")
Back to Journals » International Journal of Nanomedicine » Volume 13
Pharmacokinetics, distribution and anti-tumor efficacy of liposomal mitoxantrone modified with a luteinizing hormone-releasing hormone receptor-specific peptide
Authors Zhang L, Ren Y, Wang Y, He Y, Feng W , Song C
Received 31 August 2017
Accepted for publication 3 January 2018
Published 26 February 2018 Volume 2018:13 Pages 1097—1105
DOI https://doi.org/10.2147/IJN.S150512
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Linhua Zhang,1 Yanqing Ren,2 Yong Wang,3 Yingna He,2 Wei Feng,2 Cunxian Song1
1Key Laboratory of Biomedical Material of Tianjin, Institute of Biomedical Engineering, Peking Union Medical College and Chinese Academy of Medical Sciences, Tianjin, China; 2Hebei Key Laboratory of Chinese Medicine Research on Cardio-Cerebrovascular Disease, Pharmaceutical College, Hebei University of Chinese Medicine, Shijiazhuang City, Hebei Province, China; 3Department of Physics and Chemistry, College of Medicine, Hebei University, Baoding City, Hebei Province, China
Background: A previous study developed a novel luteinizing hormone-releasing hormone (LHRH) receptor-targeted liposome. The aim of this study was to further assess the pharmacokinetics, biodistribution, and anti-tumor efficacy of LHRH receptor-targeted liposomes loaded with the anticancer drug mitoxantrone (MTO).
Methods: Plasma and tissue distribution profiles of LHRH receptor-targeted MTO-loaded liposomes (LHRH-MTO-LIPs) were quantified in healthy mice or a xenograft tumor nude mouse model of MCF-7 breast cancer, and were compared with non-targeted liposomes and a free-drug solution.
Results: The LHRH-MTO-LIPs demonstrated a superior pharmacokinetic profile relative to free MTO. The first target site of accumulation is the kidney, followed by the liver, and then the tumor; maximal tumor accumulation occurs at 4 h post-administration. Moreover, the LHRH-MTO-LIPs exhibited enhanced inhibition of MCF-7 breast cancer cell growth in vivo compared with non-targeted MTO-loaded liposomes (MTO-LIPs) and free MTO.
Conclusion: The novel LHRH receptor-targeted liposome may become a viable platform for the future targeted treatment of cancer.
Keywords: liposome, mitoxantrone, luteinizing hormone-releasing hormone receptor, tumor targeting, gonadorelin
Introduction
Despite vast progress in the field of cancer chemotherapy, small-molecule chemotherapeutic drugs continue to be plagued with the problems of non-specific toxicity, low therapeutic index, and increasing drug resistance rates.1 These problems frequently result in suboptimal dosing, treatment delays or discontinuance, and reduced patient compliance to therapy.2 An effective solution to circumvent these problems is to deliver cancer drugs within biocompatible nanocarriers. The nanoscale drug carriers have the properties of controlled drug release, prolonged blood circulation, and superior encapsulation.3 When the surface of these carriers is modified with targeting moieties, they can be selectively delivered to the tumor region while sparing healthy tissue, thus improving their therapeutic index while reducing their side-effects.4
Liposomes are the most extensively used nanocarriers, due to their controllable size, ready modifiability, and good biocompatibility.5 Long-circulating liposomal formulations modified with polyethylene glycol (PEG) have been developed to prolong drug circulating time, enhance anti-tumor efficacy by enhanced permeability and retention (EPR) effect-based tumor drug deposition, and reduce drug toxicity by avoiding normal tissues.6–8 Active targeting can help liposomes overcome biological barriers (active transport vs non-specific endocytosis), decrease the residual toxicity of a system, and further increase the therapeutic effect.9,10 For this purpose, a variety of site-directed surface ligands, such as monoclonal antibodies, peptides, or small molecules, are used to functionalize liposomes to target malignant tumors with high affinity and specificity.11
Luteinizing hormone-releasing hormone (LHRH) is a hormonal decapeptide and one promising target ligand. It targets specific LHRH membrane receptors, which are characteristically overexpressed in many tumors, including those of the breast, ovaries, endometrium, and prostate.12,13 Conversely, normal tissues, most notably bone marrow tissue, lack LHRH receptor expression, making LHRH receptors an excellent targeting mechanism for cancer treatment.14
Mitoxantrone (MTO) is one of the most widely used chemotherapeutic agents in the clinic for the treatment of breast cancer, acute leukemia, and non-Hodgkin’s lymphoma.15 However, nonselective biodistribution and dose-limiting toxicities associated with systemic MTO chemotherapy restricted its use and therapeutic potential.16 Hence, approaches aimed at improving its therapeutic index while minimizing its side-effects have long been sought. Our previous study evaluated LHRH receptor-target MTO-loaded liposomes modified with gonadorelin, a peptide analog of LHRH with high affinity for LHRH receptors, and we found that these liposomes exhibited remarkable stability, sustained release kinetics of encapsulated MTO, and improved therapeutic efficiency in vitro.
In this report, we further tested the hypothesis that gonadorelin-modified liposomes could enhance the delivery of the anti-tumor drug MTO in vivo. The LHRH receptor-targeted MTO-loaded liposomes (LHRH-MTO-LIPs) have an average size of 120 nm, show prolonged circulation time, facilitate specific in vivo drug delivery into cancer cells, and significantly enhance the efficacy of MTO in a human breast cancer xenograft model. These results suggest that LHRH receptor-targeted liposomes may be promising systems to achieve specific drug delivery into tumor tissues.
