")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Pharmacokinetics and safety of olodaterol administered with the Respimat Soft Mist inhaler in subjects with impaired hepatic or renal function
Authors Kunz C, Luedtke D, Unseld A, Hamilton A, Halabi A, Wein M, Formella S
Received 11 August 2015
Accepted for publication 4 December 2015
Published 18 March 2016 Volume 2016:11(1) Pages 585—595
DOI https://doi.org/10.2147/COPD.S94234
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Christina Kunz,1 Doreen Luedtke,1 Anna Unseld,2 Alan Hamilton,3 Atef Halabi,4 Martina Wein,5 Stephan Formella6
1Translational Medicine and Clinical Pharmacology, 2Global Biometrics and Clinical Applications, Boehringer Ingelheim Pharma GmbH and Co KG, Biberach, Germany; 3Boehringer Ingelheim, Burlington, ON, Canada; 4CRS Clinical Research Services Kiel GmbH, Kiel, 5Drug Metabolism and Pharmacokinetics, Boehringer Ingelheim Pharma GmbH and Co KG, Biberach, 6Medicine Coordination, Boehringer Ingelheim Pharma GmbH and Co KG, Ingelheim, Germany
Purpose: In two trials, the influences of hepatic and renal impairment on the pharmacokinetics of olodaterol, a novel long-acting inhaled β2-agonist for treatment of COPD, were investigated.
Subjects and methods: The first trial included eight subjects with mild hepatic function impairment (Child–Pugh A), eight subjects with moderate impairment (Child–Pugh B), and 16 matched healthy subjects with normal hepatic function. The second trial included eight subjects with severe renal impairment (creatinine clearance <30 mL·min-1) and 14 matched healthy subjects with normal renal function. Subjects received single doses of 20 or 30 µg olodaterol administered with the Respimat Soft Mist inhaler.
Results: Olodaterol was well tolerated in all subjects. The geometric mean ratios and 90% confidence intervals of dose-normalized area under the plasma concentration-time curve from time zero to 4 hours (AUC0–4) for subjects with mild and moderate hepatic impairment compared to healthy subjects were 97% (75%–125%) and 105% (79%–140%), respectively. Corresponding values for dose-normalized maximum concentration (Cmax) were 112% (84%–151%) (mild impairment) and 99% (73%–135%) (moderate impairment). The geometric mean ratio (90% confidence interval) of AUC0–4 for subjects with severe renal impairment compared to healthy subjects was 135% (94%–195%), and for Cmax was 137% (84%–222%). There was no significant relationship between creatinine clearance and AUC0–4 or Cmax. Renal clearance of olodaterol was reduced to 20% of normal in severe renal impairment.
Conclusion: Mild to moderate hepatic function impairment or severe renal function impairment did not result in a clinically relevant increase of olodaterol systemic exposure after a single inhaled dose.
Keywords: chronic obstructive pulmonary disease, exposure, olodaterol, long-acting β2-agonist, hepatic impairment, renal impairment
Introduction
Olodaterol (Striverdi®) is a novel, once-daily, long-acting β2-adrenergic receptor agonist (LABA) with high β2-selectivity that has recently obtained regulatory approval for the maintenance therapy of patients with COPD.1 A duration of bronchodilatory action over 24 hours was demonstrated in preclinical studies,2,3 and has been confirmed in Phase III clinical trials.4,5 In sum, the Phase III program demonstrated the long-term efficacy and safety of once-daily inhaled doses of 5 and 10 μg olodaterol in patients with moderate to very severe COPD continuing with usual-care maintenance therapy, with a satisfactory safety profile.4 The inhalation device used was the Respimat® Soft Mist™ inhaler, which delivers medication as a nebulized aqueous solution.6
The pharmacokinetics of olodaterol are linear,7 with a dose-proportional increase of systemic exposure observed after single inhaled doses of 2–40 μg.8 Liver and kidney functions are important intrinsic patient factors that can potentially affect pharmacokinetics, and the consequences of their impairment need to be investigated, especially if the patient population includes renally and hepatically impaired subjects.9,10 In the case of olodaterol, hepatic metabolism was identified as the principal route of elimination, in which the parent compound is substantially metabolized by direct glucuronidation and by O-demethylation at the methoxy moiety followed by conjugation.7 Of the six metabolites identified, only the unconjugated demethylation product of olodaterol (SOM 1522) is active at β2-adrenoceptors, but is generally not detectable in plasma after chronic inhalation of the therapeutic dose of 5 μg.7 In view of the significance of the hepatic metabolism pathways for olodaterol elimination, the first trial reported here was conducted to investigate the effect of impaired liver function on pharmacokinetics and safety of olodaterol.
