")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Pharmacokinetics and tolerability of IDP-73152 mesylate after a single oral administration under fasted and fed conditions in healthy volunteers
Authors Shin D , Park SI , Lee HS, An KM, Jung J, Lee M, Yu KS
Received 19 March 2019
Accepted for publication 3 July 2019
Published 24 July 2019 Volume 2019:13 Pages 2483—2490
DOI https://doi.org/10.2147/DDDT.S209238
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Dongseong Shin,1–3 Sang-In Park,4,5 Hong-Sub Lee,6 Kyung-Mi An,6 Juyoung Jung,6 MyongJae Lee,6 Kyung-Sang Yu3
1Department of Pharmacology, Gachon University College of Medicine, Incheon, Korea; 2Clinical Trials Center, Gachon University Gil Medical Center, Incheon, Korea; 3Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Hospital, Seoul, Korea; 4Department of Clinical Pharmacology and Therapeutics, Kyung Hee University Hospital, Seoul, Korea; 5East-West Medical Research Institute, Kyung Hee University, Seoul, Korea; 6Research Laboratories ILDONG Pharmaceutical Co. Ltd, Hwaseong, Korea
Background and objective: IDP-73152 mesylate is a peptide deformylase inhibitor under investigation for the treatment of complicated skin and respiratory tract infections. The objective of this study was to investigate the pharmacokinetic (PK) profile and tolerability of IDP-73152 and the effect of food after a single oral administration.
Methods: A dose block-randomized, double-blind, placebo-controlled, dose-escalation study was conducted. A total of 56 healthy volunteers received IDP-73152 mesylate in a single oral dose of 40, 80, 160, 320, 640, or 1280 mg in the fasted and fed (640 mg only) states. Blood and urine samples for PK analysis were collected up to 48 h post dose.
Results: The area under the plasma concentration-time curve (AUC0-t) of IDP-73152 increased in a dose-proportional manner in the range of 40–320 mg. The mean terminal half-life decreased from 10.7 to 6.2 hrs as the dose increased. The fraction excreted unchanged in the urine ranged from 0.05 to 0.12. In the 640-mg dose group, food delayed the median time to peak concentration (tmax) from 0.9 to 3.5 hrs. Furthermore, the maximum plasma concentration (Cmax) were decreased by 36.2%; however, AUC0-t was not generally affected. No serious adverse event or clinically significant findings were observed.
Conclusions: The systemic exposure of IDP-73152 proportionally increased as the dose increased up to 320 mg. The rate of absorption and extent of exposure were reduced by food intake. IDP-73152 was well tolerated without clinically significant adverse effects after a single oral administration.
Keywords: phase I, pharmacokinetics, antibiotics, peptide deformylase inhibitor
Introduction
The development of antibiotics with new targets against drug-resistant pathogens has been attempted for decades. Although public health regulations or derivatives of marketed antibiotics have been effective in postponing the development of resistance, novel antimicrobial agents should be identified to control the infectious disease by pathogens resistant to multiple classes of currently used drugs.1 Acceptable targets have been suggested to be those that are found in most human pathogens but not in the human system, a part of an essential pathway for pathogen survival, highly specific for various pathogenic organisms, nontoxic for humans, and so on.1
Peptide deformylase (PDF) is a metalloprotease that catalyzes the specific removal of the N-formyl moiety at the initiation of bacterial translation using a ferrous ion (Fe2+).2,3 The initially synthesized proteins in the mitochondria of both prokaryotes and eukaryotes are N-formylated.4 However, further processing of deformylation is only essential for protein maturation and organelle function in prokaryotes.1,4 As a result, a PDF inhibitor is considered to be a reasonable candidate as it would target unique mechanisms and lack cross-resistance to existing classes of antibiotics.5 Additionally, PDF inhibitors can have bacteriocidal and bacteriostatic activities with potential proinflammatory effects by neutrophil migration and release of oxygen radicals and antimicrobial substances.6,7 Several PDF inhibitors have been suggested. However, due to safety or efficacy issues, no commercially available PDF have been approved.7–10
IDP-73152 mesylate is a novel derivative of aminopiperidine that is under development as a PDF inhibitor for the treatment of complicated bacterial skin and respiratory tract infections. Complicated skin infection represented the severe form of infectious disease that involves deeper soft tissues usually caused by Streptococcus pyogenes and Staphylococcus aureus, and respiratory tract infections included pneumonia.11 In vitro antimicrobial activity of the minimal inhibitory concentration (MIC), which was comparable to that of linezolid and vancomycin and preclinical pharmacokinetic (PK) profiles in animal models of pneumonia and systemic/skin infections support further research on this drug as a clinical candidate in humans.2,7 In this study, the PK characteristics and tolerability of IDP-73152 mesylate and the effect of food after a single oral administration from 40 to 1280 mg were investigated in healthy male subjects.