Materials and methods
Materials
Gonadorelin was purchased from ProSpec-Tany TechnoGene Ltd. (Rehovot, Israel); the sequence of gonadorelin was Pyr-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2 (MW =1,182.3). Mitoxantrone hydrochloride was obtained from Chongqing Kailin Pharmaceutical Co., Ltd. (Chongqing, China). Hydrogenated soybean phosphatidylcholine (HSPC) and N-(carbonyl-methoxypolyethylene glycol 2000)-1, 2-distearoyl-sn-glycero-3-phosphorethanolamine sodium salt (mPEG2000-DSPE) were purchased from A.V.T (Shanghai) Pharmaceutical Co., Ltd. (Shanghai, China). Cholesterol, 2-Iminothiolane, and Sephadex G10 were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide (polyethylene glycol-2000)] (ammonium salt) (Mal-PEG2000-DSPE) was purchased from Avanti Polar Lipids (Alabaster, AL, USA). Phosphate buffered saline (PBS, pH 7.4, 290 mOsm) was purchased from Tianjin Haoyang Biologicals Technology Co., Ltd. (Tianjin, China). The bicinchoninic acid (BCA) kit was purchased from Shanghai Shenergy Bicolor Bioscience & Technology Co. (Shanghai, China). Ammonium sulfate and all other chemicals used in this study were of high-performance liquid chromatography (HPLC) grade or of analytical grade.
Cell line
Human MCF-7 breast cancer cells were purchased from the Cell Resource Center of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). Cells were cultured in RPMI 1640 medium (Sigma-Aldrich Co.) supplemented with 10% fetal bovine serum (FBS) (Tianjin Haoyang Biologicals Technology Co., Ltd.). Cells were grown at 37°C in a humidified atmosphere of 5% CO2 (vol/vol) in air. All experiments were performed on cells during the exponential growth phase.
Synthesis of LHRH-MTO-LIPs
Liposomes loaded with MTO (MTO-LIPs) were produced according to a thin film hydration method combined with an ammonium sulfate gradient method described previously.17 Briefly, a mixture consisting of HSPC, cholesterol, and mPEG2000-DSPE, all dissolved in chloroform, was prepared in a mole ratio of 90:10:0.4. This lipid suspension was dried in a round-bottom bottle to form an even thin lipid film under continuous gaseous N2 supply and dried further under high vacuum overnight to remove the organic solvent completely. The lipid film obtained was hydrated in 300 mM ammonium sulfate (pH 5.5). After extrusion, the outer buffer of the obtained liposomes was changed to a sucrose (300 mM)-histidine (10 mM) buffer, pH 7.5, in a Sephadex G-50 column (Sigma-Aldrich Co.). MTO was encapsulated into the liposomes in a w/w ratio of 1:10 (MTO:HSPC) during incubation at 60°C for 10 min. Free MTO was removed in Sephadex G-50 column equilibrated with HBS buffer (25 mM HEPES, 140 mM NaCl, pH 7.4). Plain liposomes mimicking MTO-LIPs were prepared similarly, except for the omission of the MTO-loading procedure.
The LHRH receptor targeting ligand gonadorelin was coupled onto pre-manufactured MTO-LIPs using a post-insertion technique.18 Gonadorelin was first thiolated by Traut’s reagent in HBS buffer (25 mM HEPES, 140 mM NaCl, pH 7.4). Unreacted Traut’s reagent was removed by a Sephadex G10 column equilibrated with HBS buffer (25 mM HEPES, 140 mM NaCl, pH 7.4). Then, thiolated gonadorelin were coupled with Mal-PEG2000-DSPE in a 1:2 molar ratio overnight at room temperature under a nitrogen atmosphere. The formed complex was added to the preformed MTO-LIPs at a 1:1,000 a molar ratio and incubated at 60°C for 1 h. The mixture was then passed through a Sepharose CL-4B column (Sigma-Aldrich Co.) to remove unbound gonadorelin, and the content of peptide in the liposomes was determined by a BCA kit (Sigma-Aldrich Co.). As a result, LHRH receptor-targeted MTO-loaded liposomes (LHRH-MTO-LIPs) were formed.
The mean diameter, size distribution, and zeta potential of liposomes was measured by dynamic light scattering. Before analysis, each sample was diluted 20-fold in distilled water to obtain the appropriate liposomal concentration. The morphology of LHRH-MTO-LIPs was observed under transmission electron microscopy (TEM). The amount of MTO encapsulated inside the MTO-LIPs and LHRH-MTO-LIPs was determined from its absorbance at 650 nm and measured after entire liposome solubilization with 1% (v/v) Triton in water in relation to an MTO calibration curve. The loading content (LC) and encapsulation efficiency (EE) of liposomal MTO were calculated by the following equations, respectively. Measurements were carried out three times for each batch.
|
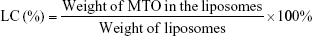
|
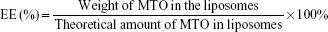
The in vitro release kinetics of MTO were assessed by a dialysis method using PBS (pH =7.4) containing 10% (w/v) plasma as the release medium. A total of 2 mL of MTO-LIPs and LHRH-MTO-LIPs (0.5 mg MTO/mL) were sealed in dialysis tubes (MWCO =10 kDa, Spectrum Laboratories Inc., Piscataway, NJ, USA). The dialysis tubes were immersed in 200 mL release medium in an Erlenmeyer flask. The containers were kept in an incubator shaker at 100 rpm and 37°C. At different time points, the concentration of MTO retained in the dialysis tubes was measured by HPLC with the detector set at 650 nm. The samples were analyzed using the method mentioned in the biodistribution studies. Values were reported as the mean from triplicate samples.
