")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Pharmacokinetics, pharmacodynamics and safety of CKD-519, a CETP inhibitor, in healthy subjects
Authors Kim CO, Oh ES, Choi CG, Kim YJ, Lee S, Kim S, Park MS
Received 22 August 2016
Accepted for publication 7 October 2016
Published 15 November 2016 Volume 2016:10 Pages 3763—3770
DOI https://doi.org/10.2147/DDDT.S120387
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Choon Ok Kim,1 Eun Sil Oh,2 Chungam Choi,1 Yeonjoo Kim,3 Sera Lee,4 Semi Kim,4 Min Soo Park1,5
1Department of Clinical Pharmacology, Severance Hospital, Yonsei University College of Medicine, Seoul, 2Department of Pharmaceutical Medicines and Regulatory Science, Colleges of Medicine and Pharmacy, Yonsei University, Incheon, 3Chong Kun Dang Clinical Research, Chong Kun Dang Pharmaceutical Corp., 4Chong Kun Dang Research Institute, Chong Kun Dang Pharmaceutical Corp., 5Department of Pediatrics, Yonsei University College of Medicine, Seoul, Republic of Korea
Abstract: CKD-519 is a selective and potent cholesteryl ester transfer protein (CETP) inhibitor being developed for the treatment of dyslipidemia to raise high-density lipoprotein cholesterol. We investigated the safety, tolerability, pharmacokinetics, and pharmacodynamics of single doses of CKD-519 in healthy adult subjects. A randomized, double-blinded, placebo-controlled, single ascending dose study was performed. Eight healthy subjects were enrolled in each CKD-519 dose group (25, 50, 100, 200, or 400 mg) and randomized to CKD-519 (n=6) or matching placebo (n=2). CKD-519 reached the maximum plasma concentration (Cmax) at 5–6 h post-dose, and had a long terminal half-life ranging between 40–70 h. The area under the plasma concentration–time curve (AUC) and Cmax increased with the dose, however, Cmax and AUC normalized by dose decreased with each incremental dose. CETP activity decreased with dose, and the maximum decrease (63%–83%) was observed at 6–8 h post-dose. A sigmoid Emax model best described the relationship between CKD-519 plasma concentrations and CETP activity with an EC50 of 17.3 ng/mL. Overall, 11 adverse events (AEs) were observed. All AEs were mild or moderate in intensity, and resolved without any complications. There were no clinically significant effects on blood pressure. In conclusion, single doses of CKD-519 up to 400 mg were well tolerated and showed potent inhibition of CETP activity.
Keywords: cholesteryl ester transfer protein inhibitor, CKD-519, pharmacokinetics, pharmacodynamics
Introduction
Cardiovascular disease, the leading cause of mortality in the world, is closely related with dyslipidemia.1 Elevated low-density lipoprotein cholesterol (LDL-C) represents a major risk for cardiovascular events.2 LDL-C lowering has been and still is a primary target to prevent or reduce cardiovascular events.3 Although each decrease of 1.0 mmol/L in LDL-C with 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) has been reported to reduce major cardiovascular events by 24%, there remain some substantial residual cardiovascular risks.4 The decreasing level of high-density lipoprotein cholesterol (HDL-C) was associated with increasing frequency of major cardiovascular events.5 A low level of HDL-C is regarded as a strong risk factor of cardiovascular disease.6–8 Even in patients who maintain very low LDL-C level with statins, a low level of HDL-C is predictive of potential risk of cardiovascular disease.5,7
Based on these findings, raising HDL-C levels has become a potential target for reducing cardiovascular events. There are several approaches known to increase HDL-C. One is stimulation of production of apolipoprotein AI, the principal protein component of HDL-C, by fibrates, statins, or glitazones.9,10 However, there are several limitations to this approach using these therapeutic agents. The fibrates and statin produce only modest elevations in HDL-C, and the use of them has been restricted by side effects.11,12 Another approach is HDL particles remodeling by inhibition of cholesteryl ester transfer protein (CETP).13 CETP is a glycoprotein that facilitates the transfer of cholesteryl esters and triglycerides from HDL-C to apolipoprotein B-containing lipoproteins, very low-density lipoprotein (VLDL), and LDL-C.14 According to this mechanism, CETP inhibition leads to HDL-C increase and LDL-C decrease as exhibited by a few CETP inhibitors.12,15 Several CETP inhibitors have failed to access the market, whereas others are still under development. Torcetrapib showed HDL-C increase and LDL-C decrease in the ILLUMINATE trial; however, the