")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Pharmacokinetics and safety of revefenacin in subjects with impaired renal or hepatic function
Authors Borin MT , Lo A , Barnes CN, Pendyala S, Bourdet DL
Received 2 February 2019
Accepted for publication 15 August 2019
Published 8 October 2019 Volume 2019:14 Pages 2305—2318
DOI https://doi.org/10.2147/COPD.S203709
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Marie T Borin,1 Arthur Lo,2 Chris N Barnes,3 Srikanth Pendyala,4 David L Bourdet2
1Department of Clinical and Translational Pharmacology, Theravance Biopharma US, Inc., South San Francisco, CA, USA; 2Department of Drug Metabolism and Pharmacokinetics, Theravance Biopharma US, Inc., South San Francisco, CA, USA; 3Department of Biostatistics, Theravance Biopharma US, Inc., South San Francisco, CA, USA; 4Department of Clinical Development, Inflammation and Immunology, Theravance Biopharma US, Inc., South San Francisco, CA, USA
Correspondence: David L Bourdet
Department of Drug Metabolism and Pharmacokinetics, Theravance Biopharma US, Inc., 901 Gateway Boulevard, South San Francisco, CA 94080, USA
Tel +1 650 808 3755
Email [email protected]
Purpose: Revefenacin, a long-acting muscarinic antagonist for nebulization, has been shown to improve lung function in patients with chronic obstructive pulmonary disease. Here we report pharmacokinetic (PK) and safety results from two multicenter, open-label, single-dose trials evaluating revefenacin in subjects with severe renal impairment (NCT02578082) and moderate hepatic impairment (NCT02581592).
Subjects and methods: The renal impairment trial enrolled subjects with normal renal function and severe renal impairment (estimated glomerular filtration rate <30 mL/min/1.73 m2). The hepatic impairment trial enrolled subjects with normal hepatic function and moderate hepatic impairment (Child-Pugh class B). Subjects received a single 175-μg dose of revefenacin through nebulization. PK plasma samples and urine collections were obtained at multiple time points for 5 days following treatment; all subjects were monitored for adverse events.
Results: In the renal impairment study, the maximum observed plasma revefenacin concentration (Cmax) was up to 2.3-fold higher and area under the concentration–time curve from time 0 to infinity (AUCinf) was up to 2.4-fold higher in subjects with severe renal impairment compared with those with normal renal function. For THRX-195518, the major metabolite of revefenacin, the corresponding changes in Cmax and AUCinf were 1.8- and 2.7-fold higher, respectively. In the hepatic impairment study, revefenacin Cmax and AUCinf were 1.03- and 1.18-fold higher, respectively, in subjects with moderate hepatic impairment compared with those with normal hepatic function. The corresponding changes in THRX-195518 Cmax and AUCinf were 1.5- and 2.8-fold higher, respectively.
Conclusion: Systemic exposure to revefenacin increased modestly in subjects with severe renal impairment but was similar between subjects with moderate hepatic impairment and normal hepatic function. The increase in plasma exposure to THRX-195518 in subjects with severe renal or moderate hepatic impairment is unlikely to be of clinical consequence given its low antimuscarinic potency, low systemic levels after inhaled revefenacin administration, and favorable safety profile.
Keywords: liver disease, kidney disease, chronic obstructive pulmonary disease, long-acting muscarinic antagonist, LAMA, revefenacin
Introduction
Chronic obstructive pulmonary disease (COPD) is a common disease with worldwide prevalence estimated at 12%, and is a leading cause of morbidity and death.1 Many patients with COPD are older or have comorbid conditions that can be associated with renal or hepatic impairment.2,3
Revefenacin is a once-daily, long-acting, lung-selective muscarinic receptor antagonist formulated as a nebulized inhalation solution for use with a standard jet nebulizer for treatment of patients with COPD. Nebulized therapies are of particular interest in patients with poor hand/breath coordination and/or respiratory muscle weakness that limits their use of dry powder inhalers and pressurized metered-dose inhalers.4–6 The clinical efficacy and safety of revefenacin have been demonstrated in patients with COPD in Phase 2 studies, in which once-daily administration of revefenacin at doses up to 700 µg significantly improved patients’ forced expiratory volume compared with placebo and was well tolerated at all dose levels.7,8 Revefenacin further demonstrated clinically and statistically significant improvements in trough forced expiratory volume in 1 second in two 12-week Phase 3 studies in patients with moderate to very severe COPD9 and was shown to be well tolerated over 1 year of treatment.10 Revefenacin inhalation solution was approved by the FDA in November 2018, and is indicated for the maintenance treatment of patients with COPD.11
The pharmacokinetic (PK) properties of inhaled revefenacin were investigated in three Phase 2 studies in patients with COPD.7,8 Plasma revefenacin concentrations following inhaled administration were low, averaging <0.6 ng/mL after daily doses up to 700 µg, and declined rapidly from the initial maximum concentration, with a slow apparent terminal elimination phase. Renal elimination of revefenacin was very low after inhaled administration, with <1% of the dose excreted in urine.8 In a Phase 1 study in healthy subjects who received revefenacin intravenously (IV) or orally, elimination was primarily via the hepatic-biliary system and fecal route; renal excretion was minimal.12 Revefenacin was extensively metabolized to its major metabolite, THRX-195518, through hydrolysis after inhaled administration in patients with COPD8 and after IV or oral administration in healthy subjects.12 THRX-195518 is approximately tenfold less potent than revefenacin and dissociates more rapidly from human M3 receptors (Theravance Biopharma, data on file).
Although PK properties of revefenacin have been described in multiple studies, PK characteristics of revefenacin and THRX-195518 have not been characterized in special patient populations, including patients with renal or hepatic insufficiency. Chronic kidney disease has been shown to affect drug disposition via multiple mechanisms, even for drugs that are not renally eliminated.3,13,14 Likewise, hepatic insufficiency and liver disease can affect the PK properties of many drugs.15–17 The effects of renal and hepatic insufficiency can cause unexpected toxicities.3 Therefore, it is important to study the PK properties and safety of specific agents in adults with renal and hepatic impairment, so PK dose modifications can be considered, if needed, in these populations.
We report findings of two multicenter, open-label, parallel-group, single-dose PK studies of revefenacin in subjects with severe renal or moderate hepatic impairment compared with subjects with normal organ function. The primary objective of the studies was to characterize the effects of severe renal impairment or moderate hepatic impairment on PK properties of revefenacin and THRX-195518 following single-dose inhaled administration. The secondary objective was to evaluate the safety and tolerability of inhaled revefenacin in subjects with severe renal impairment or moderate hepatic impairment.
Methods
Study design and conduct
Both trials were multicenter, open-label, parallel-group, single-dose, Phase 1 PK, and safety studies. The first study (ClinicalTrials.gov Identifier Number: NCT02578082) included subjects with normal renal function or severe renal impairment and was conducted between December 2015 and May 2016. The second study (ClinicalTrials.gov Identifier Number: NCT02581592) included subjects with normal hepatic function or moderate hepatic impairment and was conducted between December 2015 and April 2016.
Each study was conducted in accordance with the principles of the International Council on Harmonisation (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use Guideline for Good Clinical Practice (GCP),18 the United States Code of Federal Regulations, the principles of the World Medical Association Declaration of Helsinki,19 Ethical Principles for Medical Research Involving Human Subjects, and all applicable regulatory requirements. In each study, all subjects provided written informed consent prior to any study procedures. Subjects (or their legally authorized representatives) could have withdrawn their consent to participate in the study at any time without prejudice. The protocols for both studies were approved by an institutional review board (IntegReview IRB, 3815 S. Capital of Texas Highway, Suite 320, Austin, TX 78704, USA).
Study population
Both studies included male and female subjects, 18–80 years of age, with body mass index 19–40 kg/m2 and body weight ≥ 55 kg.
