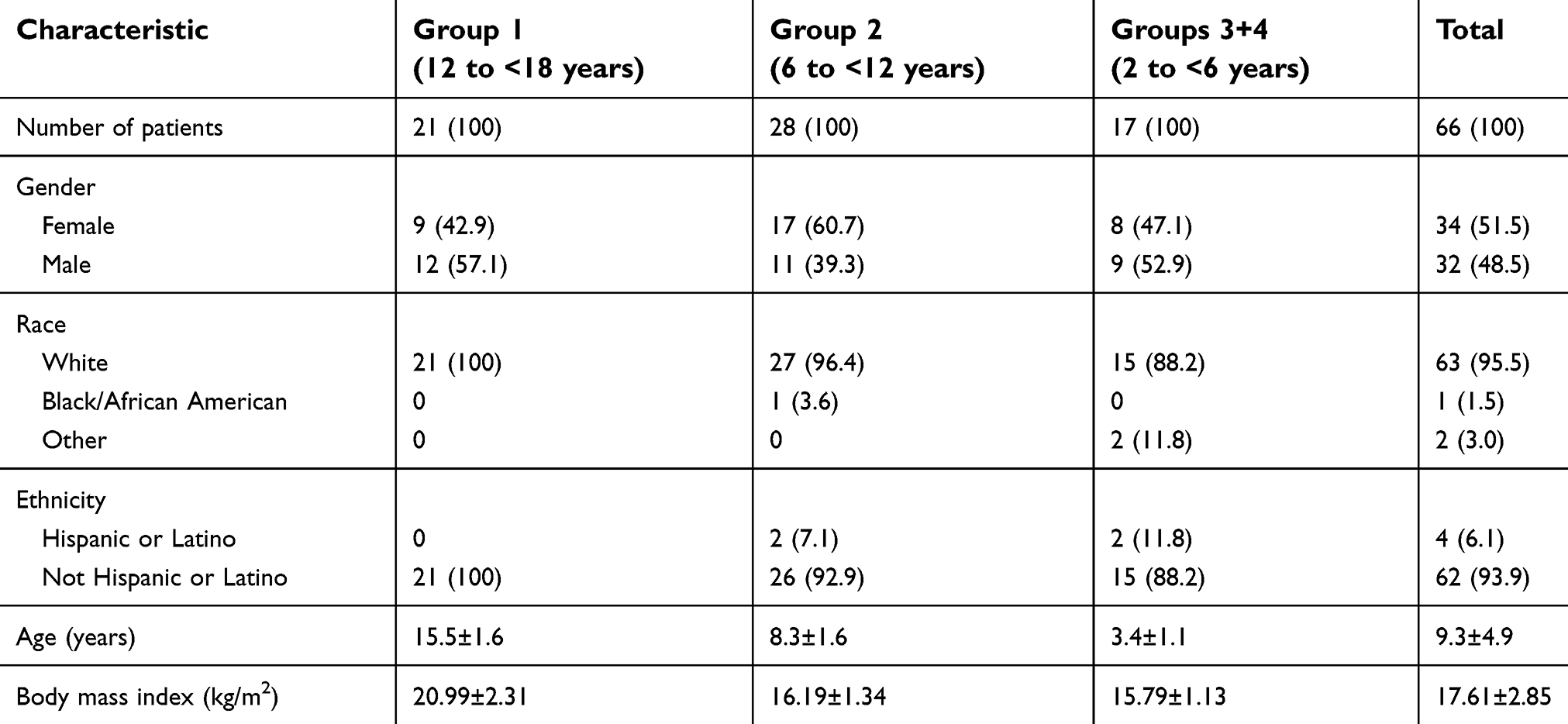 | Table 2 Patient baseline demographic characteristics |
Pharmacokinetics
Mean serum concentrations of tapentadol and tapentadol-O-glucuronide across all groups over time are summarized in Table 3. A comparison of the mean tapentadol concentration for adolescents in Group 1 (12 to <18 years) to that for adults is shown in Figure 2. The adult data were obtained in a single dose trial using 100 mg tapentadol OS (data on file). Since dose proportional increases in tapentadol exposure (Cmax and AUC) have been observed across the therapeutic dose range of 50–100 mg,19 the tapentadol serum concentrations observed for the adults were scaled by a factor of 0.8 (80/100) to derive the serum concentration profile after a single dose of 80 mg which approximates to the dose of 1 mg/kg used in the current trial in a pediatric population. This comparison clearly shows that similar mean profiles were obtained for the adolescent age group and an adult population. Individual serum concentrations for all children in all age groups in the current trial are shown overlaid in a log-linear plot in Figure 3. As shown in this plot, there is consistency in serum concentrations across an age range from 2 to <18 years, with no indication of a trend with increasing or decreasing age. Noncompartmental PK parameters (for Group 1) for tapentadol and tapentadol-O-glucuronide are summarized in Table 4.