trial was stopped due to a significantly increased risk of death and cardiac events in patients receiving torcetrapib.16 The increased mortality in the torcetrapib group could be a result of an off-target effect associated with changes in aldosterone and endhthelin-1 levels.17 Dalcetrapib did not demonstrate a significant reduction of cardiovascular events compared to placebo group, although it increased HDL-C levels.18 Evacetrapib also did not improve the efficacy in the phase 3 ACCELERATE trial. Therefore, the trial was stopped and the company discontinued development of the drug.19 Anacetrapib, the main CETP inhibitor, is still being evaluated. With not yet published results, the phase 3 double-blind clinical trial (ClinicalTrials.gov unique identifier: NCT01252953) including 30,000 patients with circulatory problems is ongoing.20
CKD-519 (Chong Kun Dang Pharmaceutical Corp., Seoul, Korea) is a potent CETP inhibitor newly developed for the treatment of dyslipidemia (Figure 1). CKD-519 inhibits the CETP-mediated transfer of cholesteryl ester in human serum (IC50 2.3 nM).21 In nonclinical studies, CKD-519 exhibited a maximum of 70%–86% inhibition of CETP activity and 25%–48% increase of HDL-C levels after two weeks of oral administration of 1, 3, or 10 mg/kg in transgenic mice expressing human CETP/apolipoprotein AI.21 In other nonclinical studies, inhibitory activity of CKD-519 was maintained until 48 h in dyslipidemic hamster model (IC50 31.4 nM) and 72 h (IC50 83 nM) in cynomolgus monkey after single administration of CKD-519 in the range from 0.1 to 30 mg/kg (Chong Kun Dang Pharmaceutical Corp., unpublished data, 2015). The differences in in vitro and ex-vivo IC50 values might have been resulted from high plasma protein binding capacity of CKD-519 (>99%).
 | Figure 1 Chemical structure of CKD-519. |
This study is the first in-human study of CKD-519, and we evaluated the pharmacokinetics (PK), pharmacodynamics (PD), safety, and tolerability of CKD-519 in healthy adult individuals.
Materials and methods
Ethics
The study protocol was approved by the Institutional Review Board of Severance Hospital, Yonsei University College of Medicine (Seoul, Korea), and conducted in accordance with the Declaration of Helsinki, the guidelines in the International Conference on Harmonization of Pharmaceuticals for Human Use-Good Clinical Practice and local regulations (IRB number: 4-2014-0113). All participants were enrolled in the study after obtaining written informed consent. This trial was registered with the identifier number NCT02156544 at ClinicalTrials.gov.
Subjects
Korean healthy adult male volunteers aged 19–55 years with a body mass index between 18.5 and 25.0 kg/m2 (body weight [kg]/height [m]2) were enrolled in this study. Both genders were eligible in this study, however, only healthy male subjects were recruited. Each subject was screened by a medical history, physical examination, 12-lead electrocardiography (ECG), and laboratory tests. Volunteers with the following criteria were excluded: a medical history of diseases that might influence the PK or PD evaluation of the CKD-519, a history of clinically significant hypersensitivity reaction to drugs or foods, systolic blood pressure (SBP) >150 mmHg or <90 mmHg, diastolic blood pressure (DBP) >100 mmHg or <50 mmHg, or positive results of urine drug screening for controlled substances (amphetamine, barbiturate, morphine, benzodiazepine, and cocaine) or serology test (hepatitis B surface antigen, anti-hepatitis C virus antibody, and anti-HIV antibody). The participants were not permitted to take any drugs or herbal medications for two weeks prior to the treatment, nor any over-the-counter drugs or supplements for one week prior to the treatment. The participants were also restricted from consuming alcohol, caffeinated beverages, or grapefruit products, as well as from smoking during this study.
Study design
A dose-block, randomized, double-blinded, placebo-controlled, single-dose, and dose-escalation study was conducted in a total of 40 subjects in five cohorts (25, 50, 100, 200, and 400 mg). Six individuals were randomly assigned to CKD-519 and two to matching placebo within each cohort. Starting with the lowest dose cohort, dose escalation to the next higher dose was proceeded after the safety profile of the dose was confirmed. CKD-519 was available in 25 mg and 50 mg tablets. In the 50 mg cohort, CKD-519 50 mg was used in accordance with the cohort dose: 50 mg ×2 tablets for 100 mg cohort, 50 mg ×4 tablets for 200 mg cohort, and 50 mg ×8 tablets for 400 mg cohort.