Renal impairment study
In the renal impairment study, enrollment of subjects with normal renal function was based on estimated creatinine clearance (CLcr) using the Cockcroft-Gault equation at screening and day –1. Subjects with normal renal function had an estimated CLcr of ≥90 mL/min at screening and day –1. Determination of renal impairment was based on estimated glomerular filtration rate (eGFR), calculated using the isotope dilution mass spectrometry–traceable Modification of Diet in Renal Disease (MDRD) equation at screening and day –1. Subjects with severe renal impairment had eGFR <30 mL/min/1.73 m2 at screening and day –1. Subjects with renal insufficiency must have had stable renal function, defined as ≤25% difference in eGFR (calculated using the MDRD equation) assessed on two occasions separated by ≥14 days. The normal renal function group was selected to be comparable with the severe renal impairment group by matching subjects for weight (±15%), sex, race, and age (±10 years).
Key exclusion criteria included a history of hypersensitivity to inhaled medication, including nebulized anticholinergics or beta 2-agonists, presence of any medical conditions that precluded the use of inhaled anticholinergics, a history of clinically significant hepatic disease, a family history of congenital long QT syndrome, and presence of an abnormal cardiovascular event.
Hepatic impairment study
In the hepatic impairment study, subjects with normal hepatic function had normal liver function tests at screening and day –1. All subjects in this study had CLcr of >70 mL/min at screening and day –1. Subjects with moderate hepatic impairment had Child-Pugh class B (7–9 points) liver disease of cryptogenic, posthepatic, hepatitis B virus, hepatitis C virus, nonalcoholic steatohepatitis, or alcoholic origin. The components that contributed to the Child-Pugh score were directly related to the underlying hepatic disease. Hepatic impairment was stable, defined as no clinically significant change in disease status within the last 30 days as documented by the subject’s recent medical history (eg, no worsening of clinical signs of hepatic impairment or no worsening of total bilirubin or prothrombin time by >50%). Subjects in the normal hepatic function and moderate hepatic impairment groups were matched according to weight (±15%), sex, race, and age (±5 years).
Key exclusion criteria included hypersensitivity to inhaled medications and the presence of any condition that could preclude the use of anticholinergics. Subjects with normal hepatic function were excluded if they had a history of porphyria, active liver disease, or unexplained elevation of serum transaminases, alkaline phosphatase, albumin, prothrombin time, or total bilirubin. Subjects with moderate hepatic impairment were excluded from the study if they had ongoing severe hepatic encephalopathy and biliary liver cirrhosis or other causes of hepatic impairment related to parenchymal disorder and/or disease of the liver. An abnormal electrocardiogram and risk of adverse cardiovascular event were also exclusionary criteria.
Treatment
Both studies were open-label. Study treatment and restrictions on concomitant medications were the same for both studies. All subjects received a single dose of revefenacin (175 µg) administered as a 3-mL solution by inhalation using the PARI LC® Sprint (PARI, Midlothian, VA, USA) jet nebulizer. Subjects were prohibited from using nephrotoxic or hepatotoxic medications for 4 weeks before study drug dosing. Permitted medications included acetaminophen, ibuprofen, milk of magnesia (magnesium hydroxide), and routine vitamins and minerals. In addition, subjects could continue a stable regimen of any medication (except those specifically prohibited) in place 30 days before day –1.
PK assessments
PK assessments were the same in both studies. Blood samples were collected at the following time points for the determination of plasma concentrations of revefenacin and THRX-195518: ≈30 mins before revefenacin dosing and at 5, 15, and 30 mins, and at 1, 2, 3, 4, 6, 8, 12 (day 1), 24, 36 (day 2), 48 (day 3), 72 (day 4), and 96 (day 5) hours post-dose (after start of nebulization).
Urine was collected over the following intervals for determination of urine concentrations of revefenacin and THRX-195518: within 60 mins before dosing (day 1); and at 0–4, 4–8, 8–12, 12–24 (day 2), 24–48 (day 3), 48–72 (day 4), and 72–96 hrs (day 5) post-dose (after start of nebulization).
Concentrations of revefenacin and THRX-195518 in plasma and urine were quantified using validated liquid chromatography with tandem mass spectrometry methods. In plasma, the lower limit of quantification (LLOQ) for revefenacin and THRX-195518 was 0.0005 and 0.005 ng/mL, respectively. In urine, the LLOQ was 0.100 ng/mL for revefenacin and THRX-195518. Quality control accuracy (% relative error) and precision (% coefficient of variation) for the plasma and urine revefenacin and THRX-195518 PK assays did not exceed 9%.
Plasma PK parameters for revefenacin and THRX-195518 were determined, where possible, by noncompartmental methods using WinNonlin version 6.4 (Certara, Princeton, NJ, USA) for each subject. Plasma PK parameters included maximum observed plasma concentration (Cmax); time to reach Cmax (Tmax); area under the concentration–time curve (AUC) from time 0 to time of last quantifiable concentration (AUCt), calculated using the linear trapezoidal method; AUC from time 0 to infinity (AUCinf); apparent terminal elimination half-life (t1/2); metabolite-to-parent ratio based on AUCt (MRAUC), calculated as the molar ratio; and metabolite-to-parent ratio based on Cmax (MRCmax), calculated as the molar ratio.
AUCinf could not be accurately computed in all subjects by noncompartmental analysis because of sparsity of PK data in the terminal phase as a result of low or unquantifiable concentrations, especially for THRX-195518. In a single-dose study, AUCinf is the most appropriate measure of total exposure; therefore, AUCinf was estimated by fitting the revefenacin and THRX-195518 PK data of all subjects in the hepatic and renal impairment studies to a previously described population PK model.20 The plasma concentrations for each individual subject were predicted using the fitted population PK model, and AUCinf was determined by noncompartmental methods using WinNonlin.
Urine PK parameters included the following: cumulative amount excreted in the urine from 0 to 96 hrs post-dose; cumulative fraction of revefenacin dose excreted in urine from 0 to 96 hrs post-dose (fe0–96h), calculated for revefenacin only and expressed as a percentage; and renal clearance (CLr).
Safety assessments
Safety assessments were the same in both studies and referred to treatment-emergent adverse events (TEAEs). The studies used the ICH E6 (R1) GCP (Section 1.2) definition of an adverse event (AE),21 and AEs were coded to the preferred terms of the Medical Dictionary for Regulatory Activities. Investigators evaluated AEs in terms of severity (mild, moderate, or severe), duration, outcome, and relationship to study drug.
Clinical laboratory measurements included hematology tests, serum chemistry tests, coagulation tests, urinalysis, serology tests, vital signs, height, weight, medical history, physical examination, and electrocardiogram, including QT interval corrected for heart rate (QTcF).
Statistical analyses
Renal impairment study
An analysis of variance (ANOVA) with renal function group as a fixed effect was performed on log-transformed values of Cmax, AUCt, and AUCinf to compare results between subjects with severe renal impairment (test group) and subjects with normal renal function (reference group). Point estimates and 90% confidence intervals (CIs) for the ratios of the geometric least-squares (LS) means of the PK parameters were calculated by applying exponential transformation to the difference between LS means and 90% CIs obtained from the ANOVA model. No formal a priori criteria for between-group differences were defined for the PK analysis. The relationship between PK parameters and renal function (eGFR and CLcr) was investigated using linear regression analysis.
Hepatic impairment study
An ANOVA using hepatic impairment group as a fixed effect was performed on log-transformed values of Cmax, AUCt, and AUCinf to compare results between subjects with moderate hepatic impairment (test group) and subjects who had normal hepatic function (reference group). Point estimates and 90% CIs for the ratios of the geometric LS means of the PK parameters were calculated as described above for the renal impairment study.
Changes to study protocols
The original study protocol for the renal impairment study was revised to align with the classification of normal renal function listed in the US Food and Drug Administration’s Guidance for Industry, Pharmacokinetics in Patients With Impaired Renal Function.22 The age-matching criteria were revised to align with the classification of normal renal function and the expected subject population.
The hepatic impairment study was conducted according to the original protocol.