In contrast, the kidneys represent only a minor elimination pathway for olodaterol, based on the findings in healthy subjects that olodaterol renal clearance accounts for less than 20% of the total plasma clearance.7,11 In addition, olodaterol has only a relatively low binding to plasma proteins of approximately 60%.7 Therefore, it can be expected that renal function impairment, which is often associated with a decrease in drug binding to plasma proteins, will not have a pronounced effect on the pharmacokinetics of olodaterol. Nevertheless, impaired renal function is known to be a concomitant disease in the patient population with COPD,12 as can also be expected from the average age of the patients participating in the Phase III trials of around 65 years.4,5 Therefore, the second trial presented here was also performed to assess the effects of renal impairment on the pharmacokinetics and safety of olodaterol. Both the hepatic and the renal impairment trials were conducted using inhaled single-dose administration via the Respimat Soft Mist inhaler, and employed trial designs recommended in current regulatory guidelines.9,10
Subjects and methods
Subjects
Hepatic impairment trial
A total of 32 males and females were eligible for inclusion, with ages in the specified range of 21–75 years. They comprised 16 subjects with impaired hepatic function and 16 healthy subjects, and were allocated to three different treatment groups. The Child–Pugh scoring system was used to evaluate the hepatically impaired subjects.13 The groups were 1) mild hepatic impairment (n=8), with Child–Pugh score 5–6 points; 2) moderate hepatic impairment (n=8), with Child–Pugh score 7–9 points; and 3) healthy subjects with normal hepatic function (n=16) who were matched to groups 1 and 2 with regard to sex, age (within ±7 years), and weight (within ±10%). The minimum creatinine clearance for inclusion was ≥70 mL·min−1 for healthy subjects and ≥40 mL·min−1 for subjects with mild or moderate hepatic impairment. The lower bound of the body mass index for inclusion was 18.5 kg·m−2, and the upper bound was 32 kg·m−2 for healthy subjects and 34 kg·m−2 for the hepatically impaired subjects.
Principal exclusion criteria were myocardial infarction 6 months before dosing, congestive heart failure of New York Heart Association grade III or IV, severe arrhythmia, cerebrovascular disorders, relevant gastrointestinal tract surgery, diseases of the central nervous system, psychiatric disorders, neurological disorders, evidence of hepatic encephalopathy related to chronic liver disease > grade 2, resting heart rate <45 or >100 bpm or systolic blood pressure <100 or >160 mmHg, diastolic pressure >95 mmHg, smokers consuming more than ten cigarettes, three cigars, or three pipes a day, excessive physical activities within 48 hours before or during the trial, asthma, or history of pulmonary hyperreactivity.
Renal impairment trial
A total of 22 males and females were eligible for inclusion, with ages in the specified range of 21–75 years. They comprised 14 healthy subjects and eight subjects with severe renal impairment, defined as creatinine clearance <30 mL·min−1 calculated by the Cockcroft–Gault formula. Eight healthy subjects were matched individually to the subjects with severe renal impairment with regard to sex, age (±7 years), and weight (±10%). The remaining six healthy subjects were matched to the average demographic data (age, sex, and weight) of the group of all enrolled subjects with renal impairment. Inclusion criteria for body mass index were as for the hepatic impairment trial. Exclusion criteria included any clinically relevant disease (not including renal impairment), any clinically relevant deviation from normal parameters in medical examinations (healthy group only), hepatic function impairment, history of asthma, and inability to refrain from smoking on trial days.