Enrolled subjects were admitted prior to dosing day. After overnight fasting, a single dose of CKD-519 or placebo with 240 mL water was administered to randomly assigned subjects. All subjects were denied drinking water before and after 1 h of dosing, and kept in fasted condition until 4 h post-dose of CKD-519 or placebo. Serial blood samples for PK were collected in sodium heparin tubes prior to dosing, and after 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 18, 24, 32, 48, 72, 96, 120, 144, and 168 h. In addition, peripheral venous blood for PD was collected in sodium heparin tubes prior to dosing, and after 0.5, 1, 2, 4, 8, 18, 24, 48, 72, 96, 120, 144, and 168 h. Each blood sample for PK and PD was centrifuged at 1,900× g for 10 min, and aliquots were stored at or below −70°C until analysis.
Safety assessment
Adverse events (AEs) were monitored throughout the study. All cohorts performed safety evaluations as follows: physical examinations, vital signs monitoring, 12-lead ECG, and laboratory tests including hematology, serum chemistry and urinalysis. SBP, DBP, and pulse rate were measured in a sitting position before drug administration and at 2.5, 4.5, 6.5, 8.5, 10.5, 12.5, 16.5, 20.5, 24.5, 32.5, 40.5, 48.5, 72.5, 96, 120, 144, and 168 h after dosing and during a follow-up visit. Twelve-lead ECG was taken prior to dosing and at 4, 8, and 72 h after dosing, and during a follow-up visit. Laboratory tests were done pre- and post-dosing.
Measurement of plasma CKD-519 concentration
The plasma concentrations of CKD-519 were measured using a validated high performance liquid chromatography assay (Shiseido Nonospace SI-2; Shiseido, Tokyo, Japan) coupled with tandem mass spectrometry (MS/MS, 4000QTRAP; AB SCIEX, Framingham, MA, USA). A 100 μL plasma sample was mixed with 10 μL internal standard working solution and 300 μL acetonitrile. After centrifugation, 3 μL of supernatant was injected into the column. The lower limit of quantification was 1.0 ng/mL. The calibration curve was linear over the concentration range of 1.0–2,000 ng/mL. The precision of the assay was less than 2.7% coefficient of variation, and the accuracy of the assay was within the range of 93.7%–113.0%.
Measurements of CETP activity
Plasma CETP activity was measured by using a fluorescent assay that was performed in microplates by using CETP activity assay kit (RB-EVAK; Roar Biomedical, New York, NY, USA). The CETP activity assay kit uses a donor molecule containing a fluorescent self-quenched neutral lipid that is transferred to an acceptor molecule in the presence of CETP. CETP-mediated transfer of the fluorescent neutral lipid to the acceptor molecule results in an increase in fluorescence (Excitation/Emission: 465/535 nm). Prepared the 87 μL of Ex Vivo Kit Reagent A per 373 μL of Ex Vivo Kit Reagent B to make Reagent C. Pipetted 100 μL of plasma samples into the wells of a black microtiter plate and added 5 μL of Reagent C to each well of the microplate containing plasma samples. Incubated the microplates for 90 minutes at 37°C and read the fluorescence intensity (excitation, 465 nm; emission, 535 nm) using FlexStation® 3 (Molecular Devices, Sunnyvale, CA, USA).
Pharmacokinetic analysis
The PK parameters of CKD-519 were calculated by non-compartmental analysis using Phoenix 64 WinNonlin 6.3 (Pharsight, Mountain View, CA, USA). The maximum plasma concentration (Cmax) and the time to reach the Cmax (tmax) were determined directly from the observed data. The area under the plasma concentration–time curve from 0 to last time (AUClast) was calculated using a linear trapezoidal rule. The terminal elimination rate constant (ke) was estimated by log-linear regression analysis. The elimination half-life (t1/2) and the apparent plasma clearance (CL/F) were calculated from the equations t1/2= ln(2)/ke and CL/F = dose/AUC, respectively. AUC from 0 to infinity (AUCinf) was obtained by summation of AUClast and Clast/ke. The apparent volume of distribution (Vd/F) was calculated from the equation, Vd/F = Dose/(ke·AUCinf).
Pharmacokinetic/pharmacodynamic (CETP) analysis
The PK/PD relationship of CKD-519 was analyzed using NONMEM version 7.3 (Icon Development Solutions, Ellicott City, MD, USA) with the GFORTRAN compiler. The PK and PD parameters were estimated by first-order conditional estimation with interaction method. Various goodness-of-fit criteria such as visual comparison of diagnostic scatter plot, likelihood ratio tests, measures of model stability and adequacy by condition number, significant digits, matrix singularity, and successful convergence were applied to select the most appropriate model. The linkage of PK and PD model was performed in a sequential manner that individual PK parameters estimated from the final PK model were fixed and added to the PD dataset.