Results
Subject disposition and baseline demographics and clinical characteristics
Sixteen subjects were enrolled and completed dosing in each study (severe renal impairment, n=8 [not on dialysis; eGFR <30 mL/min/1.73 m2]; normal renal function, n=8 [estimated CLcr ≥90 mL/min]; moderate hepatic impairment, n=8 [Child-Pugh class B]; normal hepatic function, n=8).
As expected, because of the matching process, age, sex, weight, and race were well matched between groups in the renal impairment study (Table 1). Subjects with normal renal function were slightly taller than those with severe renal impairment. In subjects with severe renal impairment, eGFR ranged from 13.6 to 27.9 mL/min/1.73 m2 and CLcr ranged from 22.8 to 44.4 mL/min.
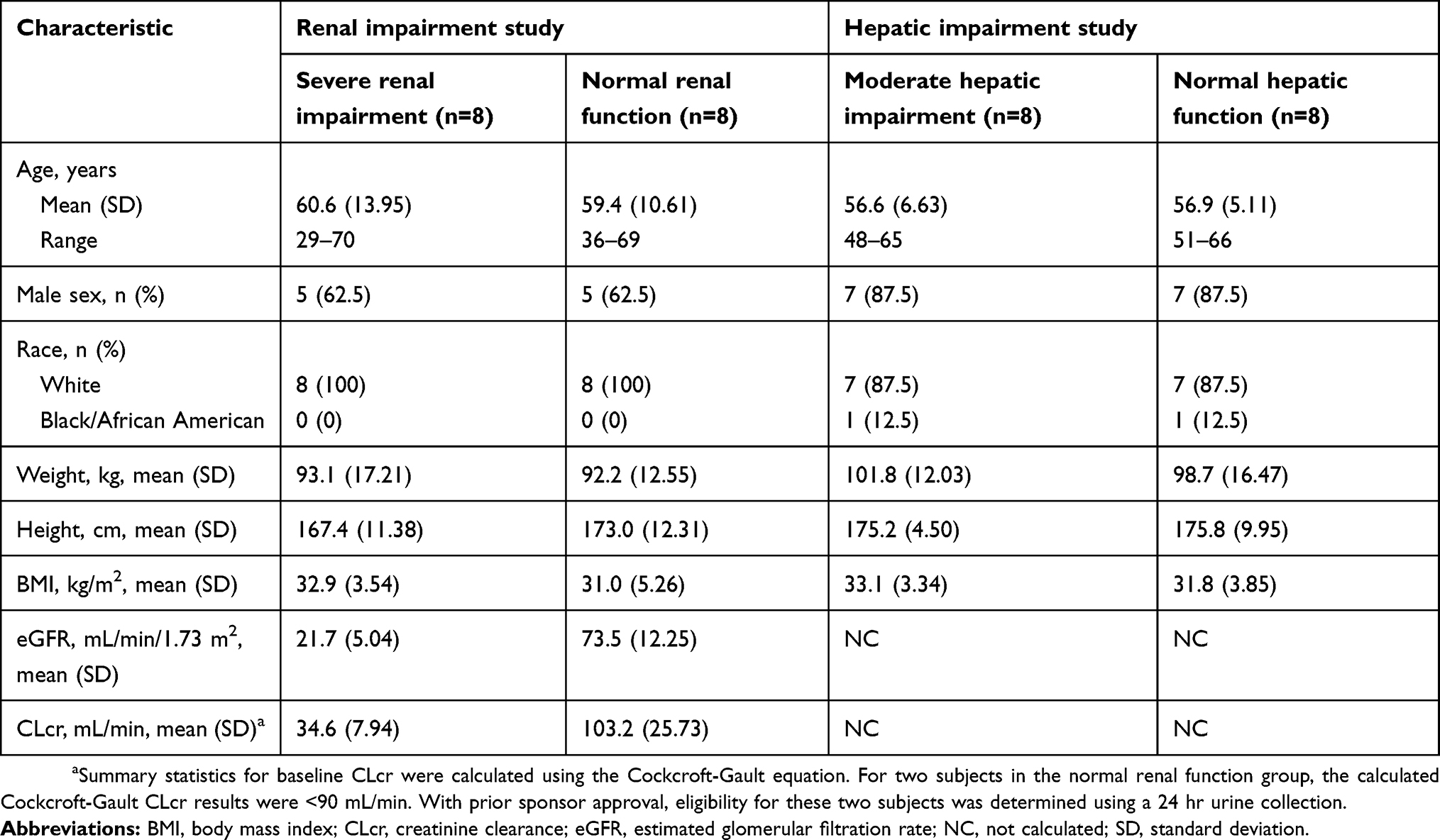 |
Table 1 Baseline demographics and clinical characteristics |
In the hepatic impairment study, baseline demographic characteristics were also well matched between groups (Table 1). Subjects with moderate hepatic impairment had a Child-Pugh B score of 7–9 and hepatic encephalopathy of grade 3 or less. Half (50%) had moderate ascites, and mean CLcr was 118.7 mL/min.
Pharmacokinetics
Renal impairment study
Mean plasma revefenacin concentrations for subjects in the severe renal impairment group were higher than those for the normal renal function group (Figure 1A) but with high between-subject variability. After excluding data from two subjects in the normal renal function group who had very low concentrations measurable to only 4 hrs and 1 hr post-dose, respectively, the mean plasma concentration–time profiles between the two renal function groups were similar (Figure S1).
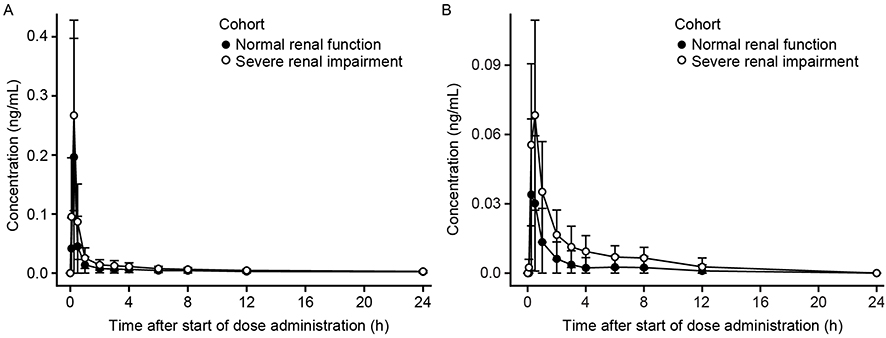 |
Figure 1 Mean ± SD plasma concentration–time profiles for revefenacin (A) and THRX-195518 (B) in subjects with and without severe renal impairment (n=8 per group). Abbreviations: h, hours; SD, standard deviation. |
In both groups, the concentration–time profile for revefenacin was characterized by a rapid absorption phase with median Tmax values of 0.250 hrs (Table 2), a rapid decline of plasma concentrations after Cmax, and then a relatively slow distribution phase and long terminal elimination phase. Mean t1/2 values were similar in subjects with normal and severely impaired renal function (Table 2). Mean revefenacin Cmax, AUCt, and population PK-predicted AUCinf values were higher in subjects with severe renal impairment than in those with normal renal function with high between-subject variability and substantial overlap in the ranges of individual values (Figure 2).
