")
Back to Journals » Clinical Ophthalmology » Volume 11
Pharmacokinetics and safety of olopatadine hydrochloride 0.77% in healthy subjects with asymptomatic eyes: data from 2 independent clinical studies
Authors Meier E , Narvekar A, Iyer GR , DuBiner HB, Vutikullird A, Wirta D, Sall K
Received 4 November 2016
Accepted for publication 16 February 2017
Published 10 April 2017 Volume 2017:11 Pages 669—681
DOI https://doi.org/10.2147/OPTH.S126690
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Edward Meier,1 Abhijit Narvekar,2 Ganesh R Iyer,2 Harvey B DuBiner,3 Apinya Vutikullird,4 David Wirta,5 Kenneth Sall6
1Apex Eye, Mason, OH, 2Alcon Research Ltd., Fort Worth, TX, 3Clayton Eye Center, Morrow, GA, 4WCCT Global, Cypress, 5Eye Research Foundation, Newport Beach, 6Sall Eye Research Medical Center, Artesia, CA, USA
Purpose: To assess the pharmacokinetics and safety of hydrochloride ophthalmic solution 0.77% olopatadine from 2 independent (Phase I and Phase III, respectively) clinical studies in healthy subjects.
Materials and methods: The Phase I, multicenter, randomized (2:1), vehicle-controlled study was conducted in subjects ≥18 years old (N=36) to assess the systemic pharmacokinetics of olopatadine 0.77% following single- and multiple-dose exposures. The Phase III, multicenter, randomized (2:1), vehicle-controlled study was conducted in subjects ≥2 years old (N=499) to evaluate long-term ocular safety of olopatadine 0.77%. Subjects received olopatadine 0.77% or vehicle once daily bilaterally for 7 days in the pharmacokinetic study and 6 weeks in the safety study.
Results: In the pharmacokinetic study, olopatadine 0.77% was absorbed slowly and reached a peak plasma concentration (Cmax) of 1.65 ng/mL following single-dose and 1.45 ng/mL following multiple-dose exposures in 2 hours (time to reach maximum plasma concentration [Tmax]). After reaching peak concentrations, olopatadine showed a similar mono-exponential decay following single and multiple doses with mean elimination half-life ranging from 2.90 to 3.40 hours. No accumulation in olopatadine exposure (Cmax and area under the plasma concentration–time curve from 0 to 12 hours) was evident after multiple doses when compared to single dose. In the safety study, treatment-emergent adverse events were reported in 26.7% and 31.4% of subjects with olopatadine 0.77% and vehicle, respectively. Blurred vision was the most frequent ocular treatment-emergent adverse event in both treatment groups (olopatadine 0.77% vs vehicle, 4.8% vs 4.1%). No deaths or serious adverse events were reported during the study.
Conclusion: Olopatadine 0.77% had minimal systemic exposure or accumulation in healthy subjects and was well tolerated in both adult and pediatric subjects.
Keywords: ocular allergy, allergic conjunctivitis, olopatadine, pharmacokinetics, safety
Introduction
Ocular allergy includes a spectrum of disorders, such as seasonal allergic conjunctivitis, perennial allergic conjunctivitis, vernal keratoconjunctivitis, and atopic keratoconjunctivitis.1 Seasonal and perennial allergic conjunctivitis, collectively known as allergic conjunctivitis, are the most common forms of ocular allergy, caused by immunoglobulin E-mediated reaction to allergens.1–3 The overall prevalence of ocular allergy is reported to be ~15%–25% in the USA.1,4 Although not life threatening, symptoms of allergic conjunctivitis, such as ocular itching, redness, eyelid swelling, chemosis, and tearing, significantly impact quality of life, particularly in the pediatric population. Therefore, multiple pathways need to be targeted to effectively alleviate these symptoms.5–7
Unlike other topical ocular medications available for the management of allergic conjunctivitis, olopatadine hydrochloride (HCl) ophthalmic solution acts through multiple pathways. Olopatadine is a selective antagonist of histamine H1 receptors as well as a mast cell stabilizer and prevents histamine-induced inflammatory cytokine production by conjunctival epithelial cells.8,9 In several clinical studies, olopatadine has consistently been shown to be well tolerated and an effective medication for the treatment of allergic conjunctivitis.10–15 Olopatadine 0.1% and 0.2% (marketed as PATANOL® and PATADAY®, respectively, in the USA, by Alcon Research Ltd., Fort Worth, TX, USA) are approved for the management of ocular itching associated with allergic conjunctivitis in >100 countries, including the USA and Canada, as twice-daily and once-daily treatments, respectively.
Because of limited aqueous solubility of olopatadine HCl at neutral pH, a new ophthalmic formulation containing olopatadine HCl at an increased concentration of 0.77% (7.76 mg/mL, which is equivalent to 0.7% [7 mg/mL] olopatadine as free base) was developed to allow olopatadine HCl to remain dissolved in a stable solution.16 In a preclinical study, olopatadine was observed at higher concentrations with prolonged presence in the target tissue (rabbit conjunctiva) following dosing with olopatadine 0.77% compared to that with olopatadine 0.2%.16 The new olopatadine 0.77% formulation has demonstrated a longer, 24-hour duration of action and superior efficacy compared to olopatadine 0.2% formulation in Phase III clinical studies.14,15 This new formulation of olopatadine 0.77% (marketed as PAZEO® by Alcon Research Ltd.) was approved by the US Food and Drug Administration (FDA) in 2015 as once-daily product for the treatment of ocular itching associated with allergic conjunctivitis. The ocular safety profile of different olopatadine formulations is well documented, including the 0.77% formulation; however, long-term ocular safety data of olopatadine 0.77% in human subjects and more importantly in the pediatric subjects, who are more prone to allergic conjunctivitis, are lacking.
Here, we describe the results from both a Phase I pharmacokinetic study and a Phase III safety study, which included subjects as young as 2 years of age.
Materials and methods
Study design
The pharmacokinetic study was a Phase I, single-center, randomized, double-masked, vehicle-controlled, parallel-group, multiple-dose study (Figure 1). The study was conducted at West Coast Clinical Trials (Cypress, CA, USA) and was approved by the IntegReview Ethical Review Board, Austin, TX, USA. Subjects received 1 drop of olopatadine 0.77% or vehicle once daily in the morning for 7 days. Pre-dose blood samples were collected 30 minutes before dosing. A ±5-minute margin from the dosing time established on Day 1 was allowed for dose administration. Post-dose blood samples were collected at pre-specified time points and analyzed for the pre-defined pharmacokinetic parameters (Figure 1).