Statistical analysis
The PK data were described as the mean ± standard deviation (SD) in each dose group except tmax, which was presented as median (range). In addition, the dose proportionality of PK parameters (Cmax, AUC) was evaluated using the power model as follows: Ln(y) = α + β*Ln[Dose].22 The equivalent limits (L, U) were derived from the following formulas: L =1+ Ln(θL)/Ln(r), U =1+ Ln(θH)/Ln(r), where r is the ratio between the highest and lowest drug dose (r=16), and 0.8 and 1.25 were used for θL and θH, respectively, based on the bioequivalence limits defined in the US Food and Drug Administration guidance. If the calculated 90% confidence interval (CI) for β (slope) was included within the predefined equivalent limits (L, U), dose proportionality was declared.
Demographic characteristics were analyzed using the Kruskal–Wallis test for comparison among the five cohorts. All analyses were conducted using SAS statistical software version 9.2 (SAS Institute Inc. Cary, NC, USA). All statistical tests were two-sided, and statistical significance was defined as P≤0.05.
Results
Study participants
A total of 40 healthy male individuals were enrolled and completed the trial. Eight participants were included in each dose group (25, 50, 100, 200, and 400 mg) and randomly assigned to active drug (CETP inhibitor, n=6) or placebo (n=2). There were no statistical differences in age, body weight, or height among the dose groups (P>0.05, Table 1).
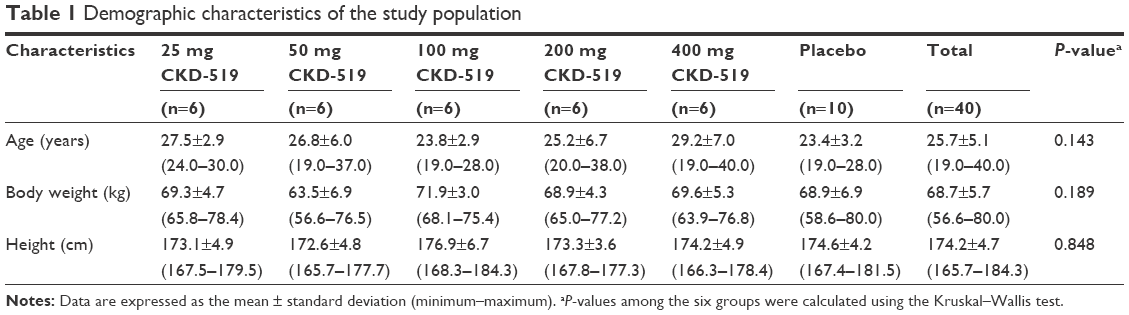 | Table 1 Demographic characteristics of the study population |
Pharmacokinetics