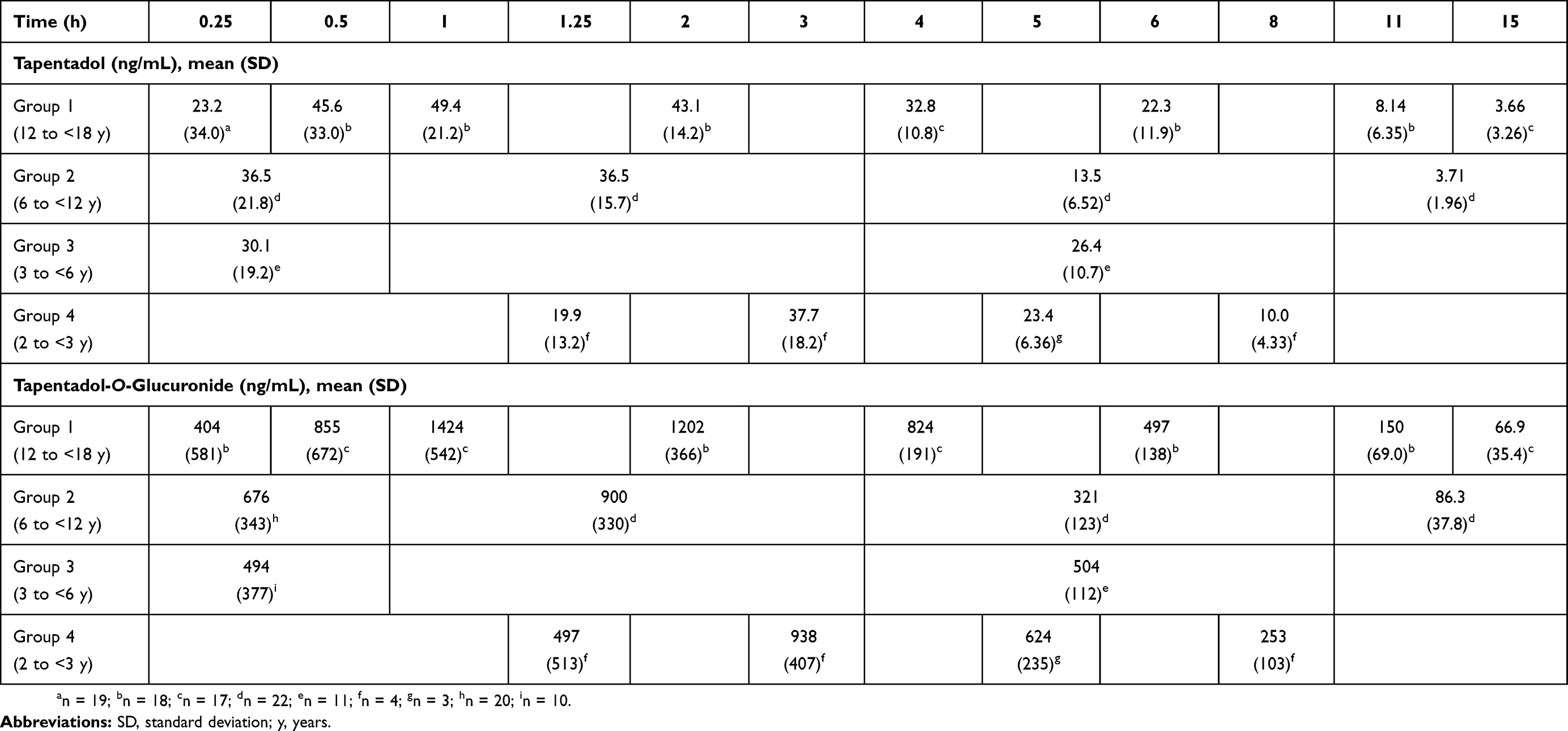 | Table 3 Mean (SD) serum concentrations of tapentadol and tapentadol-O-glucuronide for all groups |
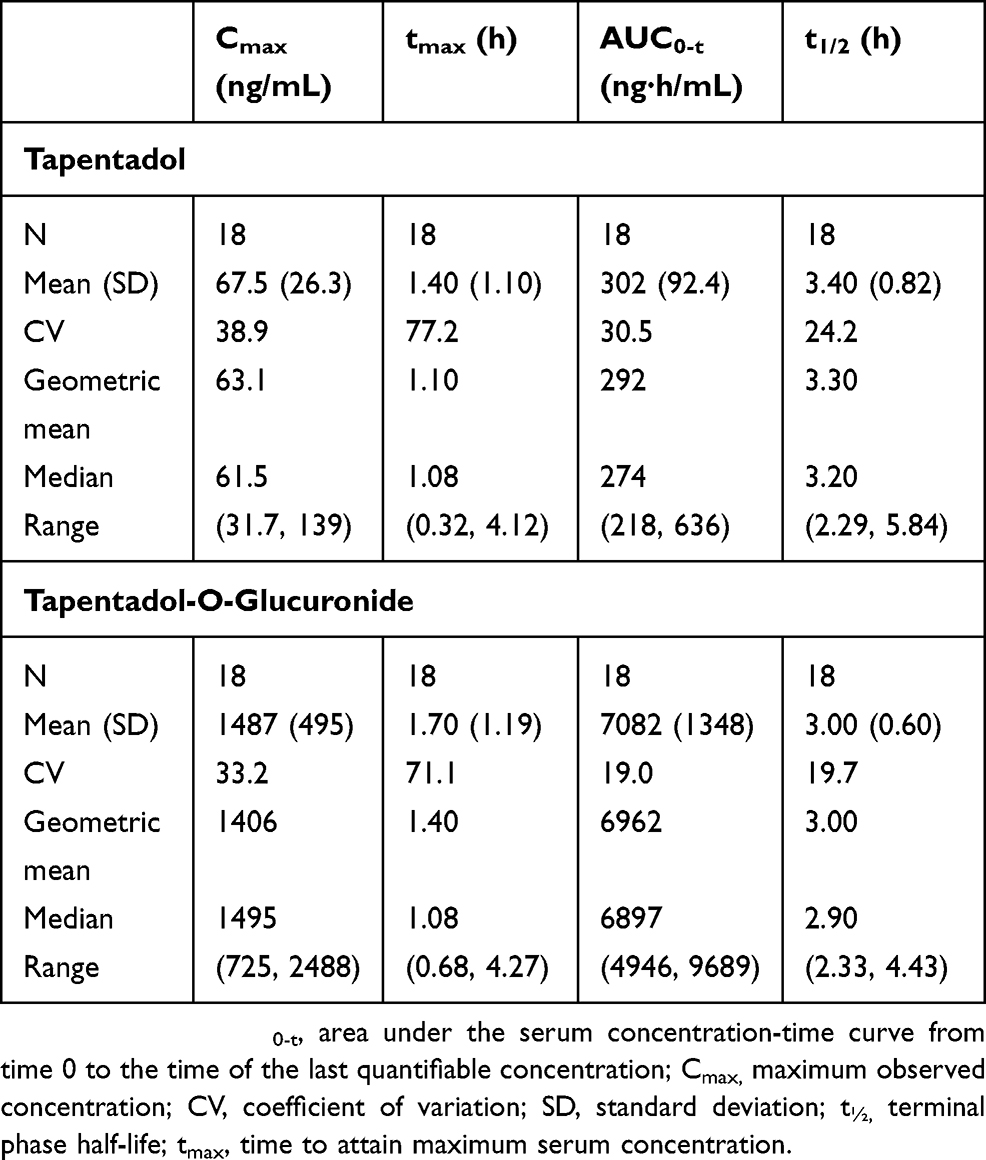 | Table 4 Noncompartmental pharmacokinetic parameters (Group 1; 12 to <18 years of age) |
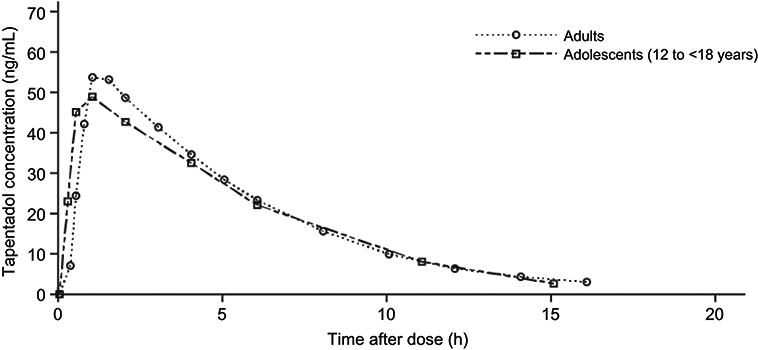 | Figure 2 Mean serum tapentadol concentration in adolescents in Group 1 (12 to <18 years of age) compared with adults who participated in an earlier trial with tapentadol oral solution.Note: Adult data have been dose-corrected to a single dose of tapentadol 80 mg, which approximates the dose of 1 mg/kg used in the current pediatric trial. |
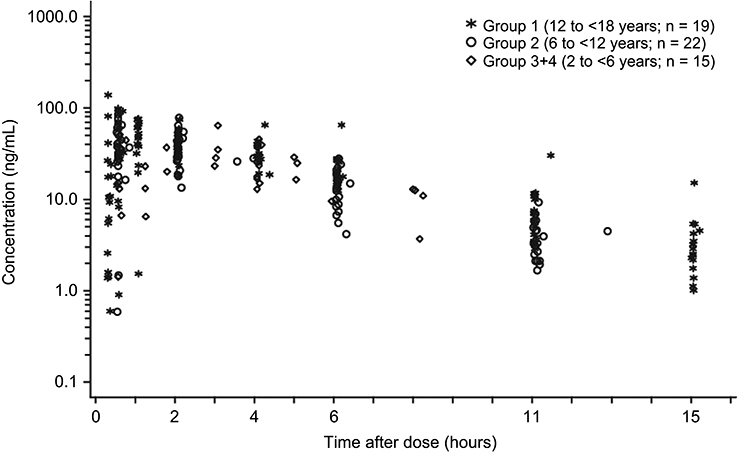 | Figure 3 Individual serum concentrations of tapentadol in pediatric patients (2 to <18 years of age). |
Efficacy
For Group 1, the mean (±SD) baseline pain intensity score on the VAS was 71.5±13.3 and decreased to 34.0±24.6 at 2 h after dosing; the mean pain intensity score then ranged from 33.4 to 43.5 through to discharge (Figure 4A). Mean pain intensity (on the CAS) over time in all patients in Group 1 and Group 2 are shown in Figure 4B. Pooling of all patients undergoing tonsillectomy in Groups 1 and 2 (n=33) showed progressive improvement in pain intensity over the first 2 h from a mean baseline score of 4.82±1.54. In patients undergoing dental surgery (n=16; all Group 1 patients), the mean baseline score of 6.95±1.5 progressively improved over time until discharge. For Groups 2 and 3, respectively, mean pain intensity scores on the FPS-R were 4.8±1.6 and 6.8±3.2 at baseline, decreasing to 3.1±2.4 and 2.7±3.1 at discharge (Figure 4C). For Groups 3 and 4, respectively, mean pain intensity scores on the FLACC were 3.8±2.2 and 5.2±1.9 at baseline and decreased to 2.2±1.9 and 1.0±1.4 at discharge (Figure 4D). The SPID was assessed as a summary measurement for pain intensity over the observation period. Pain intensity differences over 4 h from baseline and weighted for the time between the observations (SPID4) are shown for each assessment across age groups in Table 5.