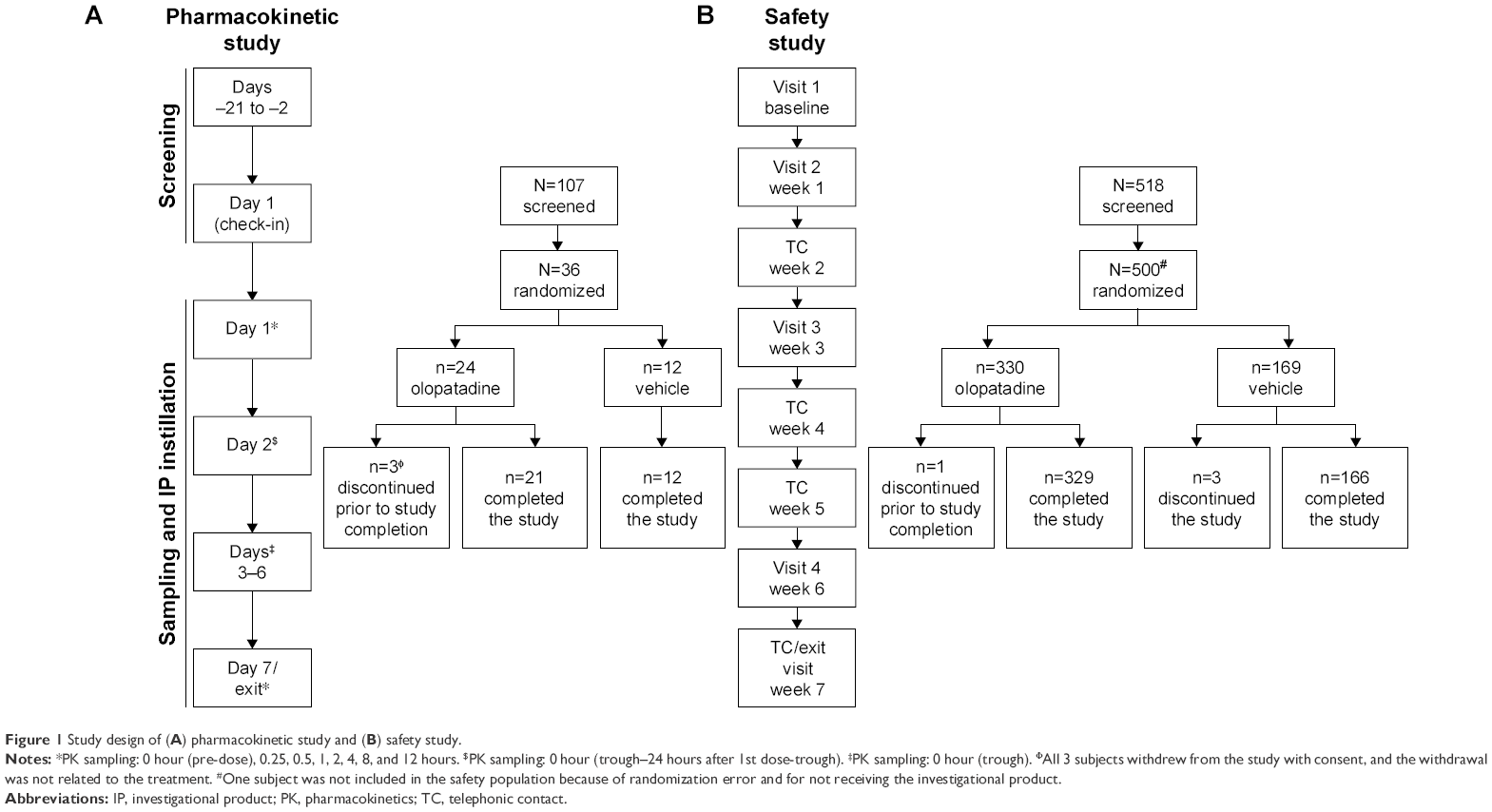 | Figure 1 Study design of (A) pharmacokinetic study and (B) safety study. |
The safety study (NCT01698814) was a Phase III, 6-week, multicenter (15 investigational sites in the USA that included eye care clinics and research centers), randomized, double-masked, vehicle-controlled, parallel-group study conducted in subjects 2 years of age and older with asymptomatic eyes (Figure 1). The study was approved by the Chesapeake Institutional Review Board, Columbia, MD, USA. Subjects received 1 drop of olopatadine 0.77% or vehicle in each eye once daily in the morning throughout the study.
In both studies, the subjects were randomized 2:1 to receive olopatadine 0.77% or vehicle. Both studies were conducted in the USA and complied with the Declaration of Helsinki 2013 and Good Clinical Practice E6 (R1) guidelines. All subjects or their parents/legal guardians (for subjects <18 years of age) provided written informed consent before entering the study.
Subjects
The Phase I pharmacokinetic study included adult subjects, at least 50% of whom were to be of Japanese ethnicity (participants of Japanese ethnicity within the third generation as proven by passport, birth certificate, or family tree) who were aged 18–65 years at the time of screening, were in good health, and agreed to comply with the study visits and dosing requirement as per study protocol. Subjects with any medical condition that may have precluded safe participation or may have affected results of the study were not enrolled. Other key exclusion criteria were history of allergy or hypersensitivity to any component of the test articles; use of any prescription or non-prescription systemic or topical medications; use of vitamins or dietary supplements within 14 days before the Day 1 visit or any prescribed drugs for psychiatric disorders within 4 months of the Day 1 visit; Fridericia-corrected QT interval >430 and >450 ms for male and female subjects, respectively, or any other significant electrocardiogram abnormality at screening visit; history of HIV, hepatitis B, or hepatitis C infection or active hepatitis A infection; values of vital signs outside of protocol-defined ranges; clinical laboratory and liver function test results at screening outside of protocol-defined ranges; systemic immunotherapy within 90 days before the Day 1 visit; body mass index <18.5 or ≥30 kg/m2; best-corrected visual acuity (BCVA) score <55 early treatment diabetic retinopathy study (ETDRS) letters; current or history of glaucoma, ocular hypertension, or intraocular pressure (IOP) <8 or >21 mmHg at Day 1 visit; current or history of chronic or recurrent severe inflammatory eye disease; current or history of severe dry eye condition in either eye; history or evidence of punctal or nasolacrimal duct stenosis or occlusion; punctal plugs in either eye; ongoing or history of clinically relevant or progressive retinal disease (eg, retinal degeneration, diabetic retinopathy, or retinal detachment) in either eye; history of ocular trauma; intraocular or laser surgery in either eye within 6 months prior to the Day 1 visit as determined by subject history and/or examination; and female subjects who were pregnant, who had a positive pregnancy test, or who were planning pregnancy during the trial period.
The Phase III safety study included subjects with asymptomatic eyes, aged ≥2 years, with BCVA ≥55 ETDRS letters at baseline (for subjects <10 years of age, a best attempt at visual acuity was made using an age appropriate measurement method in accordance with the American Academy of Pediatrics Vision Screening Guidelines), who could avoid the use of contact lens during each study visit, with no evidence of either contact lens care solution-related ocular surface damage or giant papillary conjunctivitis, and who were able and willing to comply with study protocol and follow protocol instructions. Females of child-bearing potential, who were not pregnant (negative urine pregnancy test), and were willing to adopt adequate birth control methods for the duration of the study were also eligible. Subjects with any medical condition that may have precluded safe participation or may have affected the results of the study were not enrolled. Other key exclusion criteria were any ocular infection in either eye or history of ocular infection within 30 days prior to Visit 1; use of systemic medications within 30 days prior to Visit 1; current or with history of glaucoma, ocular hypertension, or IOP <5 or >21 mmHg at Visit 1; history of retinal detachment, diabetic retinopathy, or progressive retinal disease; presence of blepharitis, active rosacea affecting the ocular adnexa, meibomian gland dysfunction, follicular conjunctivitis, IOP, or any other ophthalmic abnormality that may affect the study outcomes; corneal conditions affecting the corneal structure; history or evidence of any ocular surgical procedure within 1 year prior to Visit 1; prior (within 5 days prior to Visit 1), current, or anticipated use of ophthalmic agents other than investigational product during study participation; and known contraindications, hypersensitivities to any of the study medications, or their components.