The mean plasma CKD-519 concentration versus time profile after administration of single oral doses of CKD-519 from 25 mg to 400 mg is shown in Figure 2. The calculated PK parameters are exhibited in Table 2. Tmax was similar regardless of the dose group, with a median of 5–6 h. Cmax and AUC increased with the dose; however, Cmax and AUC normalized by dose decreased with each incremental dose. The terminal elimination phase was relatively long (39.6–70.4 h).
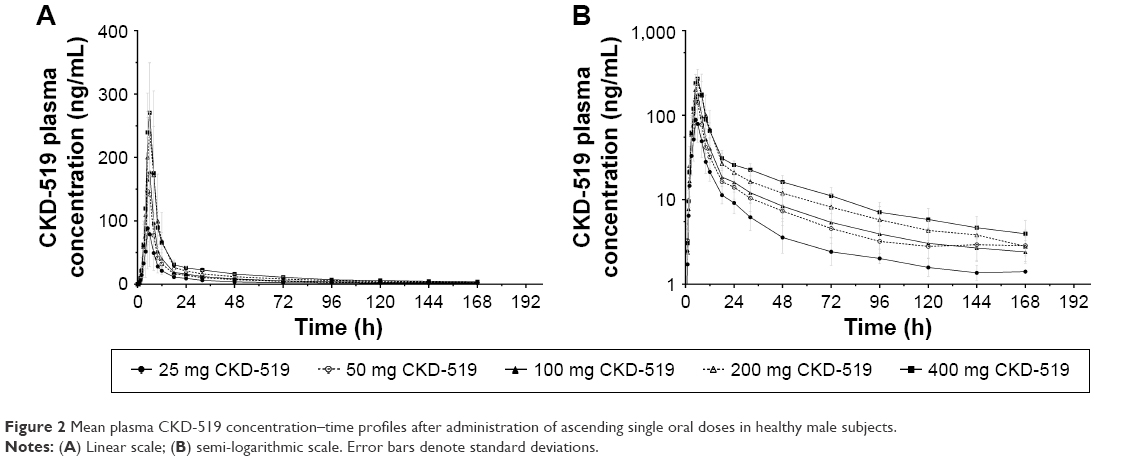 | Figure 2 Mean plasma CKD-519 concentration–time profiles after administration of ascending single oral doses in healthy male subjects. |
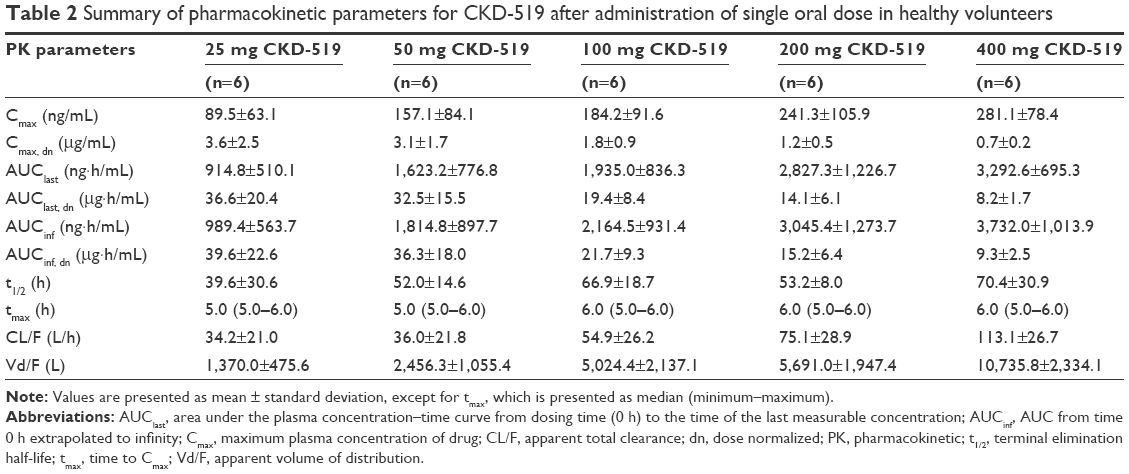 | Table 2 Summary of pharmacokinetic parameters for CKD-519 after administration of single oral dose in healthy volunteers |
Dose proportionality of PK parameters, Cmax and AUC, was analyzed with the power model. β values and 90% CI of Cmax and AUC were out of range (β values [90% CI] for Cmax, 0.46 [0.28–0.63]; for AUClast, 0.49 [0.35–0.63]; for AUCinf, 0.50 [0.35–0.65]; acceptable range for β values, 0.92–1.08).
Pharmacodynamics
The mean serum CETP activity versus time profile after administration of single oral doses of CKD-519 is shown in Figure 3. The inhibitory effect of CKD-519 on CETP activity increased with the ascending dose. The maximum inhibition of CETP activity by CKD-519 was observed at 6.3–8.3 h post-dose with approximately 65.4%–83.0% decrease from baseline: 65.4% (8.0 h) for 25 mg, 66.9% (6.3 h) for 50 mg, 78.3% (8.3 h) for 100 mg, 80.7% (7.0 h) for 200 mg and 83.0% (7.3 h) for 400 mg. The relationship between plasma concentrations of CKD-519 and inhibition of serum CETP activity is exhibited in Figure 4. There was no hysteresis in the relationship (data not shown), and it was best described using a sigmoid Emax model. The half-maximal effective concentration (EC50) was 17.3 ng/mL.