Methods
Study population and design
A dose block-randomized, double-blind, placebo-controlled, single-dose, dose-escalation clinical trial was conducted in accordance with the recommendations of the Korean Good Clinical Practice and the Declaration of Helsinki (ClinicalTrials.gov registry number: NCT01904318). The Institutional Review Board of Seoul National University Hospital reviewed and approved this study. All subjects provided voluntary written informed consent after receiving a full explanation of this study. The investigation was conducted at the Clinical Trials Center, Seoul National University Hospital.
Healthy male subjects aged from 20 to 50 years without a heart rate-corrected QT interval (QTc) >430 ms or serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) >1.25 x the normal limit were randomly assigned to sequentially escalating dose groups of 40, 80, 160, 320, 640, and 1280 mg of IDP-73152 mesylate. IDP-73152 mesylate was given in fasted state; however, 640 mg of IDP-73152 mesylate was also administered in fed state for assessment of the effect of food. In each dose group, 8 subjects were assigned to IDP-73152 mesylate or placebo (8:2 IDP-73152 mesylate to placebo). In the lower-dose groups (40 and 80 mg), 6 subjects received IDP-73152 mesylate considering anticipated therapeutic level and relatively better toxicity profiles. The effect of food was investigated in 8 subjects of 640-mg dose group through 2-period design; period 1 under fasted condition and period 2 under fed condition. The tolerability 48 hrs post-dose was used to determine dose escalation.
Bioanalytical methods
PK samples of IDP-73152 were obtained at pre-dose (0 hr) and at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 24, 36, and 48 hrs after oral administration. At each time point, 6 mL of blood was taken in heparin tubes and separated by 3000 rpm centrifugation for 10 mins at 4°C. Three aliquots of supernatant were stored below –70°C. The urine concentration and volume were measured during the following time intervals: pre-dose (0 hr) and 0–6, 6–12, 12–24, and 24–48 hrs. A 10 mL aliquot of urine was collected in polypropylene tubes and kept below –20°C. Blood and urine samples were analyzed by BioCore, Seoul, Korea.
Plasma and urine concentrations of IDP-73152 were analyzed separately using an ultra-performance liquid chromatography (ACQUITYTM UPLC system, Waters Corp., Milford, MA, US)-tandem mass spectrometry (MS/MS) (API 4000, AB SCIEX, Framingham, MA, USA) system in the positive ionization mode. Chromatographic separation was performed on a C18 column (50 mm × 2.1 mm, 3 μm) at 40°C with a 0.2 mL/min flow rate and 5 μL injection volume. The mobile phase consisted of 5 mM acetonitrile formate, acetonitrile, and formic acid (60/40/0.1, v/v/v). The lower limit of quantification of plasma was 1.0 ng/mL, and the standard curve range of plasma IDP-7315 was 1–5000 ng/mL. The intra- and inter-assay accuracies ranged from 88.9% to 110.0% and from 95.7% to 103.8%, respectively. The intra- and inter-assay precision were less than 11.2% and 8.1%, respectively. The detection range of urine IDP-7315 was 10–200,000 ng/mL. The intra- and inter-assay accuracies ranged between 88.9% and 111.4% and between 98.9% and 106.1%, respectively. The intra- and inter-assay precision were under 8.1% and 7.0%, respectively.