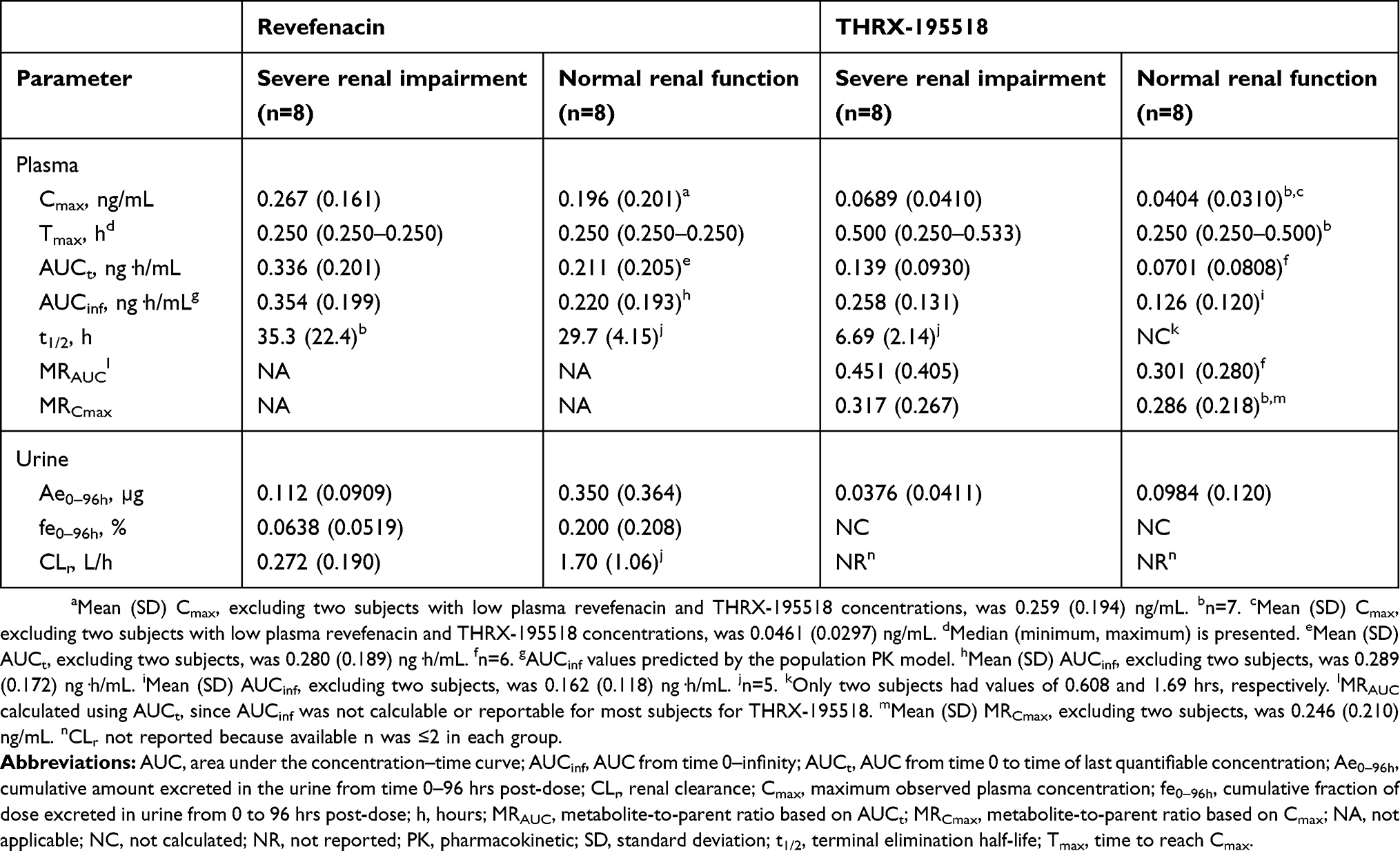 |
Table 2 Plasma and urine PK parameters (arithmetic mean [SD]) for revefenacin and THRX-195518 in renal impairment study |
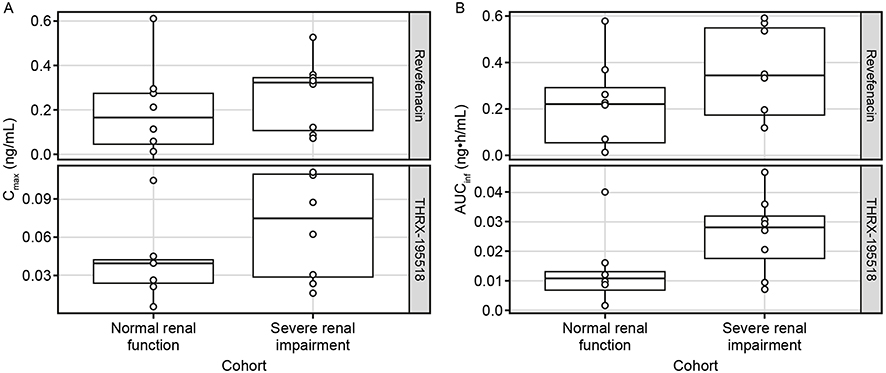 |
Figure 2 Comparison of Cmax (A) and AUCinfa (B) for revefenacin and THRX-195518 between cohorts with normal and severely impaired renal function. Notes: aAUCinf values predicted by the population PK model. Box represents the 25th–75th percentile of the data. Whiskers indicate the 1.5x interquartile range. The solid line in the box represents the median and each dot represents an individual subject.Abbreviations: AUCinf, area under the concentration–time curve from time 0 to infinity; Cmax, maximum observed plasma concentration. |
Statistical analysis showed that the ratio of geometric means (90% CI) was 234% (77–712%) for revefenacin Cmax and 337% (87–1308%) for revefenacin AUCt when comparing the severe renal impairment group with the normal renal function group. The analogous ratio of geometric means (90% CI) for revefenacin AUCinf was 244% (92–643%).
As stated previously, two subjects in the normal renal function group had very low revefenacin Cmax and AUC values. When PK data for these subjects were excluded, geometric mean ratios (90% CI) were 107% (50–227%) for Cmax, 123% (56–269%) for AUCt, and 123% (63–238%) for AUCinf.
There was no apparent relationship between renal function and revefenacin PK parameters (Figure 3). In the linear regression analyses, there were no significant correlations between eGFR and revefenacin Cmax (R2=0.107; P=0.22) or AUCinf (R2=0.121; P=0.19). Similar findings were observed for the relationship between revefenacin exposure and CLcr (Cmax: R2=0.027; P=0.54; AUCinf: R2=0.049; P=0.41).
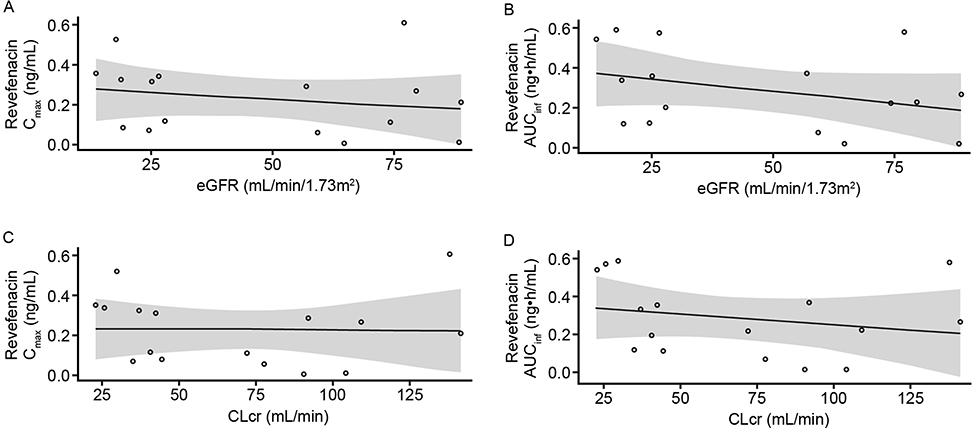 |
Figure 3 Scatter plots of revefenacin Cmax (A) and AUCinfa (B) versus eGFR. Cmax (C) and AUCinfa (D) versus CLcr in the renal impairment study. Notes: Open circles represent individual subject data. aAUCinf values predicted by the population PK model. The line indicates linear least-squares fit; the shaded area indicates 95% CI.Abbreviations: AUCinf, area under the concentration–time curve from time 0 to infinity; CI, confidence interval; CLcr, creatinine clearance; Cmax, maximum observed plasma concentration; eGFR, estimated glomerular filtration rate; PK, pharmacokinetic. |
Regarding urine PK parameters for revefenacin, mean fe0–96h was ≤0.20%, and mean CLr accounted for ≈0.3% of the total apparent clearance for subjects with normal renal function. For the severe renal impairment group, fe0–96h was lower, averaging ≈0.06%, and the mean CLr was ≈6.3-fold lower than in the normal renal function group. Overall, renal excretion of revefenacin was low in both groups, suggesting urinary excretion is a minor elimination pathway for this drug.