Pharmacokinetic and biodistribution assays
Plasma pharmacokinetic analysis was performed in normal female BALB/c mice, and MCF-7 tumor bearing BALB/c mice were used in tissue distribution studies. All animal experiments were approved by the Institutional Animal Care and Use Committee of Peking Union Medical College.
For pharmacokinetic studies, normal female BALB/c mice were injected with 0.1 mL of 1 mg/mL free MTO or MTO-mimicking liposomal formulations via the lateral tail vein. At time points of 5, 10, 15, 30, 60, 120, 240, 480, and 1,440 min post-injection, blood was collected from the submandibular vein plexus. Blood samples were immediately centrifuged at 1,000× g for 10 min to separate the plasma.
For tissue distribution analyses, human breast cancer tumors were generated by subcutaneous injection of MCF-7 breast cancer cells (4.0×106 cells/mouse) in female BALB/c nude mice (6–10 weeks old). When tumors grew to approximately 300 mm3, the mice were injected intravenously with free MTO or MTO-containing liposomes at an MTO dose of 2.5 mg/kg. At 1, 4, and 24 h post-injection, mice were sacrificed, and tissues including the tumor, liver, heart, lung, spleen, and kidney were harvested and weighed. All samples were stored at −70°C until analysis.
The concentrations of MTO in plasma and in normal and tumor tissue samples were determined using an HPLC method. Before analysis, tumor and normal tissue samples were first homogenized using a Tissue-Tearor™ equipped with a 7 mm probe (BioSpec Products, Inc., Bartlesville, OK, USA). A 10% (w/v) homogenate was prepared in a 20% ascorbic acid solution. Samples were subsequently processed for quantitative analysis using a protein precipitation method. A total of 800 μL of an extraction solution (methanol containing 0.5 M hydrochloric acid:acetonitrile [90:10, v/v]) was added to 200 μL of plasma, tumor, or normal tissue homogenate in a 2 mL microcentrifuge tube. The samples were then vortexed for 10 min and centrifuged at 10,000× g for 15 min at 4°C to remove plasma proteins. The supernatants were collected and analyzed using HPLC.
MTO was separated on a Zorbax SB C18 column (150×4.6 mm, 5 μm, Agilent Technologies, Inc., Santa Clara, CA, USA) at 35°C and quantifled by UV absorbance at 650 nm. The mobile phase consisted of acetonitrile and solutions containing 30 mM of sodium 1-heptanesulfonate and 9.0 mL/L of glacial acetic acid (37:63, v/v) at a flow rate of 1 mL/min.
The concentration of MTO in each sample was determined using a constructed calibration curve. Parameters such as half-life (t1/2), volume of distribution (Vd), area under the concentration-time curve (AUC), maximum concentration (Cmax), and clearance (CL) were determined for MTO in the plasma by noncompartmental analysis using DAS 2.0 software (the net for drug evaluation in China).
In vivo anti-tumor efficacy assay
To prepare tumor-bearing mice, 4.0×106 MCF-7 breast cancer cells were inoculated subcutaneously into the front armpit area of 4–5-week-old BALB/c female nude mice. When the tumors had developed to approximately 100 mm3, the mice were divided into the following four groups (n=7) in a way that minimized weight and tumor size differences between the groups: a control group treated with saline, a free-MTO group (2.5 mg/kg), an MTO-LIP group, and a LHRH-MTO-LIP group (2.5 mg/kg MTO-equivalent for liposomes). For all experiments, mice were treated once weekly for 2 weeks, on days 0, 7, and 14, via tail-vein injections. The body weight and tumor size were measured twice per week up to 21 days. The tumor volume was calculated using the formula: V = π/6× larger diameter × (smaller diameter)2. After 21 days, mice were sacrificed, and tissues (tumor, liver, heart, lung, spleen, and kidney) were collected for hematoxylin and eosin (H&E) staining. Our investigations were performed after approval by our local ethical committee at Peking Union Medical College, and in accordance with the Principles of Laboratory Animal Care.
Statistical methods
Results are expressed as the mean ± SD. The statistical significance of tumor size or body weight differences between drug-treated groups and the control group were calculated using one-way ANOVAs with Dunnett’s multiple comparison test, and between different drug-treated groups, using a one-way ANOVA with Tukey’s multiple comparison test. In all cases, P<0.05 was considered to be statistically significant.