The subjects of the two trials were recruited in cooperation with several local hepatology and nephrology centers and the University Hospital of Kiel, Germany. The healthy subjects were recruited from the database of the trial center.
Trial design and treatments
These were two open-label, single-dose, parallel-group trials conducted in one center (CRS Clinical Research Services Kiel GmbH). The studies were approved by the ethics committees of Schleswig-Holstein, Bad Segeberg, Germany (hepatic impairment trial) and the State Chamber of Physicians of Schleswig-Holstein, Kiel, Germany (renal impairment trial), and were conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonisation (ICH) guidelines for good clinical practice, and German drug law. All subjects provided written informed consent before entering the trial.
Trial restrictions included abstention from smoking and alcoholic beverages from 24 hours before the start of the treatment period until the last sample was collected. Methylxanthine-containing drinks or foods (coffee, tea, cola, energy drinks, chocolate, etc) were not permitted for the 48 hours preceding the administration of the trial medication and until 24 hours after the dosing. Apples, citrus fruits, in particular grapefruits and Seville oranges (sour or bitter oranges), and their juices, as well as products containing St John’s wort were not permitted for the 5 days before the administration of trial medication and until the last pharmacokinetic procedure had been performed after drug administration. Physical stress had to be avoided during the course of the trials. Women of childbearing potential had to maintain adequate contraception throughout the trials.
During a screening period from 21 to 2 days before drug administration, subjects were assessed for eligibility, and the included subjects underwent a baseline evaluation on the day before trial-drug administration. A single orally inhaled dose of olodaterol was administered with the Respimat Soft Mist inhaler on trial day 1. In the hepatic impairment trial, the doses were 30 μg for healthy subjects (six actuations of 5 μg) and 20 μg for subjects with hepatic impairment (four actuations of 5 μg). In the renal impairment trial, both the impaired and healthy subject groups received a single 30 μg dose of olodaterol as six inhaler actuations of 5 μg. All trial subjects were instructed on the correct use of the inhalation device during two training sessions: one during the screening period, and a second on the day before the administration of the first dose. Particular attention was paid to the coordination between device actuation and adequate inhalation.
In order to ensure sufficient systemic exposure after a single dose in the trial subjects without compromising safety, the dose selected for healthy subjects and renally impaired subjects (30 μg) was three times the maximum anticipated therapeutic dose of 10 μg once daily that was under clinical investigation over 48 weeks in Phase III clinical trials.4,5 A dose that was only twice as high as the maximal therapeutic dose (20 μg) was selected for the hepatically impaired subjects, in consideration of the possibilities that plasma concentrations of olodaterol in these subjects might be elevated and that baseline potassium values may be altered toward the lower end of the reference range in patients with impaired hepatic function. On days 1–4 of both trials, blood and urine samples were collected for pharmacokinetic and safety measurements. An end-of-study examination was performed within 5–14 days after dosing.
Pharmacokinetics
Blood samples (10 mL) for the measurement of plasma concentrations of olodaterol and the active metabolite SOM 1522 were taken using ethylenediaminetetraacetic acid (EDTA) as anticoagulant from a forearm vein of each subject before dosing and at 5, 10, 20, 40 minutes, 1, 2, 4, 6, 8, 12, 24, 48, and 72 hours after dosing. Immediately after collection, the blood samples were centrifuged at 4–8°C for 10 minutes at 2,000–4,000× g. Plasma was prepared as two aliquots per time point and stored at −20°C until bioanalytical measurement. Urine fractions were collected for determination of olodaterol and SOM 1522 before dosing and at 0–8, 8–12, 12–24, 24–48, and 48–72 hours after dosing. Two 3 mL aliquots of each fraction were stored at −20°C until bioanalysis.