Objectives
Primary objective of the pharmacokinetic study was to assess the pharmacokinetics and safety of single and multiple doses of olopatadine 0.77% compared with vehicle, when administered once daily in both eyes for 7 days.
Primary objective of the safety study was to evaluate the ocular safety of olopatadine 0.77% compared with vehicle, when administered once daily in both eyes for up to 6 weeks.
Assessments
In the pharmacokinetic study, plasma concentrations of olopatadine and its metabolites, N-desmethyl olopatadine and N-oxide olopatadine, were assessed on Day 1 (following the first [single] dose) and Day 7 (after multiple doses) at 0 hour before dosing and at 0.25, 0.5, 1, 2, 4, 8, and 12 hours post-dose; on Day 2 at 0 hour (24 hours after the first drop-trough sample and prior to dosing); and trough sample on Day 3 through Day 6 at 0 hour. Plasma concentrations of olopatadine and its metabolites were determined using validated high-performance liquid chromatography coupled with tandem mass spectrometry methods at Tandem Labs, Salt Lake City, UT, USA. Analytical methods were fully validated with respect to accuracy, precision, and sample stability consistent with the sample collection and storage procedures.
The single- and multiple-dose pharmacokinetic parameters estimated included peak plasma concentration (Cmax), time to reach maximum plasma concentration (Tmax), area under the plasma concentration–time curve (AUC) from 0 to 12 hours (AUC0–12), AUC from 0 to the last quantifiable concentration at time t (AUC0–t), and elimination half-life (t1/2) and the elimination rate constant (Kel). In addition, trough (24-hour pre-dose) samples were also obtained from Day 2 to Day 6. In the absence of pharmacokinetic plasma sampling beyond 12 hours after last dosing, AUC0–12 was used for assessing any accumulation following repeated daily dosing instead of AUC0–24. AUC time curve and half-life (t1/2) were estimated using non-compartmental analysis (NCA) methods.
In the safety study, the following safety variables were assessed: adverse events (AEs) and serious adverse events (SAEs) at baseline and Weeks 1–7 or at the Early Exit visit, BCVA and ocular signs (eyelids, conjunctiva, cornea, iris, anterior chamber, and lens) at baseline and Weeks 1, 3, and 6 or at the Early Exit visit, and IOP, fundus examination (optic nerve, peripheral retina, vitreous, choroid, and macula), and vital signs (blood pressure and pulse) at baseline and Week 6 or at the Early Exit visit. The safety analysis also included an evaluation of both AEs and other safety-related parameters according to age. For all AEs, an assessment of the causality (related or not related to study treatment) was determined by the investigator.
Statistical analyses
In the pharmacokinetic study, all subjects who received the test article, satisfied protocol criteria, and had ≥1 post-dose blood draw were considered evaluable for the pharmacokinetic analysis. Subjects with inadequate pharmacokinetic data, any collection, or analytical deviations that would have affected integrity of the data were excluded from pharmacokinetic analysis. Descriptive statistics were used to summarize the pharmacokinetics of olopatadine 0.77% after single and multiple doses. Non-compartmental descriptive pharmacokinetic methods and compartmental modeling methods were used for the pharmacokinetic analyses. AUC and t1/2 were estimated using NCA methods.
In the safety study, safety analysis set included all subjects who received the study treatment. Safety results were summarized descriptively. No formal statistical hypothesis testing was planned or conducted.
Results
Baseline characteristics and subject demographics
In the pharmacokinetic study, a total of 36 healthy subjects were randomized to receive either olopatadine 0.77% (n=24) or vehicle (n=12). All 24 subjects (12 Japanese and 12 non-Japanese) who received olopatadine 0.77% were included in the pharmacokinetic data, and all 36 subjects who received either olopatadine 0.77% or vehicle were included in the safety data. The rationale for recruiting at least 50% Japanese subjects to this study was that the same population has been previously evaluated for the pharmacokinetics parameters following oral administration of olopatadine. Overall, 21 (87.5%) subjects in the olopatadine 0.77% group and 12 (100%) subjects in the vehicle group completed the study. Three (8.3%) subjects discontinued study in the olopatadine 0.77% group due to consent withdrawal (Figure 1). Overall, the baseline demographic characteristics were comparable between the treatment groups (Table 1). Majority of the subjects in both treatment groups were of Asian ethnicity (≥50%).
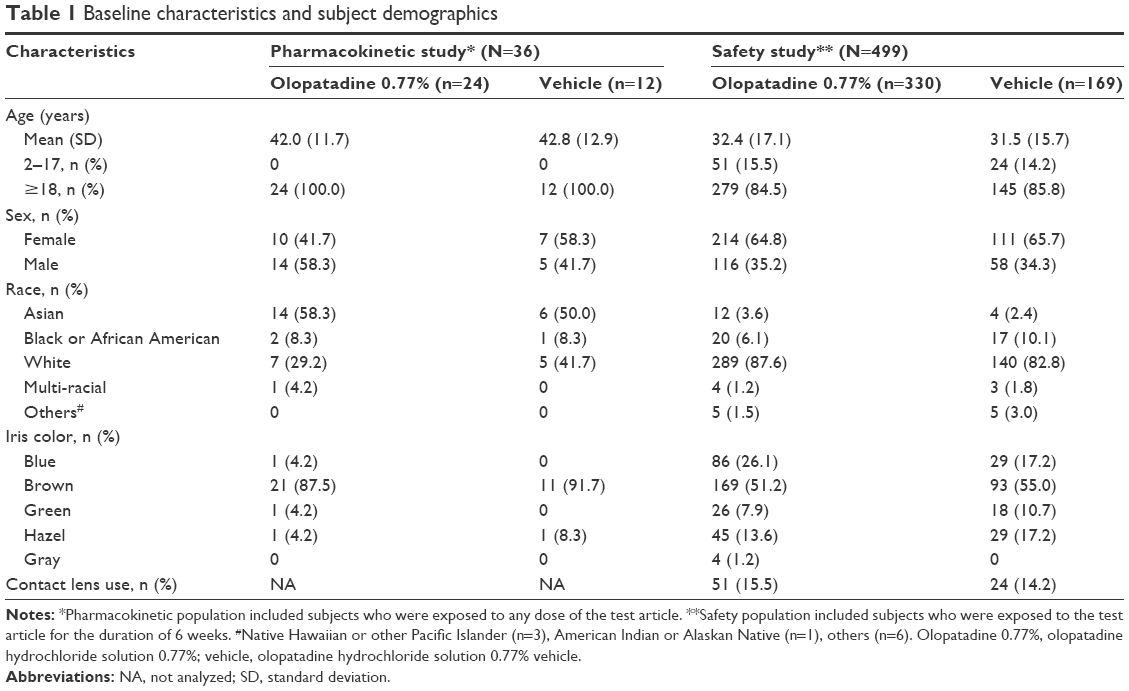 | Table 1 Baseline characteristics and subject demographics |
In the safety study, of the 500 subjects randomized, 499 (olopatadine 0.77%, n=330; vehicle, n=169) were included in the safety data. One subject in the olopatadine 0.77% group was erroneously randomized and hence not included in the safety data set. Overall, 329 (99.7%) subjects in the olopatadine 0.77% group and 166 (98.2%) in the vehicle group completed the study (Figure 1). Two (1.8%) subjects discontinued the study due to non-SAEs not related to the treatment in the vehicle group, with no discontinuations in the olopatadine 0.77% group.