PK analysis
The non-compartmental approach using WinNonlin® 8.0 (Pharsight Co, Cary, CA, USA) was selected to calculate the PK parameters. The peak plasma concentration (Cmax) after a single oral administration was directly observed from the plasma concentration-time profiles. The area under the plasma concentration–time curve from time 0 to the last detectable time point (AUC0-t) was assessed using the linear-up/log-down trapezoidal method. Renal clearance (CLR) was estimated as the cumulative amount of unchanged drug excreted in urine divided by the AUC0-t.
Safety and tolerability
The tolerability profiles included adverse events (AEs); physical examination; vital signs including blood pressure, pulse rate, and body temperature; 12-lead electrocardiogram (ECG); clinical laboratory tests; protein electrophoresis; and oxygen saturation monitoring. Continuous pulse oximetry was applied for 4 hrs after the single administration of IDP-73152 mesylate.
Statistical analysis
SPSS 22.0 (SPSS Korea, Seoul, Korea) was used for statistical analysis. Data distribution was assessed using Shapiro–Wilk test. If Shapiro–Wilk test results had a P-value greater than 0.05, the data were assumed to be normally distributed. Demographic characteristics including weight, height, and body mass index (BMI) were analyzed with ANOVA, and age was compared using Kruskal–Wallis test among dose groups. For dose proportionality evaluation, ANOVA was performed to compare the dose-normalized Cmax (Cmax/D) and dose-normalized AUC0-t (AUC0-t/D). The PK parameters (Cmax/D and AUC0-t/D) were assumed to be normally distributed. The log-transformed Cmax and AUC0-t values were tested by linear regression. If the 95% CI for the slope of the regression line included the value of 1.0, then dose proportionality was concluded to exist. The log-transformed Cmax and AUC0-t were analyzed to evaluate food effects. The mean difference between the fasted and fed states in the 640-mg dose group was back-transformed to calculate the geometric mean ratios (GMRs) and 90% CIs with a linear mixed-effects model.
The maximum QTc (QTcmax) intervals and maximum change from pre-dose in QTc (ΔQTcmax) of each dosage group were compared with the placebo using ANOVA followed by post-hoc analyses with Dunnett’s t-test. Statistical significance was defined as P<0.05.
Results
Demographic characteristics
A total of 56 subjects completed this study, and no one discontinued the study after dosing. Forty-four subjects received the active drug, IDP-73152 mesylate, and 12 subjects were administered placebo. The overall age, weight, height, and BMI were 27±6 years, 66.9±5.9 kg, 174±5 cm, and 22.2±1.5 kg/m2 (mean ± standard deviation), respectively. All demographic characteristics except height were not significantly different among the dosage groups (P=0.267, P=0.148, P=0.007, and P=0.064). Furthermore, all demographic factors were balanced over the entire dosage group for subjects who received IDP-73152 mesylate (age (P=0.251), weight (P=0.169), height (P=0.400), and BMI (P=0.126)), and subject randomization was appropriate to evaluate the PK profiles.
PK characteristics
After a single oral administration of 40–1280 mg IDP-73152 mesylate in the fasted state, plasma IDP-73152 levels increased with a median time to peak concentration (tmax) of 0.5–1.3 hrs (Figure 1). The mean terminal half-life and apparent clearance were 6.2–10.7 hrs and 14.9–33.6 L/hr, respectively (Table 1). As the dose increased, the tmax showed a tendency to delay (P=0.003), and the half-life and clearance decreased (both P<0.001). During excretion, the fraction of unchanged IDP-73152 excreted in the urine (fe) was 0.05–0.12 and differed among dosage groups (P<0.001). On the other hand, the mean CLR was not affected by the increase in the dose and ranged from 1.6–2.1 L/hr (P=0.519). In addition, the mean tmax was 0.5 hr with terminal half-life of 1.05 hrs in the mice model. The plasma clearance and volume of distribution were 1.14 L/hr kg and 1.71 L/kg, respectively.