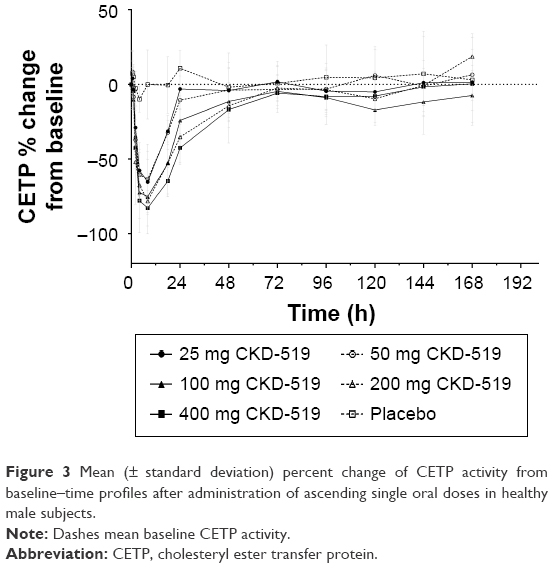 | Figure 3 Mean (± standard deviation) percent change of CETP activity from baseline–time profiles after administration of ascending single oral doses in healthy male subjects. |
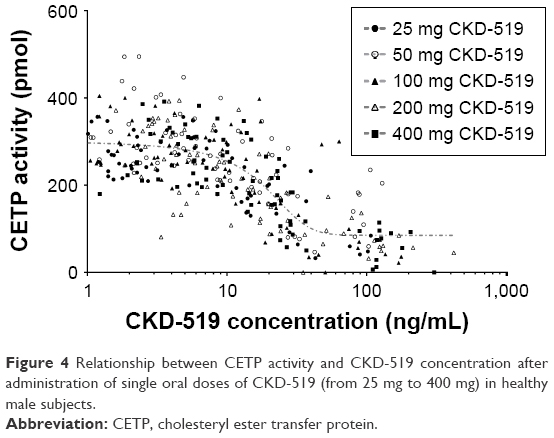 | Figure 4 Relationship between CETP activity and CKD-519 concentration after administration of single oral doses of CKD-519 (from 25 mg to 400 mg) in healthy male subjects. |
Safety
There were 13 AEs in total (two cases for 25 mg group: headache, hypertriglyceridemia; three cases for 50 mg group: diarrhea, nausea, dizziness; one case for 100 mg group: creatine phosphokinase [CPK] increased; one case for 200 mg group: dyspepsia; two cases for 400 mg group: periodontal disease, allergic rhinitis; four cases for placebo group: headache, CPK increased, dyspnea, conjunctivitis). Among the AEs, three AEs (two events of CPK increased, one event of hypertriglyceridemia) were considered to be possibly related with the study drug. However, all AEs were of mild or moderate severity, and the subjects recovered without any sequelae or complications. There were no serious drug-induced AEs in this study. A repeated ANOVA found no significant differences in SBP and DBP between the dose groups (Figure 5). In addition, there were no clinically meaningful changes in physical examinations and ECG (data not shown).
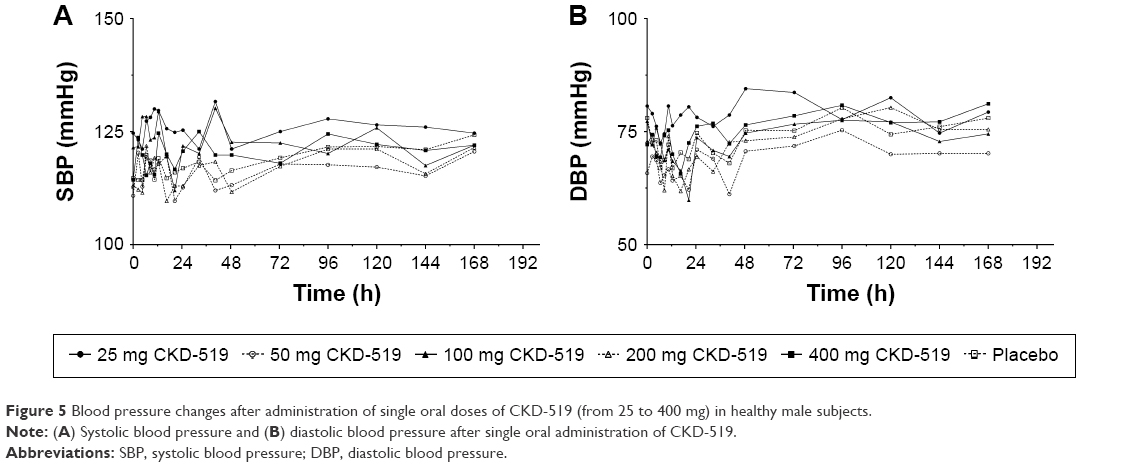 | Figure 5 Blood pressure changes after administration of single oral doses of CKD-519 (from 25 to 400 mg) in healthy male subjects. |
Discussion
This is the first clinical study in humans to evaluate the PK, PD and safety of CKD-519, a new CETP inhibitor. Based on the results, CKD-519 exhibits potent CETP inhibitory action and is tolerable without any significant AEs, including increase in blood pressure. In addition, it has PK and PD profiles appropriate for once-daily dosing regimen.
CKD-519 exhibited biphasic elimination with long terminal t1/2 (39.6–70.4 h). Most of its exposure showed within 48 h after oral administration in fasted state, followed by a slow and flat terminal elimination phase. CKD-519 has neutral lipophilic properties (LogD is 7.4, and LogP is >5.0 at pH 7.4), which explains its high affinity and accumulation in adipose tissue (Chong Kun Dang Pharmaceutical Corp., unpublished data, 2015). After single intravenous CKD-519 administration in rat, hamster, and monkey, the Vd was equal to 0.99–1.11 L/kg in all animals (Chong Kun Dang Pharmaceutical Corp., unpublished data, 2015). The Vd/F in this study was higher than the Vd of animal, and it showed a tendency to increase with the dose after being normalized with each participant’s body weight: 19.8 L/kg for 25 mg group, 38.7 L/kg for 50 mg group, 69.9 L/kg for 100 mg group, 82.6 L/kg for 200 mg group, and 154.3 L/kg for 400 mg group. This property of CKD-519 might be the reason for its long terminal t1/2 in plasma.