Overall, the demographic characteristics were comparable between treatment groups (Table 1). In the olopatadine 0.77% and vehicle groups, 51 (15.5%) and 24 (14.2%) subjects, respectively, were aged between 2 and 17 years. The proportions of subjects using contact lenses were 15.5% and 14.2% in the olopatadine 0.77% and vehicle groups, respectively.
Single- and multiple-dose systemic pharmacokinetics of olopatadine
Olopatadine was absorbed slowly and reached a Cmax in 2 hours following bilateral topical administration of olopatadine 0.77% (Table 2). The Day 1 (single-dose) and Day 7 (multiple-dose) mean olopatadine plasma concentration vs time profile is shown in Figure 2A. After reaching peak plasma concentrations, the single- and multiple-dose olopatadine showed mono-exponential decay with similar mean t1/2 (Figure 2A; Table 2). The single- and multiple-dose individual and mean AUC0–12 are given in Figure 2B and Table 2.
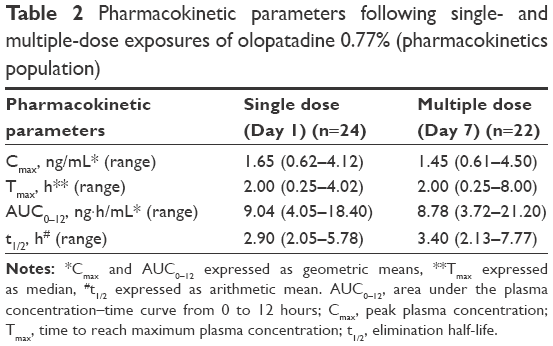 | Table 2 Pharmacokinetic parameters following single- and multiple-dose exposures of olopatadine 0.77% (pharmacokinetics population) |
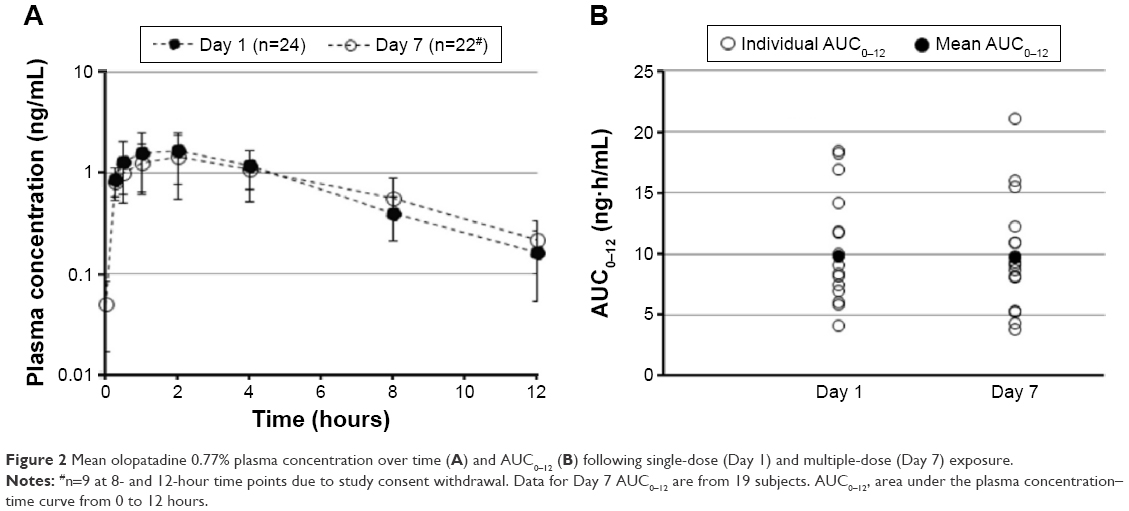 | Figure 2 Mean olopatadine 0.77% plasma concentration over time (A) and AUC0–12 (B) following single-dose (Day 1) and multiple-dose (Day 7) exposure. |
No accumulation in olopatadine exposure (Cmax and AUC0–12) was evident at Day 7 compared to Day 1 (Table 2). In addition, the maximum trough plasma concentration of olopatadine observed over the duration of treatment ranged from 0.108 to 0.247 ng/mL (individual data points not given).
The Day 1 and Day 7 pharmacokinetic parameters were comparable between the Japanese and non-Japanese subjects (Table S1). However, the Day 1 systemic olopatadine 0.77% exposure was marginally higher in the non-Japanese (Cmax, 2.07 ng/mL; AUC0–12, 10.55 ng·h/mL) than that in the Japanese subjects (Cmax, 1.75 ng/mL; AUC0–12, 9.25 ng·h/mL).
Single- and multiple-dose pharmacokinetics of olopatadine metabolites
N-desmethyl olopatadine, the minor active metabolite of olopatadine, was non-quantifiable (≤0.050 ng/mL) in the plasma samples collected from all the subjects. N-oxide olopatadine metabolite was observable up to 4 hours in 6 of 24 subjects who received olopatadine 0.77% on Day 1 and in 1 subject on Day 7. Cmax of N-oxide olopatadine observed on Day 1 and Day 7 was 0.121 and 0.174 ng/mL, respectively.
Safety data from the Phase I pharmacokinetic study
No descriptive safety data from this study have been presented here due to small sample size. In brief, no deaths or SAEs were reported during the study, and no subject discontinued the study due to an AE. No subject in the olopatadine 0.77% group reported a treatment-related AE, whereas only 1 subject in the vehicle group reported treatment-related AE (eye irritation).
Phase III safety study
Treatment-emergent adverse events (TEAEs)
Overall incidence of TEAEs was comparable between the treatment groups (Table 3). In total, 88 (26.7%) subjects from the olopatadine 0.77% group and 53 (31.4%) subjects from the vehicle group reported ≥1% TEAE (Table 3). Among the ocular TEAEs reported in the safety population, blurred vision was the most frequent (n=16; 4.8%) followed by dry eye (n=11; 3.3%) in the olopatadine 0.77% group, whereas blurred vision, corneal staining, and abnormal sensation in eye were the most frequent ocular TEAEs (n=7; 4.1% each) reported in the vehicle group. Among the non-ocular TEAEs, dysgeusia was only reported in the olopatadine 0.77% group (n=8) and had the highest incidence rate (2.4%), whereas headache, upper respiratory tract infection, and nasopharyngitis had the highest incidence rates (1.8% each) in the vehicle group.