THRX-195518 PK parameters
Mean plasma concentrations of THRX-195518, the major metabolite of revefenacin, were higher in subjects with severe renal impairment compared with those with normal renal function (Figure 1B), but with high between-subject variability (Figure 2). After reaching Cmax at a median of 0.250–0.500 hrs (Tmax), THRX-195518 concentrations appeared to decline more slowly in subjects with severe renal impairment versus those with normal renal function (Figure S2). THRX-195518 data were limited in the normal renal function group, with quantifiable concentrations of THRX-195518 observed for only two subjects beyond 3 hrs post-dose. As a result, t1/2 could not be calculated for most subjects in this group.
PK parameters for THRX-195518 are shown in Table 2. Mean systemic exposure to THRX-195518, measured by Cmax, AUCt, and population PK-predicted AUCinf, appeared higher in subjects with severe renal impairment versus those with normal renal function. However, there was high variability in these parameters, and the overall range of individual data was similar between the two study groups (Figure 2). Revefenacin was rapidly converted to THRX-195518 after oral inhalation in both study groups, with mean MRAUC and MRCmax values ranging from 0.286 to 0.451 (Table 2).
The ratio of geometric means (90% CI) was 180% (85–380%) for THRX-195518 Cmax and 206% (72–591%) for AUCt. The analogous geometric mean ratio (90% CI) for THRX-195518 AUCinf was 274% (123–613%).
There was no apparent relationship between renal function and THRX-195518 PK parameters (Figure 4). No significant correlations were found between eGFR and THRX-195518 Cmax (R2=0.149; P=0.16) or AUCinf (R2=0.201; P=0.08) in the linear regression analyses. Similar findings were observed using CLcr (Cmax: R2=0.055; P=0.40; AUCinf: R2=0.095; P=0.25).
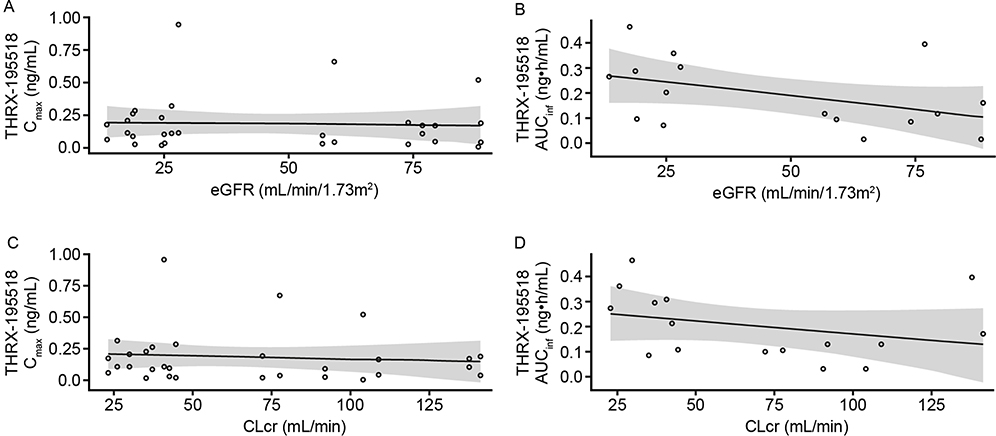 |
Figure 4 Scatter plots of THRX-195518 Cmax (A) and AUCinfa (B) versus eGFR. Cmax (C) and AUCinfa (D) versus CLcr in the renal impairment study. Notes: Open circles represent individual subject data. aAUCinf values predicted by the population PK model. The line indicates linear least-squares fit; the shaded area indicates 95% CI.Abbreviations: AUCinf, area under the concentration–time curve from time 0 to infinity; CI, confidence interval; CLcr, creatinine clearance; Cmax, maximum observed plasma concentration; eGFR, estimated glomerular filtration rate; PK, pharmacokinetic. |
In the urinary analyses, renal excretion of THRX-195518 was low in both renal function groups (Table 2). CLr was not estimable in the majority of subjects in both groups.
Hepatic impairment study
Mean plasma revefenacin concentration–time profiles were nearly superimposable between the groups with moderate hepatic impairment and normal hepatic function (Figure 5A). As in the renal function study, the profiles were characterized by a rapid absorption phase, a rapid decline in concentration after Cmax, and a relatively slow distribution and terminal elimination phase. Mean t1/2 values were similar between groups (Table 3).
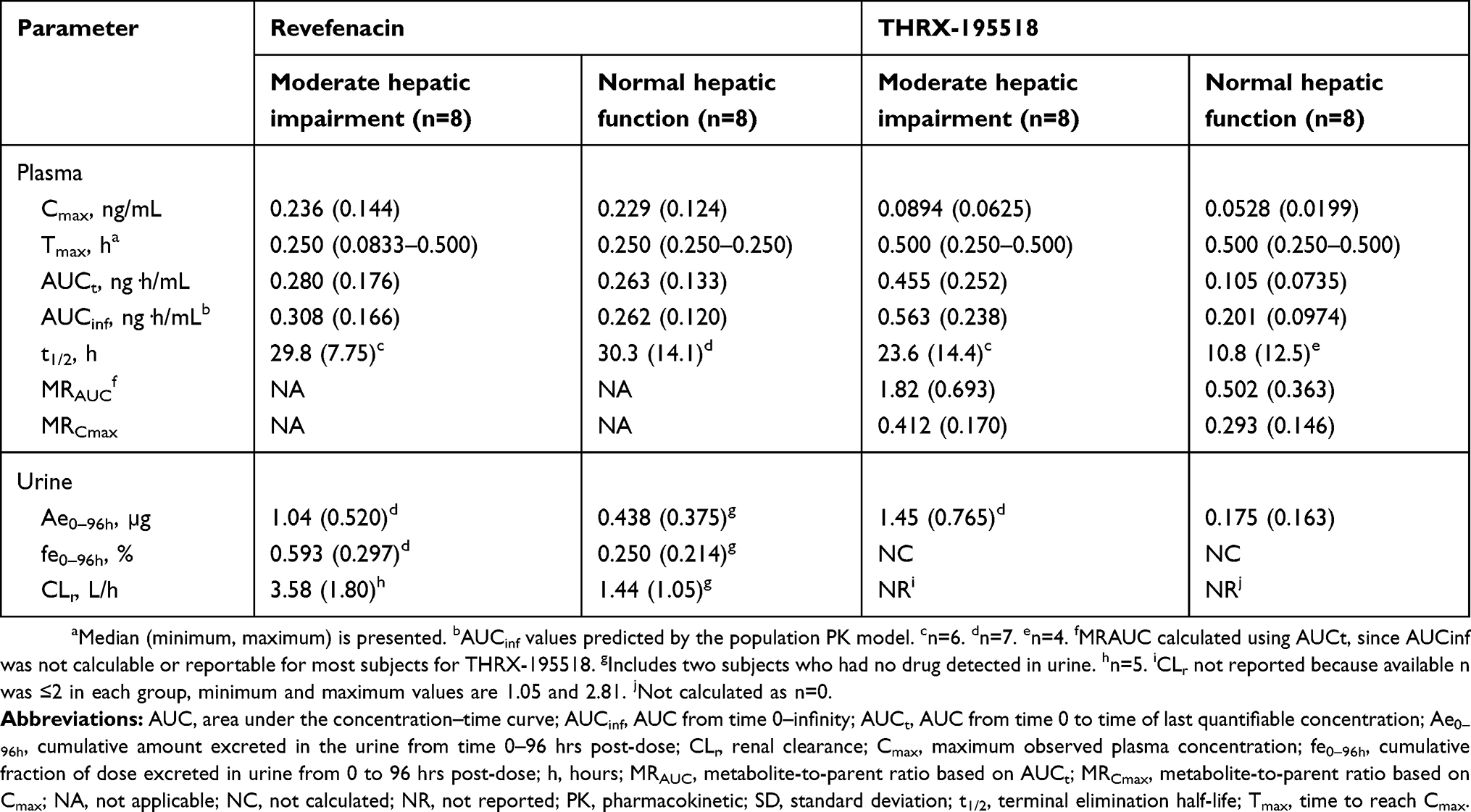 |
Table 3 Plasma and urine PK parameters (arithmetic mean [SD]) for revefenacin and THRX-195518 in hepatic impairment study |
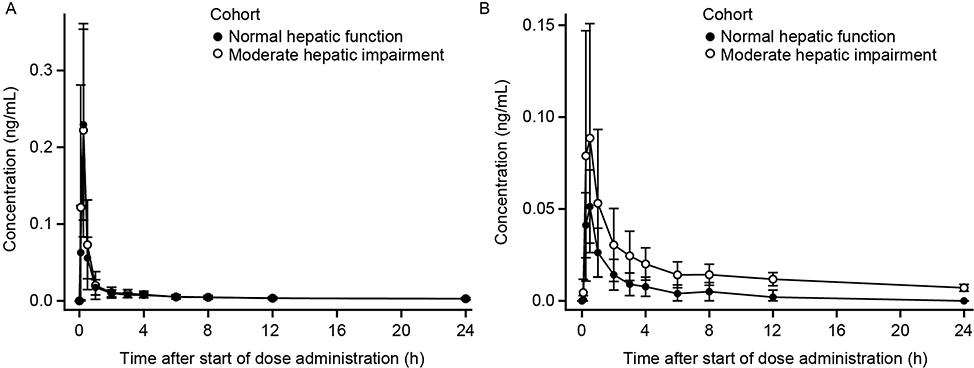 |
Figure 5 Mean ± SD plasma concentration-time profiles for revefenacin (A) and THRX-195518 (B) in subjects with and without moderate hepatic impairment (n=8 per group). Abbreviations: h, hours; SD, standard deviation. |
Systemic exposures to revefenacin (measured by mean Cmax, AUCt, and population PK-predicted AUCinf) were similar between subjects with and without hepatic impairment (Table 3), with high between-subject variability (Figure 6). The ratio of geometric means (90% CI) was 103% (57–185%) for revefenacin Cmax and 107% (60–193%) for revefenacin AUCt when comparing the moderate hepatic impairment group with the normal hepatic function group. The analogous ratio of geometric means (90% CI) for revefenacin AUCinf was 118% (72–191%).
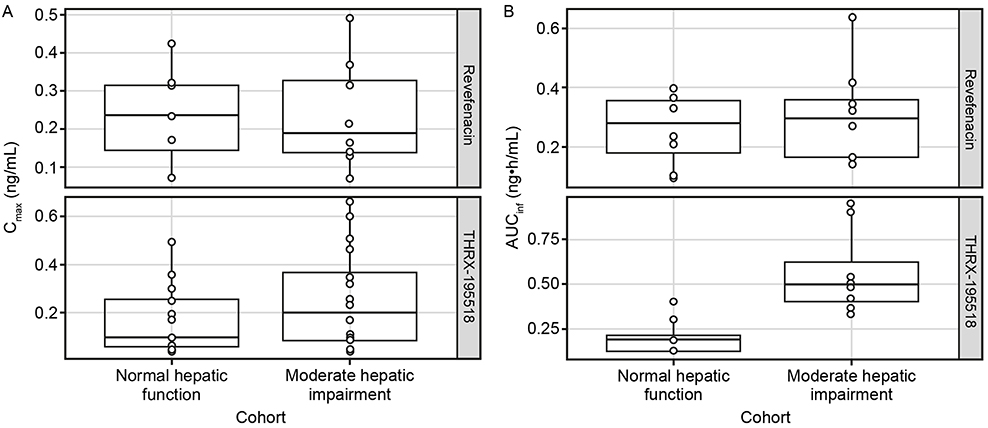 |