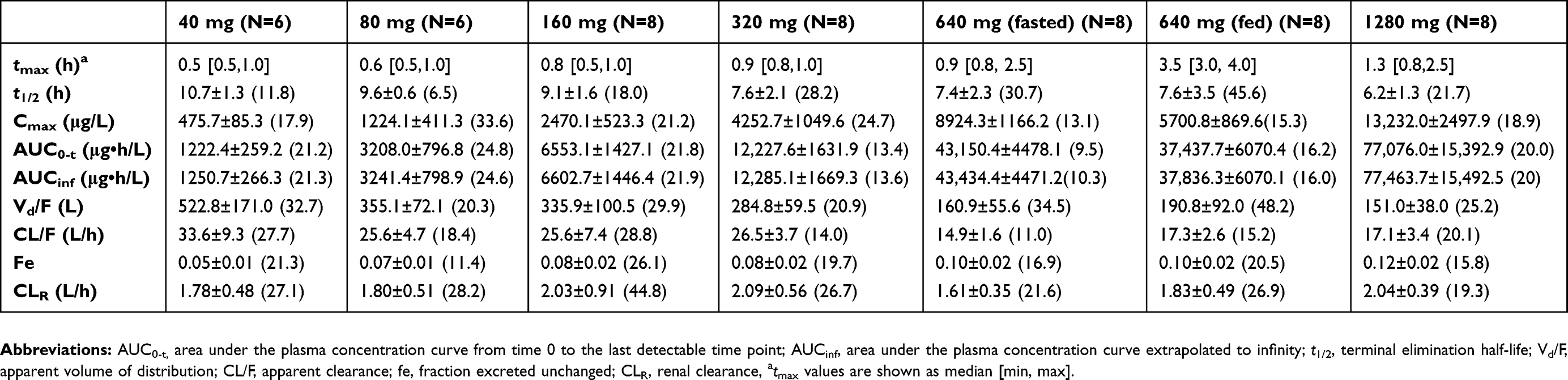 |
Table 1 Pharmacokinetic parameters of IDP-73152 after a single oral administration of IDP-73152 mesylate 40, 80, 160, 320, 640 (fasted/fed), or 1280 mg |
 |
Figure 1 Mean plasma concentration–time profiles of IDP-73152 after a single oral dose of 40, 80, 160, 320, 640, or 1280 mg of IDP-73152 mesylate. Bars represent standard deviations. (A) Plotted on a linear scale. (B) Plotted on a log scale. |
Analysis of systemic IDP-73152 mesylate exposure revealed that the mean Cmax and AUC0-t increased with a regression coefficient close to 1.0 (95% CI: 0.886–1.014 for Cmax and 1.146–1.269 for AUC0-t) across the full dose range. In particular, the 95% CIs of the slope for Cmax and AUC0-t at the lower-dose level within 40–320 mg were 0.915–1.169 and 0.987–1.211, respectively, both included 1.0 in the CI. The dose-normalized parameters were significantly different among all dose groups (P=0.015 for Cmax/D and P<0.001 for AUC0-t/D). However, the differences in Cmax/D in the groups given less than 640 mg (P=0.189) and in the AUC0-t/D in the groups given less than 320 mg (P=0.055) were not statistically significant.
After a high-fat meal in the 640-mg dose group, the systemic exposure of IDP-73152 was significantly delayed and reduced. The median tmax was 0.9 hrs in the fasted state and 3.5 hrs in the fed state (P=0.011). The GMR (with 90% CI) of Cmax and AUC0-t were 0.638 (0.563–0.723) and 0.862 (0.811–0.917), respectively. The terminal half-life was not affected by food intake (P=0.779).