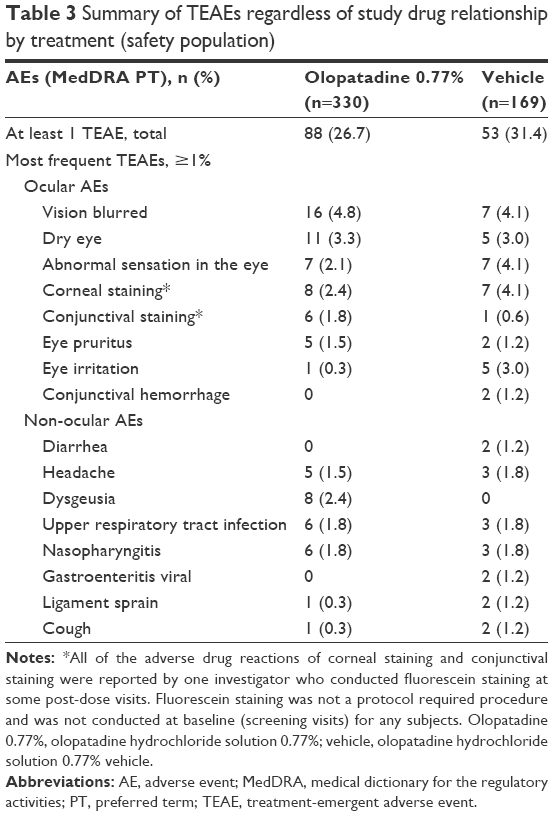 | Table 3 Summary of TEAEs regardless of study drug relationship by treatment (safety population) |
TEAEs with an incidence of ≥1% in the age groups are summarized in Table 4. Overall, the incidences of ocular TEAEs were lower in the 2–17 age group compared to the ≥18 age group in both treatment groups (Table 4). The most frequent ocular TEAEs reported in the 2–17 age group were abnormal sensation in eye and conjunctival staining (3.9% each) in the olopatadine 0.77% group and conjunctival staining, chalazion, and eye irritation (4.2% each) in the vehicle group. Among the non-ocular TEAEs reported in the 2–17 age group, upper respiratory tract infection (5.8%) and pyrexia (3.9%) were most common with olopatadine 0.77% and with vehicle, pyrexia and sinus congestion (4.2% each). In the ≥18 age group, blurred vision was the most frequent ocular TEAE (5.7%) with olopatadine 0.77%, whereas blurred vision, abnormal sensation in eye, and corneal staining were equally frequent in the vehicle group (4.8% each). No subject exposed to olopatadine 0.77% discontinued the study due to an AE, whereas 2 (1.2%) subjects in the vehicle group discontinued due to AEs not related to treatment. No deaths or other SAEs were reported during the study.
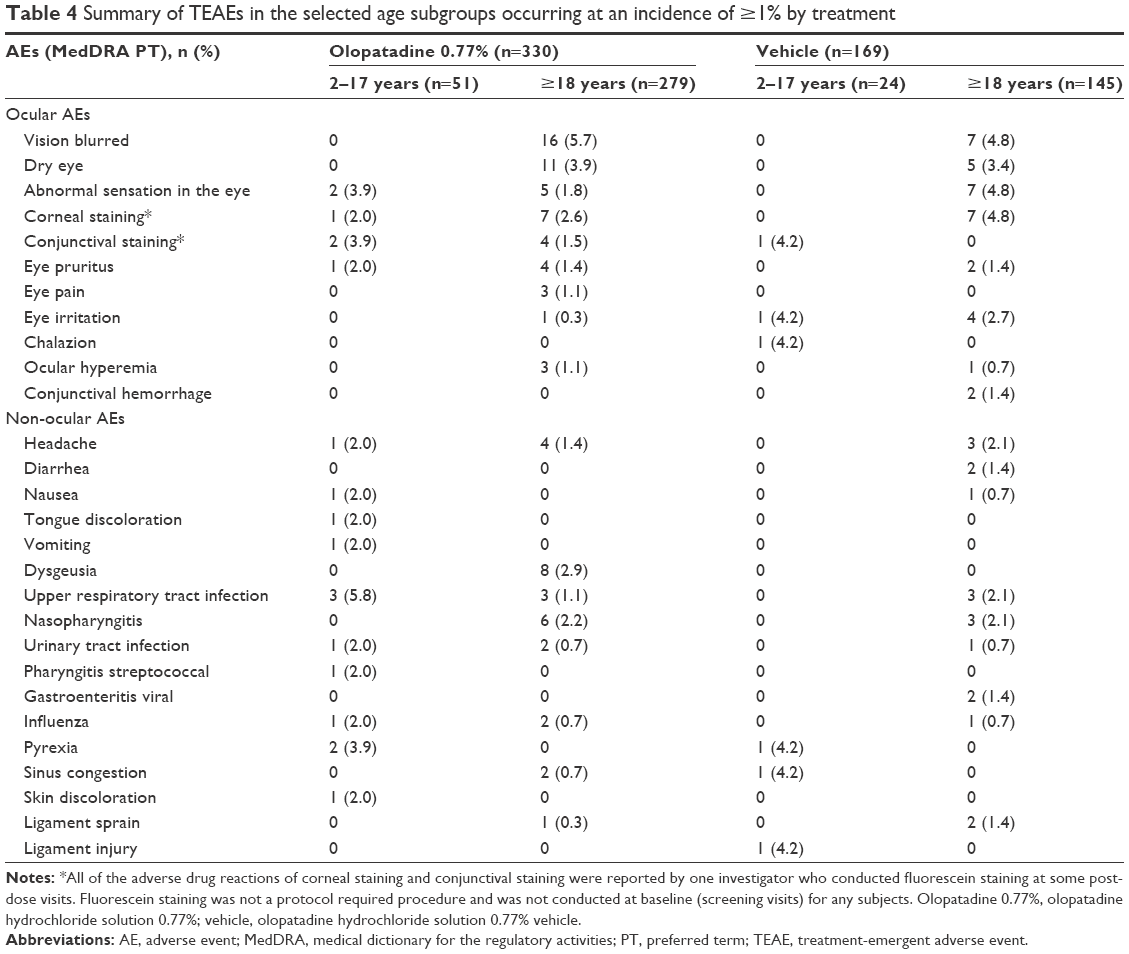 | Table 4 Summary of TEAEs in the selected age subgroups occurring at an incidence of ≥1% by treatment |
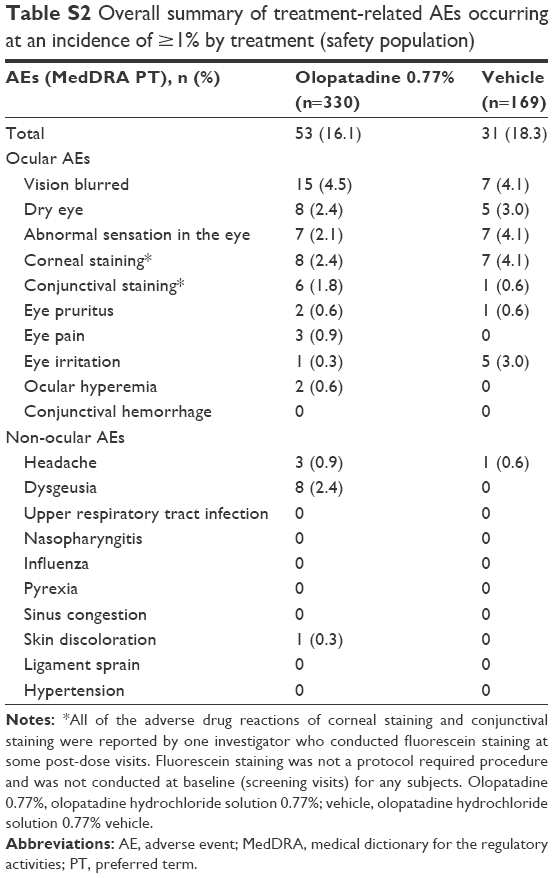
Treatment-related AEs
Overall incidence of treatment-related AEs was similar between the treatment groups: 16.1% and 18.3% with olopatadine 0.77% and vehicle, respectively (Table S2). Among the most frequent treatment-related AEs (≥1%), dysgeusia was uniquely reported with olopatadine 0.77% (n=8; 2.4%). Observed incidence of the most frequent treatment-related AEs in different age subgroups is given in Table S3. In the 2–17 age group, the most frequent ocular treatment-related AEs with olopatadine 0.77% was conjunctival staining (n=2; 3.9%), whereas with vehicle, conjunctival staining, and eye irritation were observed in 1 subject each (4.2%), as shown in Table S3.