The Cmax and AUC increased with dose, but the increase was less than proportional. In fact, CKD-519 exposure when normalized to dose decreased with dose. This can be explained by the in vivo data that CKD-519 has decreased bioavailability at higher doses (Chong Kun Dang Pharmaceutical Corp., unpublished data, 2015). In vitro studies revealed that transport-mediated uptake did not play a role in CKD-519 absorption, and the solubility of CKD-519 in gastrointestinal fluid was shown to be poor in animal studies (Chong Kun Dang Pharmaceutical Corp, unpublished data, 2015). These data suggest that gastrointestinal absorption of CKD-519 in human may get saturated at high doses.
The inhibition of CETP activity by CKD-519 increased as the dose increased. In the dose range of 25–400 mg, the single oral administration of CKD-519 achieved a maximum decrease in CETP activity by 62%–83% from baseline at about 6.3–8.3 h after dosing. CKD-519 400 mg inhibited CETP activity by 43% at 24 h post-dose. Within the range between 25 mg and 100 mg of CKD-519, CETP activity decreased sharply with dose, but then it appeared that inhibition of CETP activity was reduced with higher doses (200 and 400 mg) of CKD-519, as Cmax and AUC also showed similar pattern with ascending doses of CKD-519. The relationship between concentration of CKD-519 and CETP activity was best described as a sigmoid Emax model, with an EC50 of 17.3 ng/mL. Considering the PK and PD profiles of CKD-519, once-daily dosing regimen seems to be appropriate, and it is estimated that CKD-519 100 mg would be the most appropriate dose in an ensuing multiple doses study.
One of the main concerns in the development of CKD-519 was a possibility of it having adverse effects on the change of SBP or DBP, because the first CETP inhibitor to be developed, namely torcetrapib, was withdrawn due to its undesired effect on BP.9 Elevation of BP caused by torcetrapib was associated with a substantial increase of aldosterone, resulting in electrolyte changes and BP increase,16 which is considered an off-target effect related to the activation of mineralocorticoid receptor by aldosterone, irrespective of HDL-C increasing mechanism by CETP inhibitor.9 In this study, before investigating into a possible off-target effect of CKD-519, as described for torcetrapib, we first looked into the possibility if CKD-519 may have an effect on BP. There were no significant differences found between baseline and post-dose SBP and DBP in all dose groups, and between placebo and CKD-519 groups.
Furthermore, CKD-519 was well tolerated within the dose range of 25–400 mg by all the participants throughout the study. There were no clinically significant changes in physical examinations findings and ECG. The AEs observed in the subjects who received CKD-519 were similar to those observed in the subjects who received placebo, and there were no serious AEs.
In this study, we investigated the safety, PK, and PD of CKD-519 administered for the first time in humans. With regard to an apparently long terminal t1/2 and PK profiles suggestive of saturable absorption, we can possibly consider saturable absorption or distribution of the drug into adipose tissue. However, to better understand these PK profiles of CKD-519, other studies about absorption or disposition of CKD-519 in humans are necessary. Even though the results showed no undesirable effect on BP, it would be prudent to monitor BP closely in the subsequent studies of multiple doses, especially in patients with dyslipidemia. Evaluation of aldosterone, cortisol or intensive BP monitoring may be warranted to completely exclude a possibility of indirect effects of CETP inhibition on adrenal hormones, since adrenal gland requires cholesterol for the biosynthesis of mineralocorticoids and glucocorticoids, and human cells possess both scavenger receptor-BI and LDL receptors.23 Furthermore, the results of this study cannot be generalized to patients with dyslipidemia because this study evaluated only healthy individuals. In order to determine the safety of CKD-519, the study needs to be conducted in patients with dyslipidemia. Although there may be some limitations to the study, the results of this study, especially overall safety and tolerability as well as PD data showing clinical evidence of CETP inhibition, provide reasonable ground that enables further development of the drug.
Conclusion
Single doses of CKD-519 ranging from 25 mg to 400 mg administered to healthy individuals were well tolerated with no serious or severe AEs, and showed potent inhibition of CETP activity (maximum 83% at 400 mg dose). Therefore, CKD-519 is a safe and potent CETP inhibitor that can be administered once daily.