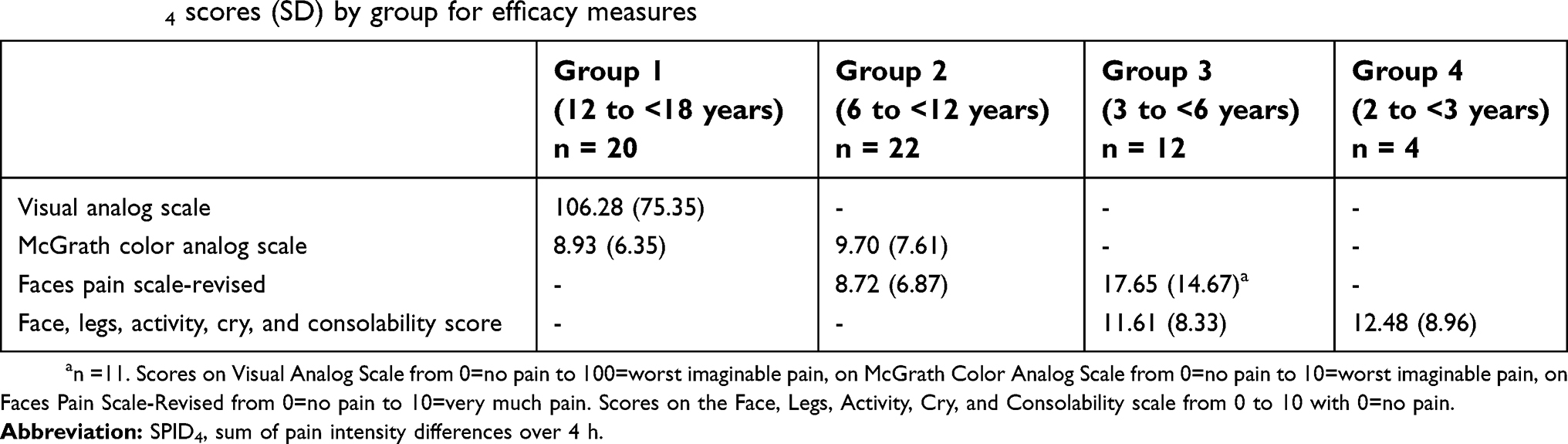 | Table 5 SPID4 scores (SD) by group for efficacy measures |
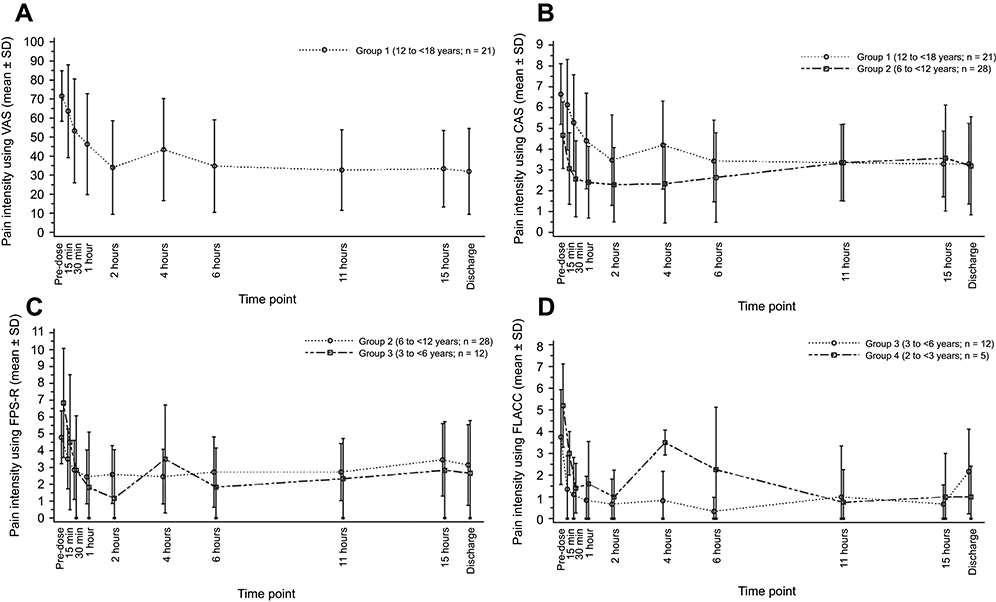 | Figure 4 Mean pain intensity over time using the (A) visual analog scale, (B) color analog scale, (C) faces pain scale-revised, and (D) face, legs, activity, cry, and consolability assessments.Abbreviations: VAS, visual analog scale; CAS, color analog scale; FPS-R, faces pain scale-revised; FLACC, face, legs, activity, cry, and consolability score. |
A total of 68.2% (45/66) of patients received supplemental analgesics during the 15-h postdose evaluation period; all these patients had received ≥1 nonopioid medication (ibuprofen in 44 patients, paracetamol in one patient), and 14 had also received ≥1 opioid medication (hydrocodone [2 patients], hydrocodone/paracetamol [12 patients]; Table 6). The median time to the first intake of any supplemental analgesic medication after tapentadol administration was 6.3 h (Figure 5).