BCVA
During the study, no discernible trend toward BCVA loss was observed in either treatment group (Table 5). Mean change in BCVA from baseline to Day 42 or at the Early Exit visit was −0.5 (±3.5) and 0.3 (±4.2) letters with olopatadine 0.77% and vehicle, respectively. No concerns were identified with respect to visual acuity change in the individual age subgroups during this study (Table 5). In the 2–17 age group, the mean BCVA change from baseline to Week 6 or at the Early Exit visit was 0.3 (±3.2) and 0.4 (±2.6) letters with olopatadine 0.77% and vehicle, respectively (Table 5).
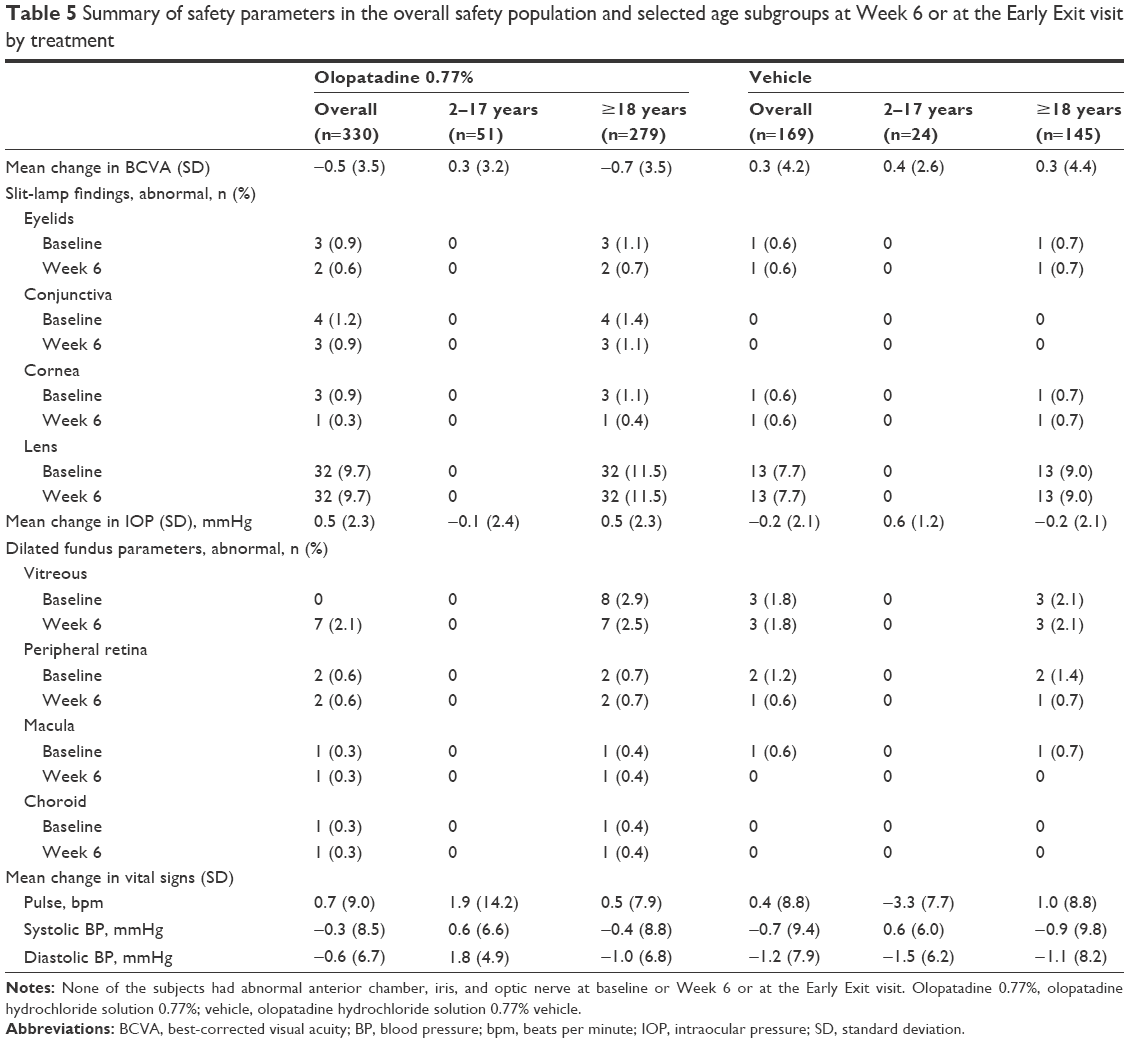 | Table 5 Summary of safety parameters in the overall safety population and selected age subgroups at Week 6 or at the Early Exit visit by treatment |
Ocular sign
No discernible trend indicating a safety concern in any ocular sign parameter was observed in the overall safety population as well as in the individual age subgroups. In the overall safety population, the number of subjects with abnormal eyelids, conjunctiva, and cornea remained largely similar from baseline to Week 6 or at the Early Exit visit in the olopatadine 0.77% group as compared to vehicle (eyelids, 0.9% vs 0.6%; conjunctiva, 1.2% vs 0.9%; cornea, 0.9% vs 0.3%; Table 5). No subject in the 2–17 age group experienced abnormal ocular sign parameters in either treatment group (Table 5).
Intraocular pressure
No clinically relevant differences were noted between olopatadine 0.77% and vehicle for changes in IOP in the overall population as well as in the individual age subgroups. Overall, mean IOP remained similar from baseline to Week 6 or at the Early Exit visit in both treatment groups, with mean changes of 0.5 (±2.3) and −0.2 (±2.1) mmHg in the olopatadine 0.77% and vehicle groups, respectively (Table 5). In the 2–17 age group, mean change in IOP from baseline to Week 6 or at the Early Exit visit was similar between the olopatadine 0.77% (−0.1 [±2.4] mmHg) and vehicle (0.6 [±1.2] mmHg) groups (Table 5).
Fundus examination
No subjects experienced a change from normal to abnormal in any dilated fundus parameters from baseline to Week 6 or at the Early Exit visit (Table 5).
Vital signs
Mean change in pulse rate and systolic and diastolic pressures by visit are summarized in Table 5. Mean change from baseline in the vital signs was minimal in both the overall population group and the age subgroups (Table 5).