Plasma and urine concentrations of olodaterol and SOM 1522 were analyzed by validated high-performance liquid chromatography–mass spectrometry (HPLC-MS/MS) assays using [D3]olodaterol or [D3]SOM 1522 as an internal standard. The assays included sample cleanup by solid-phase extraction in the 96-well plate format. Chromatography was performed on analytical reverse-phase HPLC columns with gradient elution. The substances were detected and quantified using electrospray ionization in the positive-ion mode. The lower limit of quantification (LLOQ) was 2.0 pg·mL−1 for olodaterol and 10 pg·mL−1 for SOM 1522 in plasma samples, and 10 pg·mL−1 for olodaterol and 100 pg·mL−1 for SOM 1522 in urine samples. No relevant interference of endogenous compounds was observed in human plasma or urine samples with either assay. The calibration curves of plasma samples were linear over the range of concentrations 2.00–200 pg·mL−1 for olodaterol and 10.0–200 pg·mL−1 for SOM 1522 using a sample volume of 500 μL. The calibration curves of undiluted urine samples were linear over the range of concentrations 10.0–10,000 pg·mL−1 olodaterol using 300 μL urine and 100–10,000 pg·mL−1 SOM 1522 using 50 μL urine. In-study assay validation for olodaterol at three nominal concentrations yielded assay accuracy (expressed as a deviation from the target concentration) of 0.6%–3.6% (plasma) and −2.4% to 4.0% (urine) and precision (expressed as a coefficient of variation) of 3.0%–8.2% (plasma) and 2.0%–8.7% (urine). For the SOM 1522 in-study assay validation, three nominal concentrations resulted in assay accuracy of 2.8%–6.3% (plasma) and −0.2%–8.2% (urine) and precision of 2.9%–5.6% (plasma) and 0.5%–9.4% (urine).
The plasma protein binding of olodaterol was determined in both trials. On day 1 before dosing, 15 mL of blood from each subject was collected in three vials coated with EDTA and centrifuged at 2,500× g for 10 minutes at 4°C. The plasma samples obtained were frozen at −20°C. In vitro plasma protein binding was determined by equilibrium dialysis using 3H-radiolabeled olodaterol at a concentration of 10 pmol·L−1 (3.86 pg·mL−1).
Pharmacokinetic parameters were calculated using standard noncompartmental methods with WinNonlin® Professional software (version 5.2; Pharsight Corporation, Mountain View, CA, USA). The primary end points of the hepatic impairment trial were area under the plasma concentration–time curve of olodaterol from 0 to 4 hours, divided by the dose (AUC0–4,norm) and peak plasma concentration of olodaterol, divided by the dose (Cmax,norm). The dose normalization was specified to facilitate comparison of results between the hepatically impaired subject groups that received a 20 μg dose and the healthy subject group that received a 30 μg dose. The primary end points of the renal impairment trial were area under the plasma concentration-time curve from time zero to 4 hours ( AUC0–4) and Cmax. Further pharmacokinetic parameters calculated in both trials were AUC to time of last quantifiable plasma concentration (AUC0–tz), time to reach Cmax, fraction of olodaterol dose excreted unchanged in urine to tz (fe0–tz), and renal clearance of olodaterol determined over 8 hours after dosing (CLR,0–8).
Safety and tolerability
Safety and tolerability were evaluated on the basis of adverse events, vital signs (blood pressure and pulse rate, 12-lead electrocardiography), clinical laboratory tests (hematology, coagulation, enzymes, hormones, substrates, electrolytes, urinalysis), medical examination, and investigator assessment of global tolerability. Serum potassium is known to be responsive to β2-agonists in general,14,15 in addition to its status as an established clinical safety parameter.16 Therefore, 1.2 mL of blood samples were collected for monitoring at 2 hours predose and at 30, 40 minutes, 1, 2, 3, 4, 6, and 8 hours after olodaterol dosing. Potassium was determined on the day of serum collection by an ion-selective electrode method. The reference range was 3.5–5.3 mmol·L−1.