Safety and tolerability
A total of 16 drug-related AEs were reported in 12 subjects. Fourteen AEs were observed in IDP-73152-administered subjects, and 2 occurred in the placebo group. All AEs were of mild severity and not serious. The frequently observed drug-related AEs were nausea (6 cases) and headache (2 cases) (Table 2). The AEs were mainly observed in the highest dose group of 1280 mg. None of the subjects discontinued the study due to an AE. One case of mild AST elevation (>2 times the upper limit of normal range) was reported at 48 hrs post-dose of 160 mg and spontaneously resolved in the follow-up test.
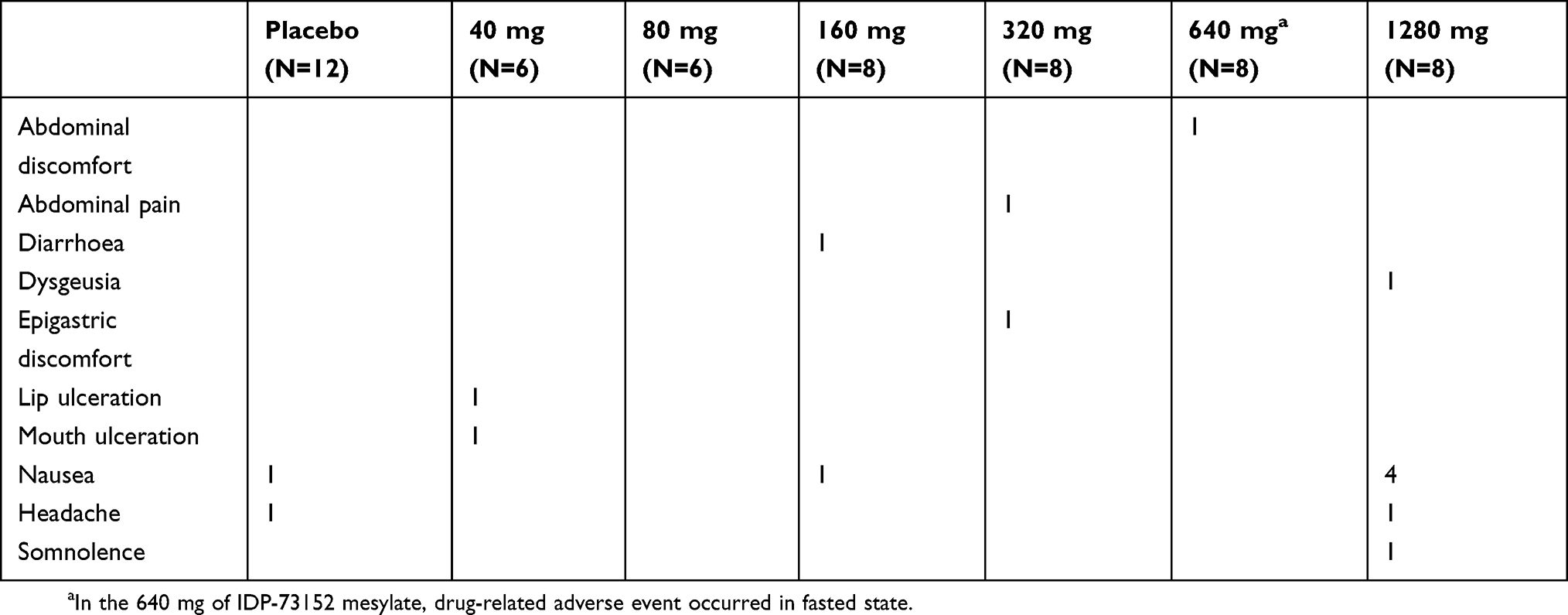 |
Table 2 Incidence of adverse events (AEs) related to IDP-73152 mesylate |
Two cases of a prolonged QTc interval (>450 ms) were found in the group given 1280 mg. The maximum QTc interval and maximum change from baseline were higher in the 1280-mg dose group than in the placebo group (both P<0.001, 95% CI of the difference, 20–48 and 13–39 ms, respectively); however, this trend was not remarkable in the other dose groups. No clinically significant changes or trends were observed in safety profiles, including physical examination, vital signs, clinical laboratory tests, or oxygen saturation monitoring.