Figure 6 Comparison of Cmax (A) and AUCinfa (B) for revefenacin and THRX-195518 between cohorts with normal and moderately impaired hepatic function. Notes: The box represents the 25th–75th percentile of the data. Whiskers indicate the 1.5x interquartile range. The solid line in the box represents the median, while each dot represents an individual subject. aAUCinf values predicted by the population PK model.Abbreviations: AUCinf, area under the concentration–time curve from time 0 to infinity; Cmax, maximum observed plasma concentration; PK, pharmacokinetic. |
In the urinary analyses for revefenacin, mean fe0–96h was <1% and mean CLr accounted for <1% of the total apparent clearance for both hepatic function groups, again indicating that urinary excretion is a minor elimination pathway for revefenacin.
Mean revefenacin fe0–96h and CLr were≈2.4 and ≈2.5 times greater, respectively, for subjects with moderate hepatic impairment versus normal hepatic function. For two subjects in the normal hepatic function group, no revefenacin was excreted in urine.
Mean plasma concentrations of THRX-195518 in subjects with moderately impaired hepatic function were higher than in subjects with normal hepatic function (Figure 5B). After reaching Cmax at a median Tmax of 0.5 hrs, plasma THRX-195518 concentrations declined relatively slowly in subjects with moderate hepatic impairment compared with those with normal hepatic function, with an approximately twofold higher mean t1/2 (Table 3).
Mean systemic exposure to THRX-195518 appeared higher in subjects with moderate hepatic impairment compared with subjects with normal hepatic function (Table 3). For population PK-predicted AUCinf, there was minimal overlap in the ranges of individual values between the two hepatic function groups (Figure 6). The mean MRAUC value was higher for the moderate hepatic impairment group than for the normal hepatic function group (Table 3).
The ratio of geometric means (90% CI) was 150% (96–233%) for THRX-195518 Cmax and 467% (283–772%) for THRX-195518 AUCt, indicating higher THRX-195518 systemic exposure following revefenacin administration in subjects with moderate hepatic impairment than in subjects with normal hepatic function. Similarly, the analogous geometric mean ratio (90% CI) for THRX-195518 AUCinf was 284% (198–408%).
Urinary excretion was low in both hepatic function groups, but a greater amount of THRX-195518 (≈8.3-fold higher) was excreted in urine among subjects with moderate hepatic impairment (Table 3).
Because AUC0–96h could not be reported (values were either not calculable or were >20% extrapolated), CLr could not be calculated for any subjects in the normal hepatic function group and for only two subjects in the moderately impaired group. Therefore, CLr cannot be compared between groups in this study.
Safety
All TEAEs in both studies were mild to moderate in severity. There were no severe TEAEs, serious TEAEs, deaths, or TEAEs leading to permanent or temporary interruption of study drug.
In the renal impairment study, TEAEs were reported by two subjects (25.0%) with normal renal function and two subjects (25.0%) with severe renal impairment. Two TEAEs of dizziness and headache in the normal renal function group and three TEAEs of upper respiratory tract infection, dizziness, and throat irritation in the severe renal impairment group were considered possibly/probably treatment related.
In the hepatic impairment study, TEAEs were reported by one subject (12.5%) with normal hepatic function and two subjects (25.0%) with moderate hepatic impairment. One TEAE of mild chest discomfort in the normal hepatic function group and occurrences of diarrhea and dizziness in the moderate hepatic impairment group were considered possibly/probably treatment related. In the moderate hepatic impairment group, a significant TEAE of dizziness led to a fall and foot fracture and avulsion fracture; only the TEAE of mild dizziness was assessed as possibly/probably related to study drug.
In each study, no clinically meaningful mean changes from baseline were noted in hematology or serum chemistry values or in urinalysis values, and no clinically notable trends were noted in vital sign measurements. No subject had a QTcF interval >500 msec or a QTcF interval change from baseline >60 msec in either study.