Statistical methods
Pharmacokinetic parameters of olodaterol were compared between the mild or moderate hepatic impairment groups (defined as “test”) and the matched healthy subject group (defined as “reference”), and between the severely renally impaired group (“test”) and the matched healthy subject group (“reference”), using analysis of variance (ANOVA). This model included the fixed effect for the corresponding subject groups with normal or impaired hepatic/renal function, depending on the study. The pharmacokinetic parameters were log-transformed before fitting the ANOVA model. The difference between the expected means for logtest – logreference was estimated by the difference in the corresponding least square means (point estimate). Two-sided 90% confidence intervals based on the t-distribution were computed. These quantities were then back-transformed to the original scale to give the point estimator (geometric mean) and interval estimates for the median intrasubject ratio between the test and reference pharmacokinetic parameters. No explicit power calculation for determination of sample size was performed, because of practical constraints on the numbers of patients with hepatic or renal dysfunction that could be recruited, especially those with moderate or severe impairment. The number of patients selected was in conformance with the regulatory guidelines.9 In the renal impairment trial, the relationship between creatinine clearance and the pharmacokinetic parameters AUC0–4 and Cmax was investigated by linear regression analysis.
Results
Trial population
Hepatic impairment trial
A total of 32 subjects were treated, with baseline demographics given in Table 1. All subjects completed the trial and were included in the pharmacokinetic, pharmacodynamic, and safety analysis. In one healthy subject, olodaterol plasma concentrations were below LLOQ (2 pg·mL−1), but the urine concentrations were measurable. In a further subject with moderate hepatic impairment, an olodaterol plasma concentration with magnitude comparable to Cmax was detected in the predose sample. As this was an implausible finding, the plasma-concentration data of this subject were excluded from the final pharmacokinetic analysis. It was however confirmed by additional analysis including the respective data that this omission did not affect the overall trial conclusions (Tables S1 and S2). Urine data of this subject were used.
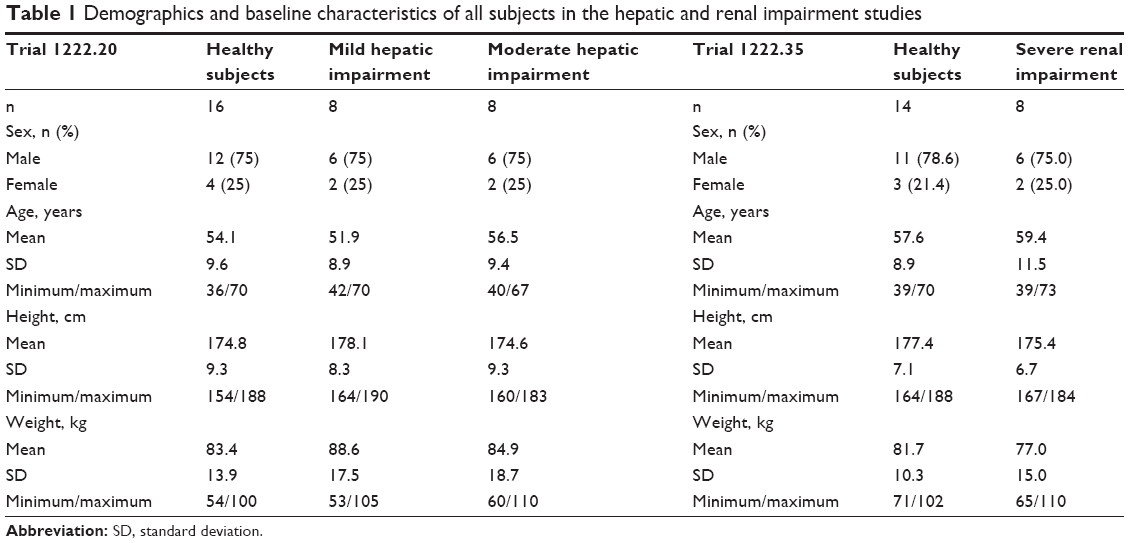 | Table 1 Demographics and baseline characteristics of all subjects in the hepatic and renal impairment studies |
Renal impairment trial
A total of 22 subjects were treated, with baseline demographics also shown in Table 1. All subjects completed the trial and were included in the pharmacokinetic and safety analysis. In one subject with severe renal impairment and one healthy subject, olodaterol plasma concentrations were below LLOQ. Olodaterol was measurable in the urine fractions of all treated subjects.