Discussion
Over the past few decades, ocular allergy, particularly seasonal and perennial allergic conjunctivitis, has shown a trend toward increased prevalence, more importantly in the pediatric population.17–22 The new ophthalmic formulation of olopatadine at an increased concentration of 0.77% was developed with the aim to increase the aqueous solubility of olopatadine at neutral pH and to provide better efficacy in the management of ocular allergy compared with the previously approved olopatadine ophthalmic formulations (0.1% and 0.2%). Superior efficacy of olopatadine 0.77% over 0.1% and 0.2% formulations was recently shown in Phase III clinical trials.14,15 In a pre-clinical study, olopatadine 0.77% formulation showed improved solubility and a 4-fold increase in the concentration of olopatadine in the aqueous humor when compared to olopatadine 0.2%, with no sign of capacity-limit kinetics.16 Likewise, in the conjunctiva, a slightly greater dose proportional increase in olopatadine was observed after an ocular dose of olopatadine 0.77% compared with olopatadine 0.2%, suggesting potentially longer anti-allergic activity.
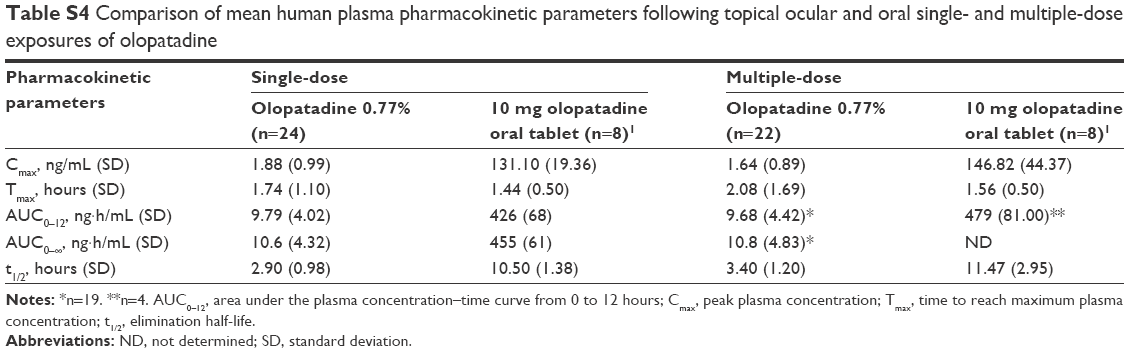
In the pharmacokinetic study, following topical ocular administration of olopatadine 0.77% once daily for 7 days, measurable olopatadine concentrations were observed at all time points after single- and multiple-dose exposures in healthy subjects. Olopatadine 0.77% was slowly absorbed in all the subjects analyzed, followed by rapid elimination from the body (single-dose geometric mean t1/2, 2.78 hours; multiple-dose geometric mean t1/2, 3.26 hours). When compared with the single- and multiple-dose oral exposure of 10 mg olopatadine, the pharmacokinetic parameters of topical ocular exposure of olopatadine 0.77% were substantially lower23 (Table S4). The mean single-dose topical ocular Cmax (1.88 ng/mL) and mean AUC0–12 (9.79 ng·h/mL) of olopatadine 0.77% were ~70- and 43.5-fold lower, respectively (mean Cmax, 131.10 ng/mL and mean AUC0–12, 426.00 ng·h/mL). Similarly, following multiple ocular doses of olopatadine 0.77%, the mean Cmax (1.64 ng/mL) and mean AUC0–12 (9.68 ng·h/mL) were ~89.5- and 49.5-fold lower than the multiple oral doses of 10 mg olopatadine, respectively (mean Cmax, 146.82 ng/mL and mean AUC0–12, 479.00 ng·h/mL).23 These data indicate that topical ocular doses of olopatadine 0.77% have a wide margin of safety since it resulted in low systemic exposure than that of oral doses of 10 mg olopatadine HCl. No statistically significant difference was observed in the pharmacokinetic parameters between Japanese and non-Japanese subjects. Plasma concentration of the metabolite N-desmethyl olopatadine was non-quantifiable and that of N-oxide olopatadine was low and sparse after topical bilateral dosing of once-daily olopatadine 0.77%.
Although the new olopatadine 0.77% ophthalmic formulation was recently shown to be well tolerated in the adult population,14,15 the safety profile of olopatadine 0.77% with long-term use in adults and safety information in the pediatric population were lacking. This safety study is the first clinical trial to describe the safety of olopatadine 0.77% with 6 weeks dosing in both the pediatric (2–17 age group) and adult (≥18 age group) populations. The overall types of AEs and their characteristics with olopatadine 0.77% were comparable with its vehicle in the overall safety population as well as in the age subgroups. Olopatadine, although administered topically, is absorbed systemically as are other topically administered medicinal products, and thus, some systemic/non-ocular side-effects are possible. The commonly reported non-ocular AEs associated with olopatadine include headache, dysgeusia, nasal dryness, and fatigue that are a known class-effect of antihistamines.24 The incidence of treatment-related non-ocular AEs with olopatadine 0.77% was low during the 6-week study period. In adults, dysgeusia, which is not an uncommon association with the instillation of eye drops and does not represent a safety concern, was the only most frequent treatment-related AE (≥1%) uniquely reported in the olopatadine 0.77% group. The similar incidence of dysgeusia (reported in 2 subjects) and other AEs reported previously following the topical ocular administration of olopatadine 0.77%14 supports the findings of this study. Other treatment-related non-ocular AEs (≥1.5%−2%) reported in this study, both with olopatadine and vehicle, were headache, nasopharyngitis, and upper respiratory tract infection. The overall incidence of AEs was comparable with the 0.1% and 0.2% ophthalmic formulations of olopatadine, albeit at different frequencies.14,15 This further supports that this new olopatadine ophthalmic formulation with almost 4-fold increased concentration than 0.2% does not raise any new safety concerns. In the 2–17 age group, the only treatment-related AEs reported were corneal staining, conjunctival staining, abnormal sensation in the eyes, and eye pruritus with no incidence of dysgeusia. Except for corneal staining and conjunctival staining, the rates of all other AEs were lower in the 2–17 age group as compared to the ≥18 age group. The AEs of corneal staining and conjunctival staining were reported without baseline measurements since fluorescein staining was not a protocol required procedure and therefore was not conducted at baseline (screening visits) for any subjects. Thus, it is unknown if this finding represents an untoward change relative to a pre-treatment baseline. Overall, these data show that olopatadine 0.77% was well tolerated in the 2–17 age group with no new safety issues identified, which is consistent with the safety profile of olopatadine 0.2% ophthalmic formulation in ≥3-year-old subjects.25,26 No discernible trends from baseline or clinically relevant differences were observed during the 6-week study period between the olopatadine 0.77% and the vehicle groups based on a review of safety parameters that included BCVA ocular sign parameters, IOP, dilated fundus parameters, and vital signs.
Conclusion
Olopatadine 0.77% after topical ocular administration of single and multiple doses has a low systemic exposure with quick clearance. Olopatadine 0.77% was well tolerated with no new safety issues after once-daily topical ocular dosing for 6 weeks in adults and in pediatric subjects as young as 2 years of age. The overall safety profile of olopatadine 0.77% is consistent with that of olopatadine 0.2%.
Acknowledgments
This study was funded by Alcon Laboratories, Inc. (Fort Worth, TX, USA). The authors thank Audesh Bhat and Usha Gutti (Global Medical and Clinical Services, Novartis Healthcare Pvt Ltd., Hyderabad, India) for their medical writing and editorial assistance toward the development of this manuscript.
Disclosure
Dr Abhijit Narvekar is an employee in Alcon Research Ltd (Fort Worth, TX, USA). The other authors report no conflicts of interest in this work.