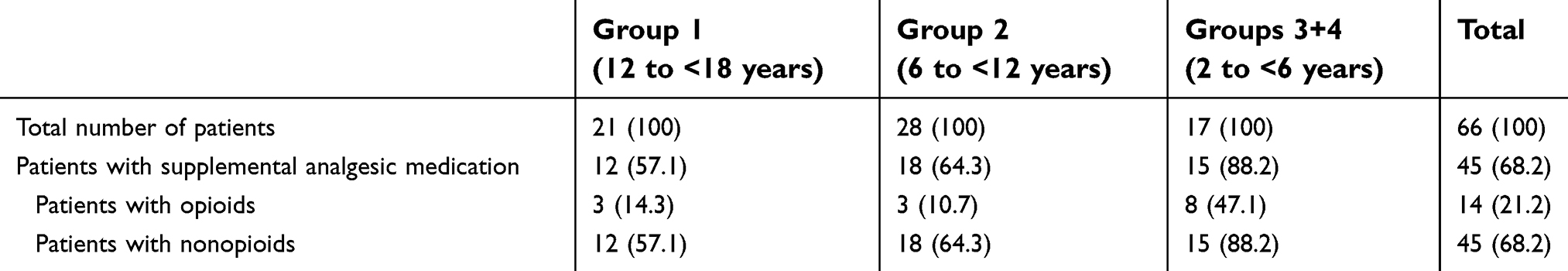 | Table 6 Patients with supplemental analgesic medication |
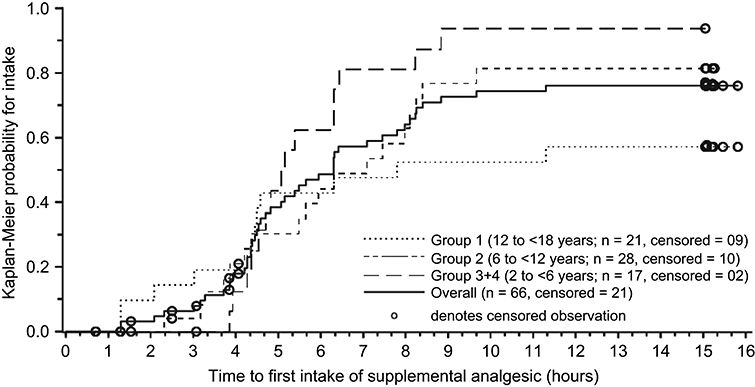 | Figure 5 Kaplan-Meier curves for time to first intake of supplemental analgesic medication by age group.Note: Censored patients are patients who did not take any supplemental analgesics until the discharge visit (censoring was at the time point of the discharge visit). |
Safety and tolerability
TEAEs were reported for a total of 57.6% (38/66) of patients (Table 7). The most common TEAEs (incidence ≥5%) were nausea (24.2% [16/66]), vomiting (16.7% [11/66]), dizziness (9.1% [6/66]), and headache (6.1% [4/66]). In the investigators’ opinion, 31.8% (21/66) of TEAEs were at least possibly related to the trial medication, 33.3% (7/21) in Group 1, 42.9% (12/28) in Group 2, and 11.8% (2/17) in combined Groups 3+4. All TEAEs were classified as either mild or moderate in intensity, and there were no deaths or serious TEAEs. One patient experienced a serious AE of postprocedural (tonsillectomy) hemorrhage; however, this AE occurred 6 days after administration of the trial drug (after therapeutic reach). This event was considered as not related to administration of tapentadol oral solution and resolved after cauterization (postsurgical bleeding is a known complication of tonsillectomy, and there was no plausible temporal relationship between tapentadol intake and the bleeding event). Six patients (all in Group 2) were discontinued early due to vomiting within 3 h after administration of trial drug, which was a prespecified discontinuation criterion for the trial.