Pharmacokinetics
Hepatic impairment trial
The plasma concentration–time profiles of the trial groups are shown in Figure 1 and the pharmacokinetic parameters in Table 2. Olodaterol plasma concentrations were quantifiable up to at least 1 hour after dose administration for the trial subjects, with the exception of the one healthy subject described in the Subjects and methods section. Cmax was reached within 7 minutes to 2 hours after inhalation. The tz was highly variable between subjects, but was generally later in the healthy subjects dosed with 30 μg olodaterol than in the subjects with hepatic impairment dosed with 20 μg (Table 2). The geometric mean ratios and 90% confidence intervals of dose-normalized Cmax and AUC0–4 for the mild and moderate hepatic impairment subject groups compared to the healthy subjects are given in Table 3. The geometric mean ratios were close to unity and the confidence intervals included unity, indicating similar systemic exposure between both subject groups with hepatic impairment and the healthy subjects.
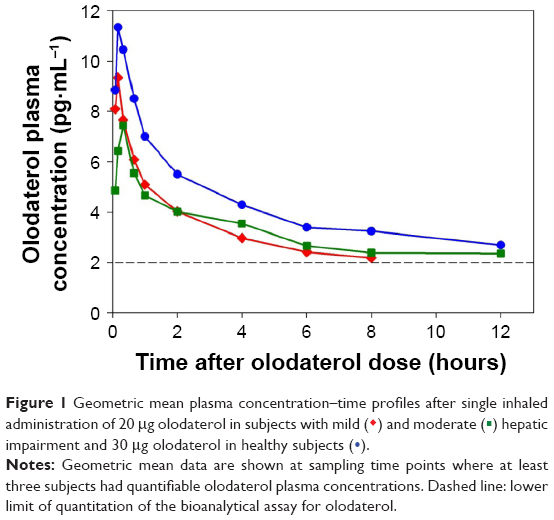 | Figure 1 Geometric mean plasma concentration–time profiles after single inhaled administration of 20 μg olodaterol in subjects with mild ( |
 ) and moderate (
) and moderate ( ) hepatic impairment and 30 μg olodaterol in healthy subjects (
) hepatic impairment and 30 μg olodaterol in healthy subjects ( ).
).
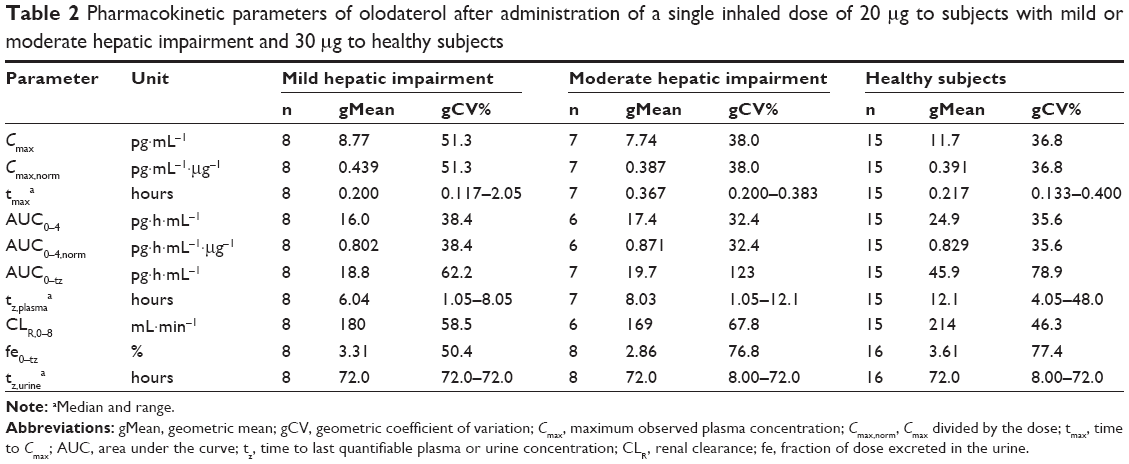 | Table 2 Pharmacokinetic parameters of olodaterol after administration of a single inhaled dose of 20 μg to subjects with mild or moderate hepatic impairment and 30 μg to healthy subjects |