Results and discussion
Preparation and characterization of liposomes
Liposomes were prepared by the thin film hydration method, followed by extrusion. The ammonium sulfate gradient method was used to load MTO into liposomes. The target ligand gonadorelin was coupled to maleimide-containing Mal-PEG2000-DSPE forming stable thioether linkages, and the obtained gonadorelin coupled DSPE-PEG2000 (DSPE-PEG2000-gonadorelin) was then transferred into the outer monolayer of preformed MTO-LIPs via post-insertion techniques to yield LHRH-MTO-LIPs. The content of gonadorelin in LHRH-MTO-LIPs was 0.67% (w/w). The loading content and encapsulation efficiency of LHRH-MTO-LIPs were 9.5% and 98.2%, respectively. TEM showed that LHRH-MTO-LIPs were monodispersed, highly soluble, and stable in aqueous solution (Figure 1A).
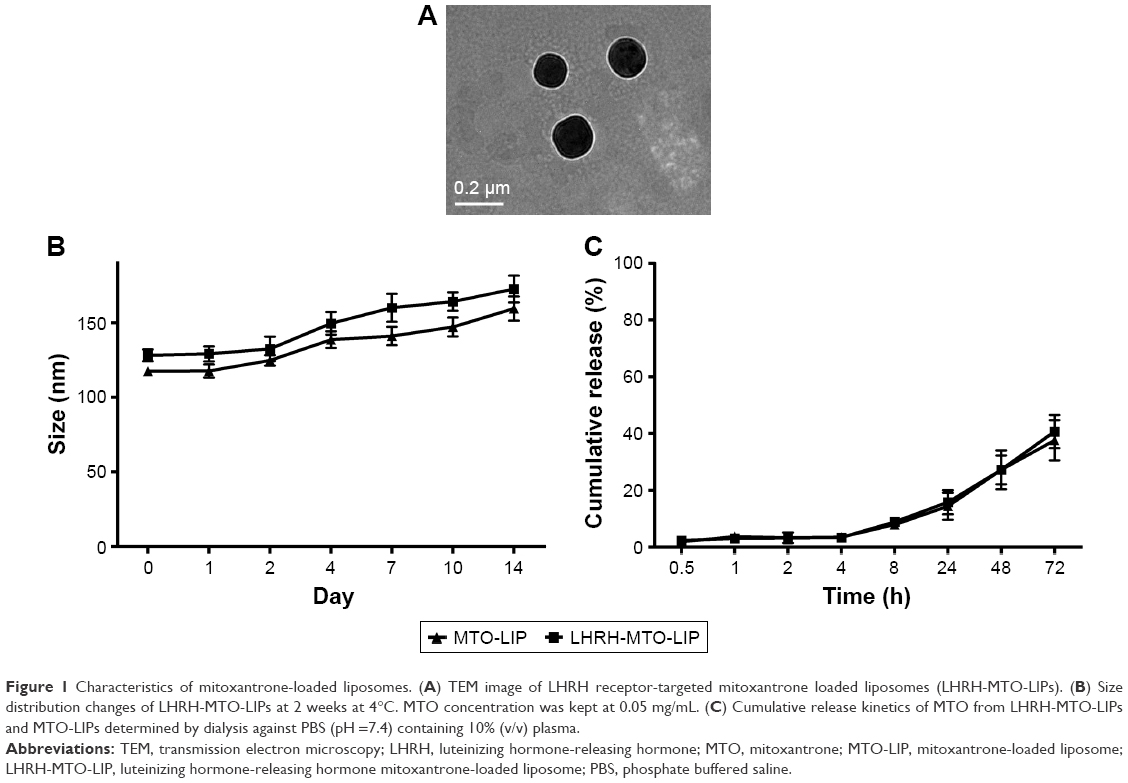 | Figure 1 Characteristics of mitoxantrone-loaded liposomes. (A) TEM image of LHRH receptor-targeted mitoxantrone loaded liposomes (LHRH-MTO-LIPs). (B) Size distribution changes of LHRH-MTO-LIPs at 2 weeks at 4°C. MTO concentration was kept at 0.05 mg/mL. (C) Cumulative release kinetics of MTO from LHRH-MTO-LIPs and MTO-LIPs determined by dialysis against PBS (pH =7.4) containing 10% (v/v) plasma. |
The mean particle size and zeta potential of LHRH-MTO-LIPs were assessed. As shown in Table 1, the mean particle size was 103.3±0.70 nm, and the zeta potential was −10.68±0.13 mV. It has been demonstrated that drug carriers with a size of approximately 100 nm and with weak negative charge could exhibit favorable in vivo behaviors, including efficient EPR effect-based tumor disposition.19 Therefore, the physicochemical properties of LHRH-MTO-LIPs were suggested to be suitable for EPR effect-based efficient tumor disposition. The particle size and zeta potential of the plain liposomes and MTO-LIPs were nearly identical, indicating that the encapsulation of MTO into the liposome did not affect the physicochemical characteristics of the liposome. Moreover, the mean particle size of LHRH-MTO-LIPs was approximately 10 nm larger than that of MTO-LIPs; this slight difference suggests that DSPE-PEG2000-gonadorelin was successfully inserted into the MTO-LIPs.