Discussion
The PK and safety profiles of a novel PDF inhibitor for primary indications of respiratory tract and complicated skin infection, IDP-73152 mesylate, were evaluated after a single ascending dose ranging from 40 to 1280 mg in healthy subjects.
After a single oral administration, IDP-73152 was rapidly absorbed and extensively bound to plasma protein (95.4%) with a large apparent volume of distribution (from 151 to 523 L). The distribution coefficients of the liver and lung in rats were approximately 6.0 and 3.5, respectively. These tissue distribution characteristics could affect the preclinical toxicology including liver toxicity after high-dose administration and antimicrobial effect (Investigator’s brochure, unpublished data). IDP-73152 was eliminated with a mean terminal half-life of 6.2–10.7 hrs. In a previous phase I study, the absorption rate of IDP-73512 was similar, but the half-life was longer, compared to other PDF inhibitors.8–10 A single dose of 1280 mg maintained the plasma level of IDP-73152 above MIC level at 12 hrs post-dose. Based on these PK properties, a twice-daily dose regimen might be applicable in the clinical setting.8,9
The AUC0-t showed a more than dose-proportional increase in the systemic exposure for all dose ranges. However, the PK linearity of Cmax and AUC0-t was confirmed within the doses from 40 to 320 mg (Figure 2). In the plasma concentration-time profiles, the elimination process was apparently different in the dose groups over 640 mg (Figure 1). The elimination in the groups given less than 320 mg was close to a biphasic process, with a rapid decrease in the initial phase and a longer terminal half-life in the lower-dose groups than in the higher-dose groups. Because statistically consistent renal clearance was shown across all dose ranges, the supra-dose-proportional increase in the higher-dose group was possibly attributed to saturation of the non-renal pathway.
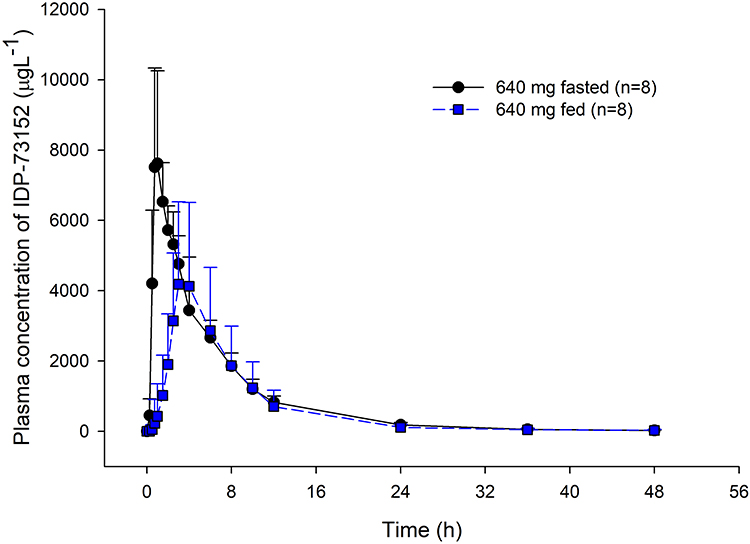 |
Figure 2 Food effect on plasma IDP-73152 concentration after single oral administrations of 640 mg IDP-73152 mesylate. Bars represent standard deviations. |
Administration of IDP-73152 mesylate with a high-fat meal led to a 36% reduction in Cmax and postponed tmax. with increased inter-individual variability (Figure 3). On the other hand, the overall systemic exposure based on the AUC0-t was comparable between the fasting and fed states. The effect of the high-fat meal on drug exposure was observed in the absorption rate due to delayed gastric emptying.10 The elimination process was not relatively affected by food, and the terminal half-life remained consistent.