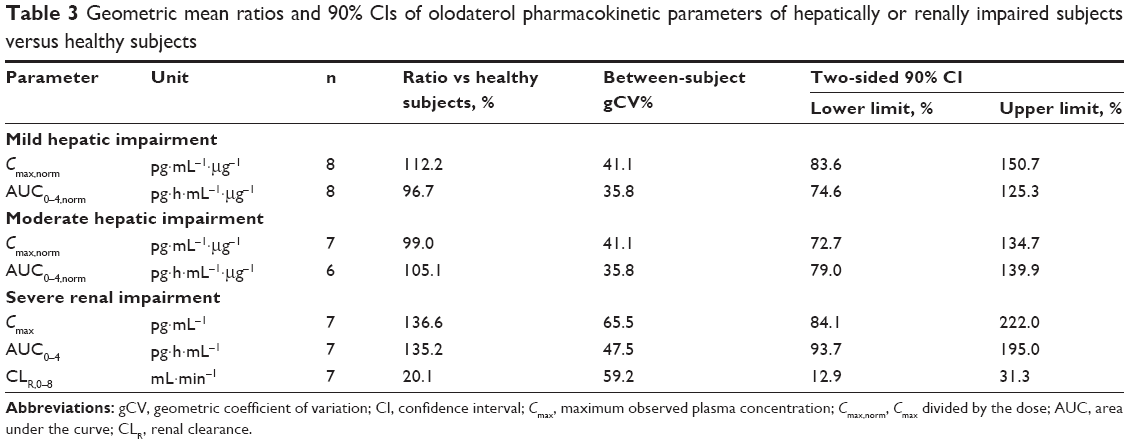 | Table 3 Geometric mean ratios and 90% CIs of olodaterol pharmacokinetic parameters of hepatically or renally impaired subjects versus healthy subjects |
Quantifiable olodaterol concentrations in urine fractions were found up to 48–72 hours after inhalation. The fe0–tz and CLR,0–8 values of the three subject groups are given in Table 2. Both of these parameters were slightly higher (geometric mean) in the healthy subjects compared to the two hepatically impaired groups, but their between-subject variability was also relatively high (Table 2).
The in vitro plasma protein binding of olodaterol was very similar between the groups: 59.8%±4.5% (mean ± standard deviation) for the healthy subjects, 62.8%±8.9% for the subjects with mild hepatic impairment, and 56.6%±6.2% for the subjects with moderate hepatic impairment. Concentrations of the metabolite SOM 1522 in plasma were below the LLOQ (10 pg·mL−1) at all plasma-sampling time points of all subjects. In urine, only very small amounts (8.0–130 ng) were detected in a very small number of the urine fractions taken from all subjects. It was thus not possible to perform a pharmacokinetic analysis on SOM 1522.
Renal impairment trial
The plasma concentration–time profiles in each group are shown in Figure 2 and the pharmacokinetic parameters in Table 4. Olodaterol plasma concentrations were below LLOQ in two subjects as described earlier, but were quantifiable up to at least 2 hours after dosing in all other subjects. Cmax was reached within 8 minutes to 1 hour after inhalation. The tz was highly variable between subjects (Table 4).