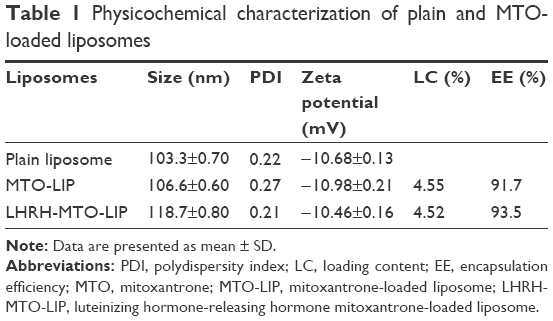 | Table 1 Physicochemical characterization of plain and MTO-loaded liposomes |
To further investigate the colloidal stability, we tested the size distribution of the same samples after 2 weeks of storage at 4°C (lipid concentration, 1 mg/mL). No aggregation was detected for all the samples (Figure 1B). This most likely resulted from the use of DSEP-PEG2000 in the assembly of the liposomes.
The release profile of MTO formulated in the MTO-LIPs was evaluated using a dialysis method. As depicted in Figure 1C, MTO formulated in MTO-LIPs exhibited sustained release kinetics. No initial burst release of MTO was observed for MTO-LIPs, indicating that an overall strong force was involved in the drug-carrier interaction. MTO formulated in LHRH-MTO-LIPs displayed a similar MTO release compared to MTO-LIPs during the entire experimental period. Decoration of MTO-LIPs with gonadorelin had a negligible impact with respect to the MTO release kinetics.
Pharmacokinetic and biodistribution assays
The concentration vs time profiles of the sum total of MTO in plasma after the administration of free MTO and the two liposomal formulations are plotted in Figure 2A. The blood retention times of MTO in both MTO liposomal formulations were significantly increased compared to that of free MTO. After the administration of free MTO, the drug was rapidly removed from circulation, and was undetectable in plasma after ~1.5 h. The pharmacokinetic parameters were obtained by fitting the blood MTO concentration vs time using a noncompartmental model, and are summarized in Table 2. The incorporation of MTO into MTO-LIPs or LHRH-MTO-LIPs led to substantially greater t1/2, AUC, and Cmax compared to free MTO. The t1/2, AUC, and Cmax of MTO-LIPs/LHRH-MTO-LIPs were 62.8/48.3, 672.6/600.7, and 25.0/25.2-fold higher, respectively, than those of free MTO. However, Vd and CL for both liposomal MTO formulations were significantly lower than those for free MTO. These data suggest that the MTO formulated in MTO-LIPs or LHRH-MTO-LIPs was well-confined within the blood circulation with significantly increased half-life values. There was no significance between the pharmacokinetic parameters of MTO administered in LHRH-MTO-LIPs and MTO-LIPs, suggesting that the accumulation of drugs at the tumor site is mainly due to the EPR effect.
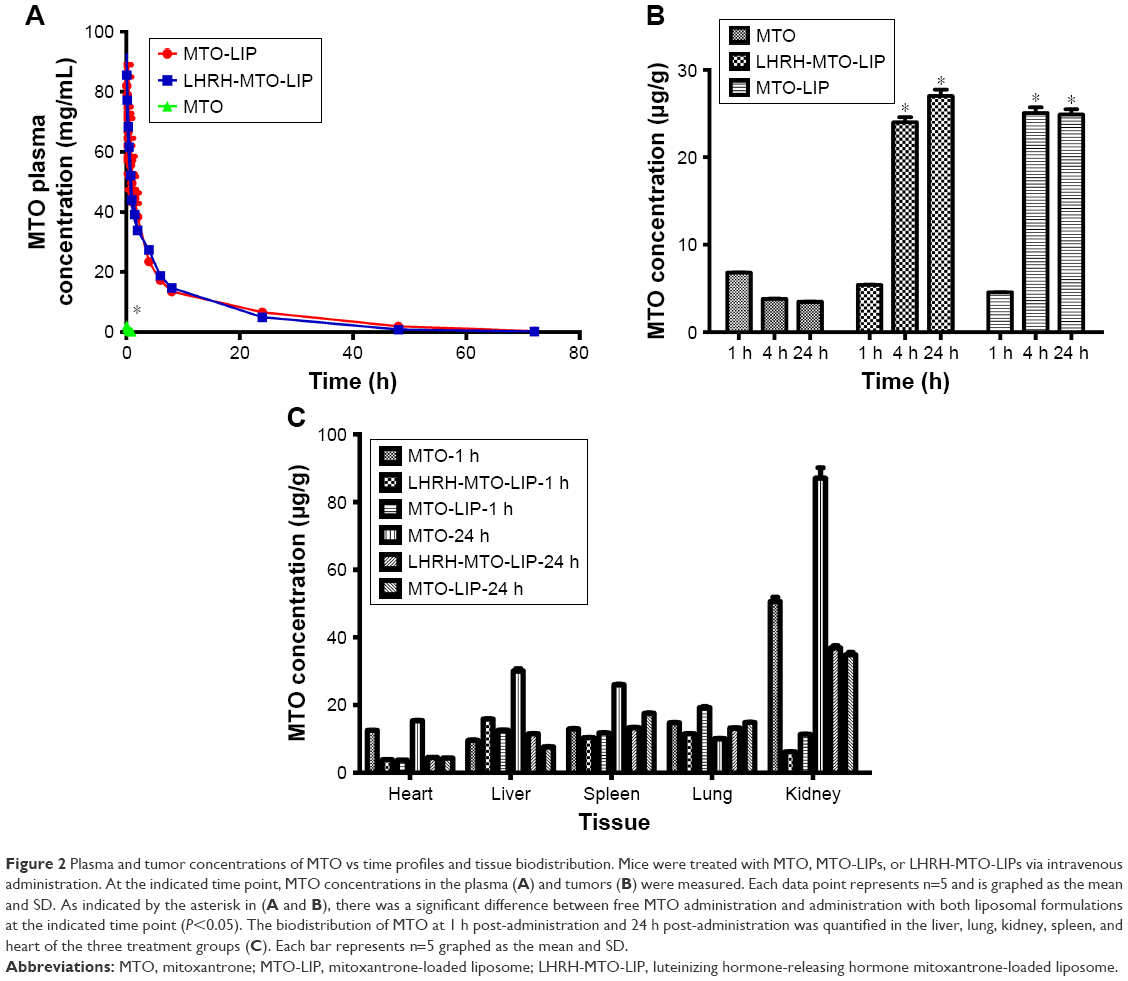 | Figure 2 Plasma and tumor concentrations of MTO vs time profiles and tissue biodistribution. Mice were treated with MTO, MTO-LIPs, or LHRH-MTO-LIPs via intravenous administration. At the indicated time point, MTO concentrations in the plasma (A) and tumors (B) were measured. Each data point represents n=5 and is graphed as the mean and SD. As indicated by the asterisk in (A and B), there was a significant difference between free MTO administration and administration with both liposomal formulations at the indicated time point (P<0.05). The biodistribution of MTO at 1 h post-administration and 24 h post-administration was quantified in the liver, lung, kidney, spleen, and heart of the three treatment groups (C). Each bar represents n=5 graphed as the mean and SD. |