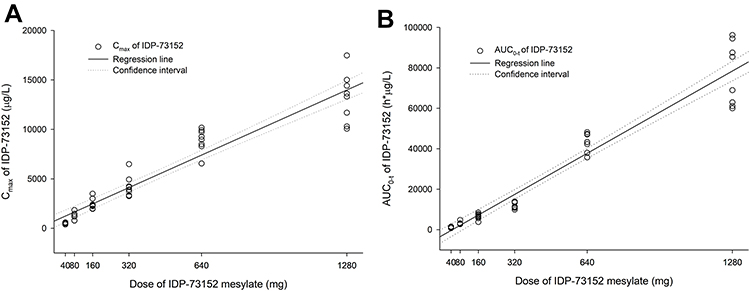 |
Figure 3 Linear regressions of IDP-73152 pharmacokinetic parameters after a single oral administration. (A) Relationship between individual Cmax and single oral dose of IDP-73152 mesylate. (B) Relationship between individual AUC0-t and single oral dose of IDP-73152 mesylate. |
The in vitro antibacterial activity of IDP-73152 against methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococci (VRE) was better or comparable to that of vancomycin or linezolid in terms of MIC. In addition, IDP-73152 presented advantageous in vivo antimicrobial effects based on the median effective dose (ED50) in VRE-infected mice compared to various PDF inhibitors (Investigator’s brochure, unpublished data). However, against Gram-negative pathogens, IDP-73152 showed an unfavorable MIC value (Investigator’s brochure, unpublished data). The antibiotic effect of IDP-73152 was observed to be AUC-dependent in a mouse model of Streptococcus pneumonia infection (Investigator’s brochure, unpublished data). Although the PDF inhibitor presented good antimicrobial activity in some Gram-negative pathogens, IDP-73152 showed more potent in vitro and in vivo activity against Gram-positive organisms in general.3,5 A previous study reported that the bacterial membrane plays a key role in modulating the antibacterial activity of the PDF inhibitor.12 Related to this property, the Gram-negative organisms had active efflux pumps and were less susceptible to PDF inhibitors.4
IDP-73152 was well tolerated after a single oral dose of up to 1280 mg. Clinically significant findings were not observed in oxygen saturation or laboratory tests, with the exception of a temporary ALT elevation in one subject. However, the ALT elevation was not meaningful and not dose-dependent. This elevation was not observed in the higher-dose group and spontaneously resolved in a sequential test. The reported drug-related AEs were mild and most frequently occurred at a dose of 1280 mg, especially nausea. Not including the 1280-mg dose group, there was no trend related to AE frequency associated with an increase in the dose, including in the placebo group. In a preclinical study, IDP-73152 induced a concentration-dependent inhibition of the hERG current (Investigator’s brochure, unpublished data). In this single-dose study, two cases of a QTc interval >450 ms and a significant increase in QTc interval from baseline after administration of 1280 mg were found compared to the placebo group. Because this result was not obtained from performing the so-called “Thorough QT/QTc Study,” assessment of the QTc interval in this study was limited to the conclusions about QTc prolongation. As a result, further investigation in clinical settings considering various risk factors such as gender (female), old age, baseline QT prolongation, bradycardia, and hepatic and renal dysfunction are required for early detection of proarrhythmic risk.13
Conclusion
We investigated the PK profile and safety/tolerability of IDP-73152 as a new PDF inhibitor. After a single oral administration, IDP-73152 showed favorable PK properties for clinical application based on a twice-daily dose regimen. Systemic exposure increased in a dose-proportional manner within the range from 40 to 320 mg. IDP-73152 was well tolerated without clinically significant adverse effects after a single oral administration. These results were obtained in a small number of healthy volunteers after a single oral dose. For further clinical application, additional large-scale clinical assessments based on the multiple doses of novel antibiotics with promising mechanisms of action against multidrug-resistant pathogens are required.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
Acknowledgments
This study was sponsored by Research Laboratories ILDONG Pharmaceutical Co., Ltd, Korea. This study was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) and funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI14C2750).