Acknowledgments
The abstract of the study was posted at the annual meeting of Korean Society of Clinical Pharmacology and Therapeutics (KSCPT; December 03–04, 2015; Seoul, Korea). This study was supported by Korea Drug Development Fund (KDDF) funded by Ministry of Science, ICT, and Future Planning (MSIP); the Ministry of Trade, Industry, and Energy (MOTIE); the Ministry of Health and Welfare (MOHW) (Grant No KDDF-201406-01, Republic of Korea), supported by a grant of the Korean Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (HI14C1062). The authors thank the staff at Severance Hospital Clinical Trials Center for their generous cooperation.
Disclosure
YJ Kim, S Lee, and S Kim are full time employees of Chong Kun Dang Pharmaceutical Corp. The other authors report no conflicts of interest in this work.
References
Ohira T, Iso H. Cardiovascular disease epidemiology in Asia: an overview. Circ J. 2013;77(7):1646–1652. | ||
Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486–2497. | ||
Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110(2):227–239. | ||
Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366(9493):1267–1278. | ||
Barter P, Gotto AM, LaRosa JC, et al. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med. 2007;357(13):1301–1310. | ||
Sharrett AR, Ballantyne CM, Coady SA, et al. Coronary heart disease prediction from lipoprotein cholesterol levels, triglycerides, lipoprotein(a), apolipoproteins A-I and B, and HDL density subfractions: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2001;104(10):1108–1113. | ||
Jafri H, Alsheikh-Ali AA, Karas RH. Meta-analysis: statin therapy does not alter the association between low levels of high-density lipoprotein cholesterol and increased cardiovascular risk. Ann Intern Med. 2010;153(12):800–808. | ||
Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62(5):707–714. | ||
Kontush A, Guérin M, Chapman MJ. Spotlight on HDL-raising therapies: insights from the torcetrapib trials. Nat Clin Pract Cardiovasc Med. 2008;5(6):329–336. | ||
Dullens SP, Plat J, Mensink RP. Increasing apoA-I production as a target for CHD risk reduction. Nutr Metab Cardiovasc Dis. 2007;17(8):616–628. | ||
Barter P, Kastelein J, Nunn A, Hobbs R. Future Forum Editorial Board. High density lipoproteins (HDLs) and atherosclerosis; the unanswered questions. Atherosclerosis. 2003;168(2):195–211. | ||
Clark RW, Sutfin TA, Ruggeri RB, et al. Raising high-density lipoprotein in humans through inhibition of cholesteryl ester transfer protein: an initial multidose study of torcetrapib. Arterioscler Thromb Vasc Biol. 2004;24(3):490–497. | ||
de Grooth GJ, Kuivenhoven JA, Stalenhoef AF, et al. Efficacy and safety of a novel cholesteryl ester transfer protein inhibitor, JTT-705, in humans: a randomized phase II dose-response study. Circulation. 2002;105(18):2159–2165. | ||
Tall AR. Plasma cholesteryl ester transfer protein. J Lipid Res. 1993;34(8):1255–1274. | ||
Miyares MA. Anacetrapib and dalcetrapib: two novel cholesteryl ester transfer protein inhibitors. Ann Pharmacother. 2011;45(1):84–94. | ||
Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109–2122. | ||
Forrest MJ, Bloomfield D, Briscoe RJ, et al. Torcetrapib-induced blood pressure elevation is independent of CETP inhibition and is accompanied by increased circulating levels of aldosterone. Br J Pharmacol. 2008;154(7):1465–1473. | ||
Schwartz GG, Olsson AG, Abt M, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367(22):2089–2099. | ||
Zhao HP, Dai Y, Xiang BR. Discontinued cardiovascular drugs in 2015. Expert Opin Investig Drugs. 2016;25(9):1093–1101. | ||
McLain JH, Alsterda AJ, Arora RR. Cholesteryl ester transfer protein inhibitors: trials and tribulations. J Cardiovasc Pharmacol Ther. 2016;pii:1074248416662349. | ||
Lee S. Abstract 11228: a novel and potent CETP inhibitor, CKD-519 exerts strong HDL increment and anti-atherosclerotic effects. Circulation. 2013;128(Suppl 22):A11228. | ||
Smith BP, Vandenhende FR, DeSante KA, et al. Confidence interval criteria for assessment of dose proportionality. Pharm Res. 2000;17(10):1278–1283. | ||
Martin G, Pilon A, Albert C, et al. Comparison of expression and regulation of the high-density lipoprotein receptor SR-BI and the low-density lipoprotein receptor in human adrenocortical carcinoma NCI-H295 cells. Eur J Biochem. 1999;261(2):481–491. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.