 | Table 2 Pharmacokinetic parameters of MTO in different formulations |
The tumor concentrations of MTO administered as free-MTO, in LHRH-MTO-LIPs, and in MTO-LIPs, are depicted in Figure 2B. At 1 h post-administration, there were low concentrations of MTO for each treatment. At 4 h post-administration, there was an obvious increase in MTO concentrations in both LHRH-MTO-LIPs and MTO-LIPs, whereas 1 h was the Tmax for free MTO, and, by 4 h post-administration, there was a decline in MTO concentration. As indicated by the asterisk in Figure 2B, there was significance between the tumor concentration of MTO administered as a free-MTO solution and that of MTO administered in LHRH-MTO-LIPs or in MTO-LIPs at the 4 h and 24 h time point. There was no significant difference, however, between either liposomal formulation. From this data, it appears that both the non-targeted MTO-LIPs and the targeted LHRH-MTO-LIPs are effective in increasing the tumor concentration of MTO relative to the unformulated drug.
The biodistribution of MTO at 1 h post-administration and 24 h post-administration is illustrated in Figure 2C. The organs analyzed include the liver, lung, kidney, spleen, and heart. For each time point, there does not appear to be a significant difference between the biodistributions of LHRH-MTO-LIPs and MTO-LIPs, showing that targeting does not contribute to increased drug uptake in this condition. The first target site of accumulation is the kidney, followed by the liver, and then the tumor mass.
In vivo anti-tumor efficacy assay
Although liposomal MTO drugs are widely studied in human carcinoma cells in vitro, there are very few reports on MTO drug therapy in vivo. A LHRH receptor-overexpressing breast cancer (MCF-7) model was selected in this study to assess the therapeutic efficacy of LHRH-MTO-LIPs in comparison to free MTO and MTO-LIPs. Tumor-bearing mice were divided into the following four groups (n=7) in a way that minimized weight and tumor size differences between the groups: a control group (saline), a free-MTO group (2.5 mg/kg), a MTO-LIP group, and a LHRH-MTO-LIP group (2.5 mg/kg MTO-equivalent for liposomes). Therapy was continued once per week through tail vein injection for 3 weeks (injection points: day 0, 7, and 14).
Uncontrolled tumor growth was shown in the saline-treated group, which was consistent with the aggressive nature of the MCF-7 tumor model. Compared with the control group, tumor volumes in all treatment groups were significantly reduced (P<0.05). Importantly, the tumor volume in the LHRH-MTO-LIPs-treated group was significantly smaller than that of the free MTO (P<0.05) or MTO-LIPs (P<0.05) groups (Figure 3A), suggesting that the active targeting mechanism most likely contributes to the enhanced anti-tumor activity of MTO.