Discussion
We conducted two studies to assess PK properties and safety of inhaled revefenacin in subjects with renal or hepatic impairment. In the renal impairment study, mean Cmax, AUCt, and AUCinf values for revefenacin and its metabolite THRX-195518 were higher in subjects with severe renal impairment than in subjects with normal renal function. However, between-subject variability was high and considerable overlap in individual subject PK parameter values between the two groups was observed, suggesting a minimal effect of renal impairment on revefenacin PK.
The finding of higher mean revefenacin systemic exposure among the renally impaired subjects may have been driven by the very low Cmax and AUC values in two subjects with normal renal function, whose revefenacin concentrations were measurable to only 4 hrs and 1 hr post-dose, respectively. Although there was no documented cause for these low drug levels, it is possible that the subjects were underdosed due to improper oral inhalation technique with the jet nebulizer. Exclusion of these two subjects resulted in mean Cmax, AUCt, and AUCinf values for the normal renal function group similar to those in the impaired renal function group. Renal excretion of revefenacin was low (mean fe0–96h ≤0.2%) in both groups. There were no apparent correlations between baseline renal function (measured by eGFR or CLcr) and the revefenacin or THRX-195518 PK parameters Cmax or AUC.
In the hepatic impairment study, systemic exposure to revefenacin, measured by mean Cmax, AUCt, and AUCinf, was similar for subjects with moderate hepatic impairment and normal hepatic function. However, systemic exposure of THRX-195518 was somewhat higher in the moderate hepatic impairment group than in the normal hepatic function group. Urinary excretion of revefenacin was very low (mean fe0–96h ≤0.6%) in both groups.
Similar to previous PK studies of revefenacin in COPD patients,7,8 the concentration–time profile of revefenacin in healthy subjects with normal organ function in the current studies was characterized by a rapid absorption phase and rapid initial decline of plasma concentrations after Cmax, followed by a relatively slow distribution phase and long terminal elimination phase. Revefenacin appeared to be rapidly converted to its metabolite THRX-195518.
Mean Cmax and AUC values for revefenacin in COPD patients were approximately 60% lower than in healthy subjects in these organ impairment studies at the same dose level (175 µg), whereas mean THRX-195518 Cmax and AUC values were approximately twofold higher in COPD patients than in healthy subjects.7,8 This finding of lower revefenacin exposure in COPD patients is consistent with observations for other inhaled LAMAs (umeclidinium, tiotropium), the corticosteroids budesonide and fluticasone propionate, and the LABAs olodaterol and AZD3199.23–26 The observed increased exposure to the metabolite THRX-195518 in COPD patients relative to healthy subjects may be due to a difference in elimination and/or possibly the formation of THRX-195518.7,8 Mean t1/2 for revefenacin was approximately 30 hrs across groups in these studies, within the range of values reported in COPD patients.7,8
Overall, these results show a modest effect of severe renal impairment or moderate hepatic impairment on systemic exposure to inhaled revefenacin, as measured by Cmax and AUC. Exposure to THRX-195518, based on estimated AUCinf, was approximately twofold higher for renally impaired subjects and threefold higher for hepatically impaired subjects compared with those with normal renal and hepatic function. This suggests that elimination of THRX-195518 is somewhat decreased due to severe renal or moderate hepatic impairment.
As in previous clinical studies of revefenacin among patients with COPD,7,8 TEAEs were relatively uncommon in these two studies (occurring in ≤25% of the subjects in each group) and were mild to moderate in severity. There was little difference between groups in occurrence of TEAEs. Dizziness occurred in 3 of 32 subjects and was considered significant in one case. Headache, the most common AE in the previous studies,7,8 occurred in one subject in these studies.
Revefenacin has exhibited a favorable clinical safety profile over a wide range of doses (up to 700 µg for 7 days) with no incidence of antimuscarinic AEs.7,8,27 The observed increase in THRX-195518 exposure due to hepatic or renal impairment is within that observed in prior studies at four times the therapeutic dose (700 µg)8 and is not expected to result in a clinically meaningful increase in AEs. Further, the systemic levels of THRX-195518 are low after inhaled administration of revefenacin, and the antimuscarinic potency of THRX-195518 is weaker as compared with revefenacin. Therefore, the observed increase in THRX-195518 exposure due to hepatic or renal impairment is unlikely to be of clinical consequence.
The safety of revefenacin has not been evaluated in COPD patients with hepatic impairment and therefore its use in this population is not recommended.11 However, exploratory population PK analyses of the effect of intrinsic and extrinsic factors on the PK of revefenacin and THRX-195518 in COPD patients receiving 88 or 175 μg daily dose of inhaled revefenacin showed no effect of elevated plasma aspartate aminotransferase (AST), alanine aminotransferase (ALT), or bilirubin levels on the PK parameters of revefenacin and THRX-195518.20 Mild hepatic impairment was defined as plasma ALT, AST, or total bilirubin levels above the upper limit of normal (ALT ≥44 IU/mL for males and ≥32 IU/mL for females; AST ≥40 IU/mL or total bilirubin ≥21 µMol/L) at the start of treatment. Revefenacin and THRX-195518 steady-state AUC values in patients with mild hepatic impairment (n=59) were ≤15% higher than in patients with normal hepatic function (n=749) (Theravance Biopharma, data on file).
There are limitations to the interpretation and applicability of the data from these studies. Although the sample size was small for both studies, it is consistent with recommendations in regulatory guidelines,22,28–30 and considered sufficient to meet study objectives. It is assumed that the findings in otherwise healthy subjects with organ impairment are representative of those in COPD patients, despite the observed difference in exposure between COPD patients and healthy subjects. Additionally, the effect of mild and severe hepatic impairment on revefenacin PK was not studied, and our findings from subjects with moderate hepatic impairment may not extend to these populations.
Conclusion
In the two studies, systemic exposure to revefenacin was modestly increased by severe renal impairment, while exposure to its major metabolite, THRX-195518, was approximately twofold higher than in healthy subjects. In subjects with moderate hepatic impairment, systemic exposure to revefenacin was similar as compared with the normal hepatic function group, while exposure to THRX-195518 was approximately threefold higher. Collectively, the results from these clinical pharmacology studies, along with the low antimuscarinic potency and low systemic levels of THRX-195518 after inhaled revefenacin administration, the short-term safety data collected in COPD patients at doses providing plasma THRX-195518 exposures similar to those seen in healthy subjects with moderate hepatic impairment, and the population PK covariate analyses assessing clinical signs of renal and hepatic function impairment in COPD patients suggest that the observed increase in THRX-195518 exposure in subjects with severe renal or moderate hepatic impairment is unlikely to be of clinical consequence.
Abbreviations
AE, adverse event; Ae0–96h, cumulative amount excreted in the urine from time 0–96 hrs post dose; ANOVA, analysis of variance; AUC, area under the concentration–time curve; AUCinf, AUC from time 0 to infinity; AUCt, from time 0 to time of last quantifiable concentration; CI, confidence interval; CLcr, creatinine clearance; CLr, renal clearance; Cmax, maximum observed plasma concentration; COPD, chronic obstructive pulmonary disease; eGFR, estimated glomerular filtration rate; fe0–96h, cumulative fraction of dose excreted in urine from 0 to 96 hrs post dose; GCP, Good Clinical Practice; ICH, International Council on Harmonisation; IV, intravenous; LLOQ, lower limit of quantification; LS, least-squares; MDRD, Modification of Diet in Renal Disease; t1/2, terminal elimination half-life; MRAUC, metabolite-to-parent ratio based on AUCt; MRCmax, metabolite-to-parent ratio based on Cmax; PK, pharmacokinetic; QTcF, QT interval corrected for heart rate; SD, standard deviation; Tmax, time to reach Cmax; TEAE, treatment-emergent AE.
Data sharing statement
Theravance Biopharma (and its affiliates) will not be sharing individual de-identified participant data or other relevant study documents.