References
Gomes PJ. Trends in prevalence and treatment of ocular allergy. Curr Opin Allergy Clin Immunol. 2014;14(5):451–456. | ||
Calonge M. Classification of ocular atopic/allergic disorders and conditions: an unsolved problem. Acta Ophthalmol Scand Suppl. 1999;(228):10–13. | ||
Bonini S. Atopic keratoconjunctivitis. Allergy. 2004;59(suppl 78):71–73. | ||
Friedlaender MH. Ocular allergy. Curr Opin Allergy Clin Immunol. 2011;11(5):477–482. | ||
Chigbu DI. The pathophysiology of ocular allergy: a review. Cont Lens Anterior Eye. 2009;32(1):3–15. | ||
Azari AA, Barney NP. Conjunctivitis: a systematic review of diagnosis and treatment. JAMA. 2013;310(16):1721–1729. | ||
Ridolo E, Montagni M, Caminati M, Senna G, Incorvaia C, Canonica GW. Emerging drugs for allergic conjunctivitis. Expert Opin Emerg Drugs. 2014;19(2):291–302. | ||
Yanni JM, Stephens DJ, Miller ST, et al. The in vitro and in vivo ocular pharmacology of olopatadine (AL-4943A), an effective anti-allergic/antihistaminic agent. J Ocul Pharmacol Ther. 1996;12(4):389–400. | ||
Baba A, Tachi M, Maruyama Y, Kazama I. Olopatadine inhibits exocytosis in rat peritoneal mast cells by counteracting membrane surface deformation. Cell Physiol Biochem. 2015;35(1):386–396. | ||
Berdy GJ, Spangler DL, Bensch G, Berdy SS, Brusatti RC. A comparison of the relative efficacy and clinical performance of olopatadine hydrochloride 0.1% ophthalmic solution and ketotifen fumarate 0.025% ophthalmic solution in the conjunctival antigen challenge model. Clin Ther. 2000;22(7):826–833. | ||
Spangler DL, Bensch G, Berdy GJ. Evaluation of the efficacy of olopatadine hydrochloride 0.1% ophthalmic solution and azelastine hydrochloride 0.05% ophthalmic solution in the conjunctival allergen challenge model. Clin Ther. 2001;23(8):1272–1280. | ||
Abelson MB, Gomes PJ, Vogelson CT, et al. Effects of a new formulation of olopatadine ophthalmic solution on nasal symptoms relative to placebo in two studies involving subjects with allergic conjunctivitis or rhinoconjunctivitis. Curr Med Res Opin. 2005;21(5):683–691. | ||
Vogelson CT, Abelson MB, Pasquine T, et al. Preclinical and clinical antiallergic effect of olopatadine 0.2% solution 24 hours after topical ocular administration. Allergy Asthma Proc. 2004;25(1):69–75. | ||
McLaurin E, Narvekar A, Gomes P, Adewale A, Torkildsen G. Phase 3 randomized double-masked study of efficacy and safety of once-daily 0.77% olopatadine hydrochloride ophthalmic solution in subjects with allergic conjunctivitis using the conjunctival allergen challenge model. Cornea. 2015;34(10):1245–1251. | ||
Torkildsen G, Narvekar A, Bergmann M. Efficacy and safety of olopatadine hydrochloride 0.77% in patients with allergic conjunctivitis using a conjunctival allergen-challenge model. Clin Ophthalmol. 2015;9:1703–1713. | ||
Iyer GR, Cason MM, Womble SW, Li G, Chastain JE. Ocular pharmacokinetics comparison between 0.2% olopatadine and 0.77% olopatadine hydrochloride ophthalmic solutions administered to male New Zealand white rabbits. J Ocul Pharmacol Ther. 2015;31(4):204–210. | ||
Bielory L. Allergic and immunologic disorders of the eye. Part II: ocular allergy. J Allergy Clin Immunol. 2000;106(6):1019–1032. | ||
Asher MI, Montefort S, Bjorksten B, et al; ISAAC Phase Three Study Group. Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC Phases One and Three repeat multicountry cross-sectional surveys. Lancet. 2006;368(9537):733–743. | ||
Aït-Khaled N, Pearce N, Anderson HR, et al; ISAAC Phase Three Study Group. Global map of the prevalence of symptoms of rhinoconjunctivitis in children: The International Study of Asthma and Allergies in Childhood (ISAAC) Phase Three. Allergy. 2009;64(1):123–148. | ||
Rosario N, Bielory L. Epidemiology of allergic conjunctivitis. Curr Opin Allergy Clin Immunol. 2011;11(5):471–476. | ||
Leonardi A, Bogacka E, Fauquert JL, et al. Ocular allergy: recognizing and diagnosing hypersensitivity disorders of the ocular surface. Allergy. 2012;67(11):1327–1337. | ||
Mallol J, Crane J, von Mutius E, et al. The International Study of Asthma and Allergies in Childhood (ISAAC) Phase Three: a global synthesis. Allergol Immunopathol (Madr). 2013;41(2):73–85. | ||
Tsunoo M, Momomura S, Masuo M, Iizuka H. Phase 1 clinical study on KW-4679, an antiallergic drug: safety and pharmacokinetics in the single and repeated administration study in healthy subjects. Kiso To Rinsho. 1995;29:18. | ||
Lichtenstein SJ, Pasquine TA, Edwards MR, Wells DT, Gross RD, Robertson SM. Safety and tolerability of olopatadine 0.2% in children and adolescents. J Ocul Pharmacol Ther. 2007;23(4):366–371. | ||
Food and Drug Administration. Pazeo Labeling; 2015. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206276Orig1s000SumR.pdf. Accessed February 8, 2017. | ||
Endre L, DuBuske LM. The safety of olopatadine in children with seasonal allergic conjunctivitis. J Allergy Clin Immunol. 2009;123(suppl S128):489. |
Supplementary materials
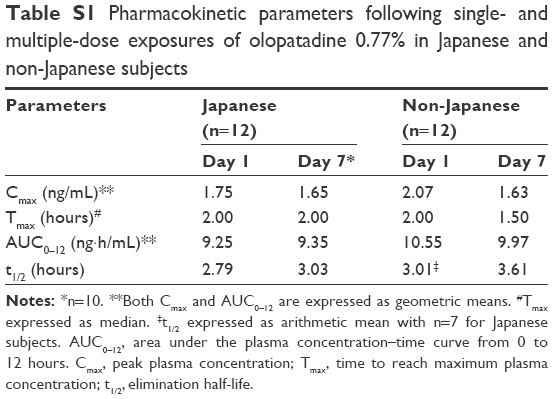 | Table S1 Pharmacokinetic parameters following single- and multiple-dose exposures of olopatadine 0.77% in Japanese and non-Japanese subjects |
 | Table S2 Overall summary of treatment-related AEs occurring at an incidence of ≥1% by treatment (safety population) |
 | Table S3 Summary of TEAEs in selected age subgroups occurring at an incidence of ≥1% by treatment (safety population) |
 | Table S4 Comparison of mean human plasma pharmacokinetic parameters following topical ocular and oral single- and multiple-dose exposures of olopatadine |
Reference
Tsunoo M, Momomura S, Masuo M, Iizuka H. Phase 1 clinical study on KW-4679, an antiallergic drug: safety and pharmacokinetics in the single and repeated administration study in healthy subjects. Kiso To Rinsho. 1995;29:18. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.