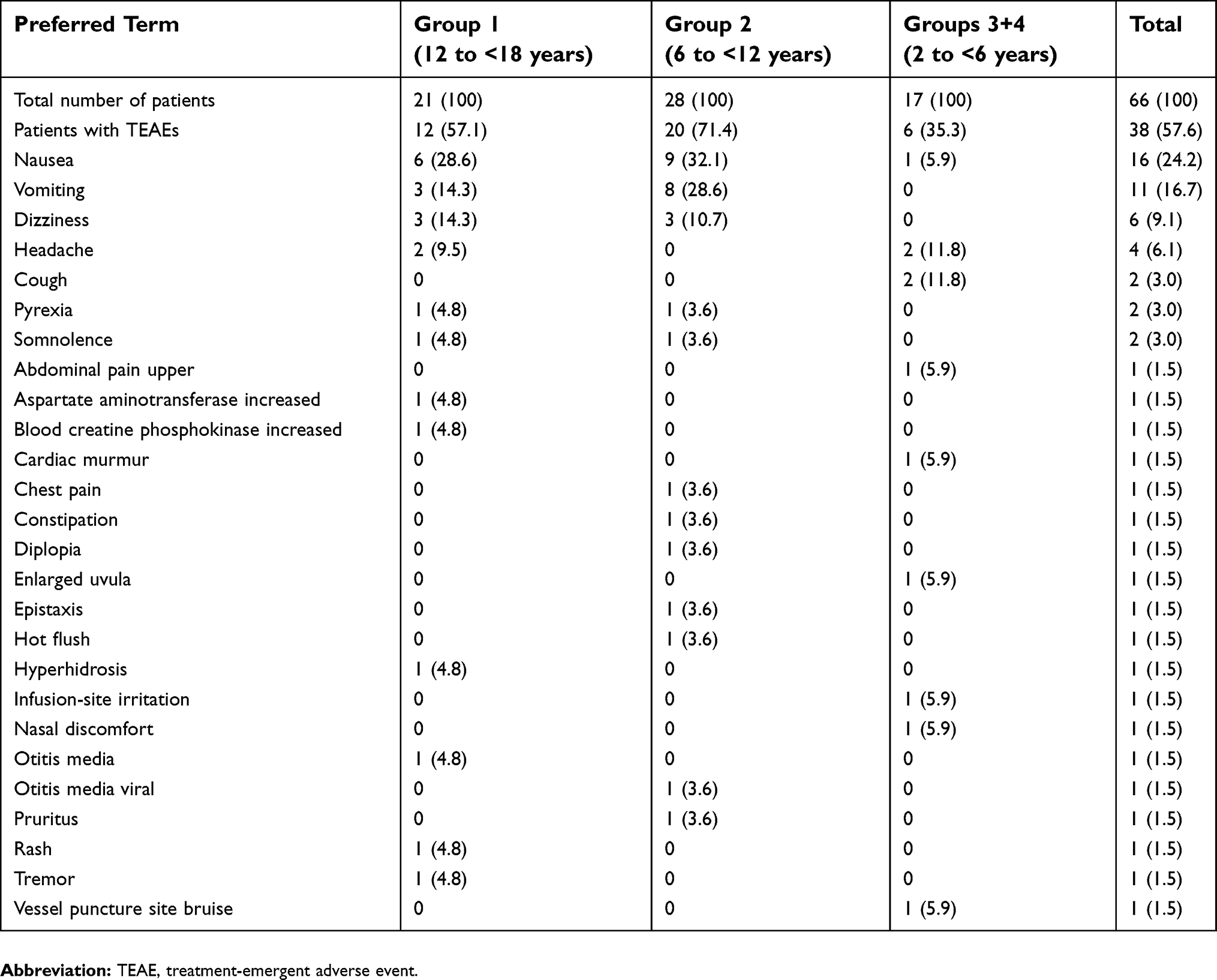 | Table 7 Treatment-emergent adverse events by preferred term |
No clinical laboratory test changes representing a safety concern were observed in this trial. Two laboratory results were reported as TEAEs, aspartate aminotransferase increase and creatine kinase increase, both of which occurred in a 17-year old male patient with influenza B in the preceding days. Three abnormal physical findings were reported as TEAEs (cardiac murmur, enlarged uvula, and skin irritation at an intravenous infusion site); all were of mild intensity and considered unlikely or not related to trial treatment. No abnormal vital signs or 12-lead ECG findings were reported as TEAEs. No abnormally low oxygen saturation was reported.
Discussion
Results from this open-label phase 2 trial indicate that a single dose of tapentadol OS (1 mg/kg) administered to pediatric patients (2 to <18 years of age) resulted in serum tapentadol and tapentadol-O-glucuronide concentrations that were generally within the range or, in the case of the metabolite, lower than concentrations observed in adults after administration of a single oral dose of tapentadol IR (50–100 mg).23–25 These findings confirm the results obtained from the first tapentadol single dose evaluation in a different patient population aged 6 to <18 years26 and thereby further extend the PK database for children in this age group. In addition, the first data in a younger patient population (2 to <6 years) were obtained in the current investigation.
A cross-study comparison was performed using data for adult patients (n=631) from single dose phase 1 biopharmaceutical and clinical pharmacology trials across all tapentadol IR doses (data on file). When the dose was adjusted to 80 mg (which approximates to the 1-mg/kg dosing regimen used in the current pediatric trial), mean maximum serum concentration (Cmax) for tapentadol in adult patients was 72.1±29 ng/mL, which was very similar to that observed in the adolescent group (12 to <18 years; 67.5±26.3 ng/mL) who participated in the current pediatric trial. The mean Cmax for tapentadol-O-glucuronide at an 80-mg dose ranged from 2619 to 3097 ng/mL across adult trials23–25 as compared with 1487 ng/mL in the adolescent group of the current pediatric trial. One explanation for the generally higher exposure to tapentadol-O-glucuronide in adults is attributed to the polar characteristics of the glucuronide metabolite and the changing body composition with age – for younger subjects, the percentage of body water increases, presumably resulting in an increased volume of distribution for the polar glucuronide and consequent lower systemic plasma concentrations. Since the tapentadol-O-glucuronide metabolite does not contribute to the analgesic effects of tapentadol,8 the lower concentrations of that metabolite observed in the current pediatric population have no therapeutic consequences.