Acknowledgment
This study was funded by Theravance Biopharma R&D, Inc. (George Town, Cayman Islands). Mylan Inc. (Canonsburg, PA, USA) and Theravance Biopharma US, Inc. (South San Francisco, CA, USA) funded medical writing support. The authors would like to thank Thomas C Marbury (Orlando Clinical Research Center, Orlando, FL, USA) and William B Smith (New Orleans Center for Clinical Research, Knoxville, TN, USA) for their support in the conduct of the trials. The authors acknowledge Lisa Baker, PhD, and Ritu Pathak, PhD, for medical writing and Paula Stuckart for editorial assistance in the preparation of the manuscript (Ashfield Healthcare Communications, Middletown, CT, USA).
Author contributions
All authors provided significant contributions to the conception and/or design of the work or the acquisition, analysis, or interpretation of data for the work, drafted the work or revised it critically for important intellectual content, provided final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
MTB is a consultant for Theravance Biopharma US, Inc. AL and DLB are employees of Theravance Biopharma US, Inc. CNB and SP were employees of Theravance Biopharma US, Inc. at the time this study was conducted. The authors report no other conflicts of interest in this work.
References
1. Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease; (2018 report). Available from: http://goldcopd.org/wp-content/uploads/2017/11/GOLD-2018-v6.0-FINAL-revised-20-Nov_WMS.pdf.
2. Mejza F, Gnatiuc L, Buist AS, et al. Prevalence and burden of chronic bronchitis symptoms: results from the BOLD study. Eur Respir J. 2017;50(5):1700621. doi:10.1183/13993003.00711-2017
3. Naud J, Nolin TD, Leblond FA, Pichette V. Current understanding of drug disposition in kidney disease. J Clin Pharmacol. 2012;52(1 Suppl):10s–22s. doi:10.1177/0091270011413588
4. Bonini M, Usmani OS. The importance of inhaler devices in the treatment of COPD. COPD Res Pract. 2015;1(1):9. doi:10.1186/s40749-015-0011-0
5. Dhand R, Dolovich M, Chipps B, Myers TR, Restrepo R, Farrar JR. The role of nebulized therapy in the management of COPD: evidence and recommendations. COPD. 2012;9(1):58–72. doi:10.3109/15412555.2011.630047
6. Tashkin DP. A review of nebulized drug delivery in COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:2585–2596. doi:10.2147/COPD.S114034
7. Pudi KK, Barnes CN, Moran EJ, Haumann B, Kerwin E. A 28-day, randomized, double-blind, placebo-controlled, parallel group study of nebulized revefenacin in patients with chronic obstructive pulmonary disease. Respir Res. 2017;18(1):182. doi:10.1186/s12931-017-0647-1
8. Quinn D, Barnes CN, Yates W, et al. Pharmacodynamics, pharmacokinetics and safety of revefenacin (TD-4208), a long-acting muscarinic antagonist, in patients with chronic obstructive pulmonary disease (COPD): results of two randomized, double-blind, phase 2 studies. Pulm Pharmacol Ther. 2018;48:71–79. doi:10.1016/j.pupt.2017.10.003
9. Ferguson GT, Feldman G, Pudi KK, et al. Improvements in lung function with nebulized revefenacin in the treatment of patients with moderate to very severe COPD: results from two replicate phase III clinical trials. Chronic Obstr Pulm Dis. 2019;6:154–165. doi:10.15326/jcopdf.6.2.2018.0152
10. Donohue JF, Kerwin E, Sethi S, et al. Revefenacin, a once-daily, lung-selective, long-acting muscarinic antagonist for nebulized therapy: safety and tolerability results of a 52-week phase 3 trial in moderate to very severe chronic obstructive pulmonary disease. Respir Med. 2019;153:38–43.
11. YUPELRI (revefenacin) Inhalation Solution, for Oral Inhalation [prescribing Information]. Morgantown, WV: Mylan Speciality L.P.; 2018. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210598s000lbl.pdf. Accessed November 16, 2018.
12. Bourdet D, Yeola S, Colson P-J, Pendyala S, Barnes C, Borin M. Absorption, distribution, metabolism, and excretion of revefenacin, a long-acting muscarinic antagonist, in healthy male subjects.
13. Dreisbach AW. The influence of chronic renal failure on drug metabolism and transport. Clin Pharmacol Ther. 2009;86(5):553–556. doi:10.1038/clpt.2009.163
14. Zhang Y, Zhang L, Abraham S, et al. Assessment of the impact of renal impairment on systemic exposure of new molecular entities: evaluation of recent new drug applications. Clin Pharmacol Ther. 2009;85(3):305–311. doi:10.1038/clpt.2008.208
15. Lin S, Smith BS. Drug dosing considerations for the critically ill patient with liver disease. Crit Care Nurs Clin North Am. 2010;22(3):335–340. doi:10.1016/j.ccell.2010.04.006
16. Palatini P, De Martin S. Pharmacokinetic drug interactions in liver disease: an update. World J Gastroenterol. 2016;22(3):1260–1278. doi:10.3748/wjg.v22.i37.8314
17. Verbeeck RK, Horsmans Y. Effect of hepatic insufficiency on pharmacokinetics and drug dosing. Pharm World Sci. 1998;20(5):183–192. doi:10.1023/A:1008656930082
18. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Integrated addendum to ICH E6(R1): Guideline for Good Clinical Practice. E6 (R2); June 11, 2015. Available from: https://goo.gl/CFOmR3.
19. World Medical Association. World medical association declaration of helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–2194. doi:10.1001/jama.2013.281053
20. Lo A, Borin MT, Bourdet DL. Sequential modeling of the population pharmacokinetics of revefenacin and its major metabolite in COPD patients.
21. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Guideline for Good Clinical Practice E6(R1); June 10, 1996. Available from: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf.
22. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry: Pharmacokinetics in Patients with Impaired Renal Function — Study Design, Data Analysis, and Impact on Dosing and Labeling; March, 2010. Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM204959.pdf.
23. Bjermer L, Kuna P, Jorup C, Bengtsson T, Rosenborg J. Clinical pharmacokinetics of AZD3199, an inhaled ultra-long-acting beta2-adrenoreceptor agonist (uLABA). Drug Des Devel Ther. 2015;9:753–762. doi:10.2147/DDDT.S66049
24. Borghardt JM, Weber B, Staab A, Kunz C, Kloft C. Model-based evaluation of pulmonary pharmacokinetics in asthmatic and COPD patients after oral olodaterol inhalation. Br J Clin Pharmacol. 2016;82(3):739–753. doi:10.1111/bcp.12975
25. Brutsche MH, Brutsche IC, Munawar M, et al. Comparison of pharmacokinetics and systemic effects of inhaled fluticasone propionate in patients with asthma and healthy volunteers: a randomised crossover study. Lancet. 2000;356(9229):556–561. doi:10.1016/S0140-6736(00)02581-2
26. Dalby C, Polanowski T, Larsson T, Borgström L, Edsbäcker S, Harrison TW. The bioavailability and airway clearance of the steroid component of budesonide/formoterol and salmeterol/fluticasone after inhaled administration in patients with COPD and healthy subjects: a randomized controlled trial. Respir Res. 2009;10:104. doi:10.1186/1465-9921-10-31
27. Ferguson G, Pudi KK, Feldmann G, et al. Efficacy and safety of revefenacin (REV), a long-acting muscarinic antagonist for nebulization: results of replicate randomized, double-blind, placebo-controlled, parallel-group phase 3 trials in participants with moderate to very severe COPD. Eur Respir J. 2017;50(Suppl 61):PA1812. doi:10.1183/13993003.00711-2017
28. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry: Pharmacokinetics in Patients with Impaired Hepatic Function — Study Design, Data Analysis, and Impact on Dosing and Labeling; May, 2003. https://www.fda.gov/media/71311/download.
29. European Medicines Agency. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function; 2014; https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-evaluation-pharmacokinetics-medicinal-products-patients-decreased-renal-function_en.pdf.
30. European Medicines Agency. Guideline on the Evaluation of the Pharmacokinetics of Medicinal Products in Patients with Impaired Hepatic Function; 2005; https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-evaluation-pharmacokinetics-medicinal-products-patients-impaired-hepatic-function_en.pdf.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.