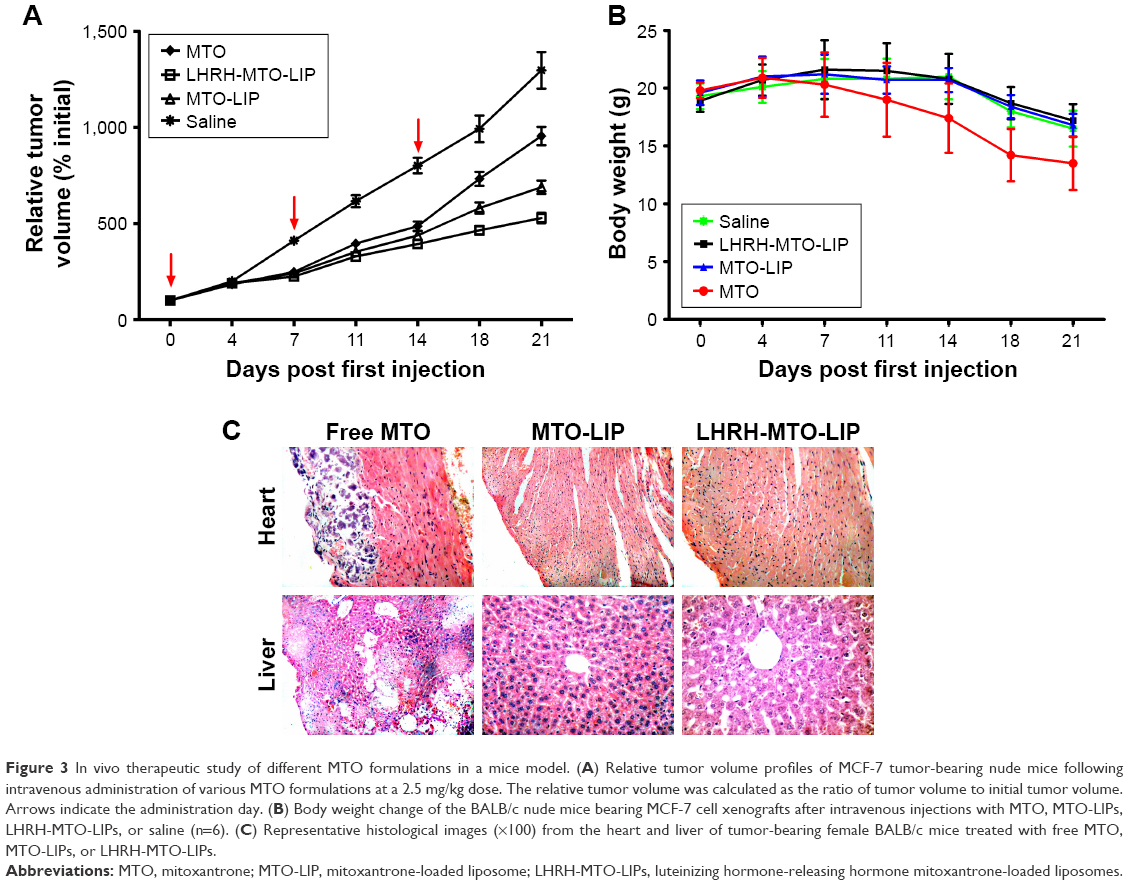 | Figure 3 In vivo therapeutic study of different MTO formulations in a mice model. (A) Relative tumor volume profiles of MCF-7 tumor-bearing nude mice following intravenous administration of various MTO formulations at a 2.5 mg/kg dose. The relative tumor volume was calculated as the ratio of tumor volume to initial tumor volume. Arrows indicate the administration day. (B) Body weight change of the BALB/c nude mice bearing MCF-7 cell xenografts after intravenous injections with MTO, MTO-LIPs, LHRH-MTO-LIPs, or saline (n=6). (C) Representative histological images (×100) from the heart and liver of tumor-bearing female BALB/c mice treated with free MTO, MTO-LIPs, or LHRH-MTO-LIPs. |
Taken together, our results suggest that MTO-LIPs and LHRH-MTO-LIPs are comparable in overall tumor accumulation, but LHRH-MTO-LIPs display enhanced antitumor activity in vivo. This may be attributed to distinct intracellular delivery mechanisms. The passively targeted EPR effect, rather than active ligand targeting, drives LHRH-MTO-LIPs blood circulation, extravasation, and accumulation in tumors. The LHRH ligand (gonadorelin) receptor interaction occurs after delivery by blood circulation and extravasation; targeting the ligand gonadorelin facilitates the entry and internalization of the payload into LHRH receptor-positive tumor cells.20
More importantly, we observed that, while treatment with free MTO suppressed tumor growth initially, after 2 weeks of treatment, tumors no longer responded to treatments and grew rapidly. This could have resulted from the tumor cells developing drug resistance to MTO. Unlike the free MTO group, the LHRH-MTO-LIPs group did not show this resurgence in tumor growth rate after the same period of treatment. This is strong evidence that LHRH-MTO-LIPs are much more effective than free MTO in treating tumors and are less susceptible to drug resistance, which is a very common occurrence in patients clinically treated with MTO.21,22
Body weight was monitored twice a week (Figure 3B). Weight loss provides a reliable indicator of drug-mediated toxicity.3 Free-MTO treatment resulted in significant weight loss over the study period, reaching a maximum of ~30% by 3 weeks. In contrast, the group of mice treated with liposomal MTO suffered smaller body weight declines throughout the treatment period, indicative of protection from drug-mediated toxicity. At day 21, the histopathologic changes in major organs, such as the liver, spleen, kidney, heart, and lung, from the mice in all treatment groups, were examined by H&E staining (Figure 3C). Free MTO had a certain influence on the cardiomyocytes and liver, while a slight toxicity in these tissues was observed in MTO-LIPs- and LHRH-MTO-LIPs-treated groups.
Conclusions
In summary, our novel-targeted liposomes, LHRH-MTO-LIPs, significantly facilitated the specific delivery of MTO to LHRH receptor-overexpressing tumor cells, and demonstrated strongly enhanced in vivo efficacy when compared with free MTO, as well as untargeted MTO-LIPs. This leads us to believe that using targeted liposomes as a specific and efficient drug delivery system is a promising strategy to treat human cancers.