This single dose trial is part of the global pediatric clinical development program of tapentadol; the PK data obtained here and from the first tapentadol evaluation26 were subsequently used in population PK analyses and a physiologically based PK model, which will be published separately. These analyses have provided the basis for the dose selection for subsequent trials designed to evaluate the PK, safety, and efficacy of both the oral solution and intravenous formulation of tapentadol in pediatric patients.
The trial was primarily designed to evaluate the PK profile of tapentadol in children and adolescents but also provided some efficacy and tolerability/safety data. Administration of a single dose of tapentadol OS improved moderate to severe pain intensity after surgery across the age groups. It should, however, be noted that 68% of the patients received supplemental analgesic medications which might have biased the findings. The most commonly used nonopioid analgesic in this trial was ibuprofen. Hydrocodone alone or in combination with paracetamol were occasionally administered. Owing to its metabolic degradation pathway and lack of active metabolites27 tapentadol has a low drug-drug interaction potential.28 This has been demonstrated in a series of phase 1 drug-drug interaction trials.23–25 The possibility of a PK interaction with tapentadol in this trial was therefore minimal. Tapentadol OS was generally well tolerated in this small patient population with a safety profile that was consistent with the known safety profile for tapentadol as observed in trials in adult patients.9–11,29
Two pain models were used in this trial to provide an age appropriate trial design for different age groups. Tonsillectomy is a common, painful pediatric surgical procedure14 that can be used for clinical pain research in young children from 2 to less than 6 years of age. Dental surgery generally results in consistent levels of pain lasting from 24 to 48 h,15 and can serve as a model for analgesic research in adult and pediatric populations.16 Across all pain scales in the current study, relatively high baseline pain intensity was observed. The mean baseline pain intensity observed on the FLACC scale for patients 2 to <6 years of age in the current trial (3.8–5.2) was consistent or slightly higher than that observed in a previous trial (~3) in patients 3–12 years of age undergoing tonsillectomy.14 Comparisons of pain scores between the current trial and other trials are limited by the range of surgeries permitted in the current trial and differences in the pain scales used to assess postsurgical pain.
Due to the limitations of the trial design, including the small number of patients, single dose administration, lack of comparator or control, allowance of concomitant analgesics, differing sample sizes among age groups, and differing surgeries among age groups, comparisons of age-specific safety and efficacy data should be interpreted with caution.
Conclusion
Tapentadol oral solution (1 mg/kg) has a similar PK profile in children aged 2 to <18 years and in adult patients. The medication demonstrated good tolerability and safety; within the limitations of the trial design, improvements in postsurgical pain intensity were observed across the age groups. Tapentadol may provide a new treatment option in the management of moderate to severe pediatric pain.
Abbreviations
AE, adverse event; AUC, area under the serum concentration-time curve; CAS, color analog scale; Cmax, maximum serum concentration; C-SSRS, Columbia-suicide severity rating scale; ECG, electrocardiogram; FLACC, face, legs, activity, cry, and consolability assessment; FPS-R, 6-point faces pain scale-revised; IR, immediate release; OS, oral solution; PID, pain intensity difference; PK, pharmacokinetics; SPID4, sum of pain intensity differences over 4 h; TEAE, treatment-emergent adverse event; VAS, visual analogue scale.
Data sharing statement
The authors will share upon request the final clinical trial report. They will also consider on a case-by-case basis requests for access to other documents and/or data including individual patient data. There is no limitation regarding document/data availability.
Acknowledgments
The authors thank all patients, parents/legal guardians, investigators, and the trial site team involved in this investigation. The trial was funded by Grünenthal GmbH. Editorial support for the writing of this manuscript was provided by Megan Knagge, PhD, of MedErgy, and was funded by Grünenthal GmbH. The authors retained full editorial control over the content of the manuscript.
Disclosure
E. Tarau is an employee of Grünenthal USA Inc., C. Lefeber, M. Sohns, and J. Goldberg are employees of Grünenthal GmbH. M. Brett and R. Rosenburg were employees of Grünenthal GmbH at the time the trial was conducted. R Rosenburg was the international clinical lead for the pediatric project at Grünenthal until Sep 2017. The authors report no other conflicts of interest in this work.
References
1. Taddio A, Katz J, Ilersich AL, Koren G. Effect of neonatal circumcision on pain response during subsequent routine vaccination. Lancet. 1997;346:599–603. doi:10.1016/S0140-6736(96)10316-0
2. Macrae WA. Chronic post-surgical pain: 10 years on. Br J Anaesth. 2008;101(1):77–86. doi:10.1093/bja/aen099
3. Williams G, Howard RF, Liossi C. Persistent postsurgical pain in children and young people: prediction, prevention, and management. Pain Rep. 2017;2(5):e616. doi:10.1097/PR9.0000000000000616
4.
5. Regulation (EC) No 1901/2006 of the European parliament and of the council of 12 December 2006 on medicinal products for paediatric use, OJ L 378. December 27, 2006. Available from:
6.
7. Tzschentke TM, Christoph T, Kögel B, et al. (-)-(1R,2R)-3-(3-dimethylamino-1-ethyl-2-methyl-propyl)-phenol hydrochloride (tapentadol HCl): a novel mu–opioid receptor agonist/norepinephrine reuptake inhibitor with broad-spectrum analgesic properties. J Pharmacol Exp Ther. 2007;323:265–276. doi:10.1124/jpet.107.126052
8. Tzschentke TM, De Vry J, Terlinden R, et al. Tapentadol hydrochloride. Analgesic, mu-opioid receptor agonist, noradrenaline reuptake inhibitor. Drugs Future. 2006;31:1053–1061.