Disclosure
MyongJae Lee reports personal fees from Research Laboratories ILDONG Pharmaceutical Co., Ltd., during the conduct of the study and personal fees from Research Laboratories ILDONG Pharmaceutical Co., Ltd., outside the submitted work. Hong-Sub Lee, Kyung-Mi An, Juyoung Jung and MyongJae Lee are employees of Research Laboratories ILDONG Pharmaceutical Co., Ltd. The authors report no other conflicts of interest in this work.
References
1. Giglione C, Pierre M, Meinnel T. Peptide deformylase as a target for new generation, broad spectrum antimicrobial agents. Mol Microbiol. 2000;36(6):1197–1205.
2. Lee HY, An KM, Jung J, et al. Identification of novel aminopiperidine derivatives for antibacterial activity against gram-positive bacteria. Bioorg Med Chem Lett. 2016;26(13):3148–3152. doi:10.1016/j.bmcl.2016.04.086
3. Lofland D, Difuntorum S, Waller A, et al. In vitro antibacterial activity of the peptide deformylase inhibitor BB-83698. J Antimicrob Chemother. 2004;53(4):664–668. doi:10.1093/jac/dkh129
4. Johnson KW, Lofland D, Moser HE. PDF inhibitors: an emerging class of antibacterial drugs. Curr Drug Targets Infect Disord. 2005;5(1):39–52.
5. Gross M, Clements J, Beckett RP, et al. Oral anti-pneumococcal activity and pharmacokinetic profiling of a novel peptide deformylase inhibitor. J Antimicrob Chemother. 2004;53(3):487–493. doi:10.1093/jac/dkh108
6. Fu H, Dahlgren C, Bylund J. Subinhibitory concentrations of the deformylase inhibitor actinonin increase bacterial release of neutrophil-activating peptides: a new approach to antimicrobial chemotherapy. Antimicrob Agents Chemother. 2003;47(8):2545–2550. doi:10.1128/aac.47.8.2545-2550.2003
7. Lee M, Kim D, Shin J, et al. Quantification of IDP-73152, a novel antibiotic, in plasma from mice, rats and humans using an ultra-high performance liquid chromatography/tandem mass spectrometry method for use in pharmacokinetic studies. J Pharm Biomed Anal. 2017;145:364–371. doi:10.1016/j.jpba.2017.06.066
8. Naderer OJ, Dumont E, Zhu J, Kurtinecz M, Jones LS. Single-dose safety, tolerability, and pharmacokinetics of the antibiotic GSK1322322, a novel peptide deformylase inhibitor. Antimicrob Agents Chemother. 2013;57(5):2005–2009. doi:10.1128/AAC.01779-12
9. Ramanathan-Girish S, McColm J, Clements JM, et al. Pharmacokinetics in animals and humans of a first-in-class peptide deformylase inhibitor. Antimicrob Agents Chemother. 2004;48(12):4835–4842. doi:10.1128/AAC.48.12.4835-4842.2004
10. Rolan P, Sun H, Macleod C, Bracken K, Evans TG. Pharmacokinetics and unexpected safety issues of LBM415, a novel oral peptide deformylase inhibitor. Clin Pharmacol Ther. 2011;90(2):256–262. doi:10.1038/clpt.2011.101
11. Leong HN, Kurup A, Tan MY, Kwa ALH, Liau KH, Wilcox MH. Management of complicated skin and soft tissue infections with a special focus on the role of newer antibiotics. Infect Drug Resist. 2018;11:1959–1974. doi:10.2147/IDR.S172366
12. Mamelli L, Petit S, Chevalier J, et al. New antibiotic molecules: bypassing the membrane barrier of gram negative bacteria increases the activity of peptide deformylase inhibitors. PLoS One. 2009;4(7):e6443. doi:10.1371/journal.pone.0006443
13. Nachimuthu S, Assar MD, Schussler JM. Drug-induced QT interval prolongation: mechanisms and clinical management. Ther Adv Drug Saf. 2012;3(5):241–253. doi:10.1177/2042098612454283
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.