Acknowledgments
The authors are grateful to the Natural Science Foundation of China (No 81601588), the Program of Excellent Innovative Talents in Hebei Provincial Institution of Higher Education (No SLRC2017048), the Hebei Provincial Natural Science Foundation, China (No H2014206359 and No B2015201085), and the Hebei 333 Talents Engineering Program (A201500149) for funding this work.
Disclosure
The authors report no conflicts of interest in this work.
References
Peters C, Brown S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep. 2015;35(4):ii. | ||
Graham SM, Carlisle R, Choi JJ, et al. Inertial cavitation to non-invasively trigger and monitor intratumoral release of drug from intravenously delivered liposomes. J Control Release. 2014;178:101–107. | ||
Pérez-Herrero E, Fernández-Medarde A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm. 2015;93:52–79. | ||
Zununi Vahed S, Salehi R, Davaran S, Sharifi S. Liposome-based drug co-delivery systems in cancer cells. Mater Sci Eng C Mater Biol Appl. 2017;71:1327–1341. | ||
Li J, Guo C, Feng F, et al. Co-delivery of docetaxel and palmitoyl ascorbate by liposome for enhanced synergistic antitumor efficacy. Sci Rep. 2016;6:38787. | ||
Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. | ||
Drummond DC, Noble CO, Hayes ME, Park JW, Kirpotin DB. Pharmacokinetics and in vivo drug release rates in liposomal nanocarrier development. J Pharm Sci. 2008;97(11):4696–4740. | ||
Yan Z, Yang Y, Wei X, et al. Tumor-penetrating peptide mediation: an effective strategy for improving the transport of liposomes in tumor tissue. Mol Pharm. 2014;11(1):218–225. | ||
Jakoby J, Beuschlein F, Mentz S, Hantel C, Süss R. Liposomal doxorubicin for active targeting: surface modification of the nanocarrier evaluated in vitro and in vivo: challenges and prospects. Oncotarget. 2015;6(41):43698–43711. | ||
Cordeiro Pedrosa LR, van Tellingen O, Soullié T, et al. Plasma membrane targeting by short chain sphingolipids inserted in liposomes improves anti-tumor activity of mitoxantrone in an orthotopic breast carcinoma xenograft model. Eur J Pharm Biopharm. 2015;94:207–219. | ||
Abu Lila AS, Ishida T. Liposomal delivery systems: Design optimization and current applications. Biol Pharm Bull. 2017;40(1):1–10. | ||
Ghanghoria R, Kesharwani P, Tekade RK, Jain NK. Targeting luteinizing hormone-releasing hormone: A potential therapeutics to treat gynecological and other cancers. J Control Release. 2018;269:277–301. | ||
Zhu S, Wang Q, Jiang J, Luo Y, Sun Z. A conjugate of methotrexate and an analog of luteinizing hormone releasing hormone shows increased efficacy against prostate cancer. Sci Rep. 2016;6:33894. | ||
Engel JB, Schally AV, Buchholz S, Seitz S, Emons G, Ortmann O. Targeted chemotherapy of endometrial, ovarian and breast cancers with cytotoxic analogs of luteinizing hormone-releasing hormone (LHRH). Arch Gynecol Obstet. 2012;286(2):437–442. | ||
Hu T, Cao H, Yang C, et al. LHD-modified mechanism-based liposome coencapsulation of mitoxantrone and prednisolone using novel lipid bilayer fusion for tissue-specific colocalization and synergistic antitumor effects. ACS Appl Mater Interfaces. 2016;8(10):6586–6601. | ||
Ling G, Zhang T, Zhang P, Sun J, He Z. Nanostructured lipid-carrageenan hybrid carriers (NLCCs) for controlled delivery of mitoxantrone hydrochloride to enhance anticancer activity bypassing the BCRP-mediated efflux. Drug Dev Ind Pharm. 2016;42(8):1351–1359. | ||
He Y, Zhang L, Song C. Luteinizing hormone-releasing hormone receptor-mediated delivery of mitoxantrone using LHRH analogs modified with PEGylated liposomes. Int J Nanomedicine. 2010;5:697–705. | ||
He Y, Zhang L, Zhu D, Song C. Design of multifunctional magnetic iron oxide nanoparticles/mitoxantrone-loaded liposomes for both magnetic resonance imaging and targeted cancer therapy. Int J Nanomedicine. 2014;9:4055–4066. | ||
Araki T, Kono Y, Ogawara K, et al. Formulation and evaluation of paclitaxel-loaded polymeric nanoparticles composed of polyethylene glycol and polylactic acid block copolymer. Biol Pharm Bull. 2012;35(8):1306–1313. | ||
Huang W, Zhang J, Dorn HC, Zhang C. Assembly of bio-nanoparticles for double controlled drug release. PLoS One. 2013;8(9):e74679. | ||
Hou L, Feng Q, Wang Y, et al. Multifunctional hyaluronic acid modified graphene oxide loaded with mitoxantrone for overcoming drug resistance in cancer. Nanotechnology. 2016;27(1):015701. | ||
Ling G, Zhang T, Zhang P, Sun J, He Z. Synergistic and complete reversal of the multidrug resistance of mitoxantrone hydrochloride by three-in-one multifunctional lipid-sodium glycocholate nanocarriers based on simultaneous BCRP and Bcl-2 inhibition. Int J Nanomedicine. 2016;11:4077–4091. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.