9. Daniels S, Casson E, Stegmann JU, et al. A randomized, double-blind, placebo-controlled phase 3 study of the relative efficacy and tolerability of tapentadol IR and oxycodone IR for acute pain. Curr Med Res Opin. 2009;25:1551–1561. doi:10.1185/03007990902952825
10. Daniels SE, Upmalis D, Okamoto A, Lange C, Haeussler J. A randomized, double-blind, phase III study comparing multiple doses of tapentadol IR, oxycodone IR, and placebo for postoperative (bunionectomy) pain. Curr Med Res Opin. 2009;25:765–776. doi:10.1185/03007990902728183
11. Hartrick C, Van Hove I, Stegmann J-U, Oh C, Upmalis D. Efficacy and tolerability of tapentadol immediate release and oxycodone HCl immediate release in patients awaiting primary joint replacement surgery for end-stage joint disease: a 10-day, phase III, randomized, double-blind, active- and placebo-controlled study. Clin Ther. 2009;31:260–271. doi:10.1016/j.clinthera.2009.02.009
12. Berde C, Walco G, Krane E, et al. Pediatric analgesic clinical trial designs, measures, and extrapolation: report of an FDA scientific workshop. Pediatrics. 2012;129:354–364. doi:10.1542/peds.2010-3591
13. Eerdekens M, Beuter C, Lefeber C, Van den Anker J. The challenge of developing pain medications for children: therapeutic needs and future perspectives. J Pain Res. 2019;12:1649–1664. doi 10.2147/JPR.S195788
14. Hosseini Jahromi SA, Hosseini Valami SM, Hatamian S. Comparison between effect of lidocaine, morphine and ketamine spray on post-tonsillectomy pain in children. Anesth Pain Med. 2012;2:17–21. doi:10.5812/aapm.4092
15. Urquhart E. Analgesic agents and strategies in the dental pain model. J Dent. 1994;22:336–341.
16. Hyllested M, Jones S, Pedersen JL, Kehlet H. Comparative effect of paracetamol, NSAIDs or their combination in postoperative pain management: a qualitative review. Br J Anaesth. 2002;88:199–214.
17. McGrath PA, Seifert CE, Speechley KN, Booth JC, Stitt L, Gibson MC. A new analogue scale for assessing children’s pain: an initial validation study. Pain. 1996;64:435–443.
18. Posner K, Brent D, Lucas C, et al. Columbia-Suicide Severity Rating Scale (C-SSRS). Available from:
19.
20. Hicks CL, von Baeyer CL, Spafford PA, van Korlaar I, Goodenough B. The faces pain scale-revised: toward a common metric in pediatric pain measurement. Pain. 2001;93:173–183.
21. Miró J, Huguet A. Evaluation of reliability, validity, and preference for a pediatric pain intensity scale: the Catalan version of the faces pain scale–revised. Pain. 2004;111:59–64. doi:10.1016/j.pain.2004.05.023
22. Merkel SI, Voepel-Lewis T, Shayevitz JR, Malviya S. The FLACC: a behavioral scale for scoring postoperative pain in young children. Pediatr Nurs. 1997;23:293–297.
23. Mangold B, Oh C, Jaeger D, Terlinden R, Upmalis D. The pharmacokinetics of tapentadol are not affected by omeprazole: results of a 2-way crossover drug-interaction study in healthy subjects. Pain Pract. 2007;7:55.
24. Smit J, Oh C, Lannie C, Naessens I, Rengelshausen J, Upmalis D. Effects of probenecid on tapentadol pharmacokinetics: results of an open-label, crossover, drug-drug interaction study. J Clin Pharmacol. 2009;49:1104.
25. Smit JW, Oh C, Rengelshausen J, et al. Effects of acetaminophen, naproxen, and acetylsalicylic acid on tapentadol pharmacokinetics: results of two randomized, open-label, crossover, drug-drug interaction studies. Pharmacotherapy. 2010;30:25–34. doi:10.1592/phco.30.1.25
26. Finkel J, Goldberg J, Rosenburg R, et al. First evaluation of tapentadol oral solution for the treatment of moderate to severe acute pain in children aged 6 to <18. J Pain Res. In press 2019.
27. Terlinden R, Kogel BY, Englberger W, Tzschentke TM. In vitro and in vivo characterization of tapentadol metabolites. Methods Find Exp Clin Pharmacol. 2010;32:31–38. doi:10.1358/mf.2010.32.1.1434165
28. Kneip C, Terlinden R, Beier H, Chen G. Investigations into the drug-drug interaction potential of tapentadol in human liver microsomes and fresh human hepatocytes. Drug Metab Lett. 2008;2:67–75.
29. Hale M, Upmalis D, Okamoto A, Lange C, Rauschkolb C. Tolerability of tapentadol immediate release in patients with lower back pain or osteoarthritis of the hip or knee over 90 days: a randomized, double-blind study. Curr Med Res Opin. 2009;25:1095–1104. doi:10.1185/03007990902816970
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.