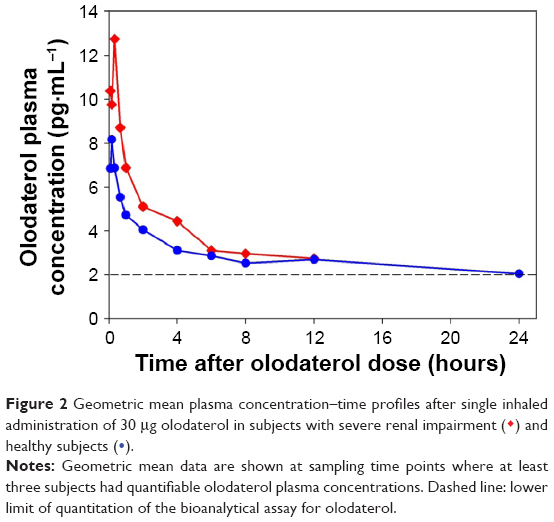 | Figure 2 Geometric mean plasma concentration–time profiles after single inhaled administration of 30 μg olodaterol in subjects with severe renal impairment ( |
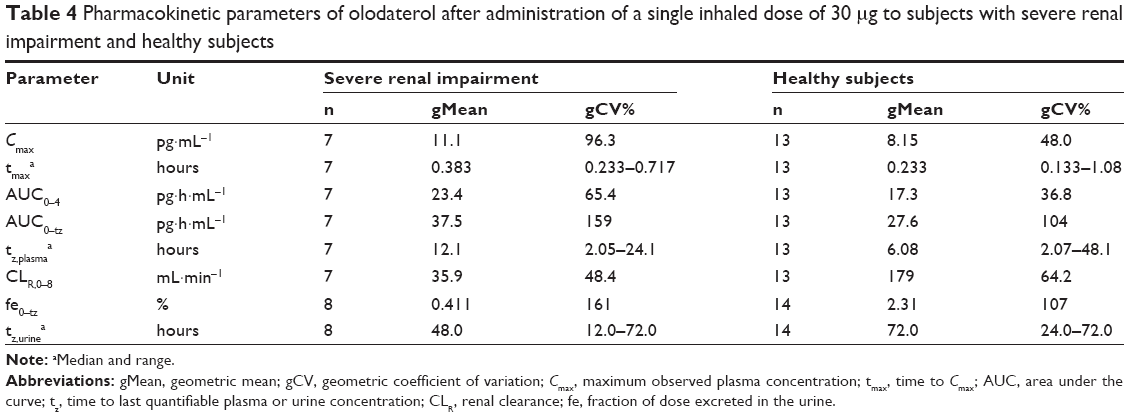 | Table 4 Pharmacokinetic parameters of olodaterol after administration of a single inhaled dose of 30 μg to subjects with severe renal impairment and healthy subjects |
Quantifiable olodaterol concentrations in urine fractions were found up to at least 12 hours after inhalation for subjects with severe renal impairment and up to 72 hours for healthy subjects. The fe0–tz and renal clearance CLR,0–8 values of the subject groups are given in Table 4. Geometric mean fe0–tz was decreased by a factor of 5.6 in the severely renally impaired subjects (0.411%) compared to the healthy subjects (2.31%).
The geometric mean ratios and 90% confidence intervals of Cmax, AUC0–4, and CLR,0–8 for the severely renally impaired subject group compared to the healthy subjects are given in Table 3. The results show 36.6% higher average maximum systemic exposure (Cmax) and 35.2% higher average total systemic exposure (AUC0–4) in the severely renally impaired group, but with wide confidence intervals that include unity. CLR,0–8 in the severely renally impaired group was only 20.1% of the magnitude in healthy subjects, with narrower confidence intervals not including unity. Linear regression analysis of AUC0–4 and Cmax on creatinine clearance yielded no significant relationship, with very low coefficients of determination (R2) of 0.160 and 0.117, respectively.
The in vitro plasma protein binding of olodaterol was very similar between groups: 60.1%±3.8% (mean ± standard deviation) for the healthy subjects and 63.7%±3.6% for the subjects with severe renal dysfunction. Concentrations of the metabolite SOM 1522 in plasma were below LLOQ at all plasma-sampling time points of all subjects. In urine, very small amounts (43.8–209 ng) were detected in a small number of the urine fractions from the healthy subjects only. It was thus not possible to perform a pharmacokinetic analysis on SOM 1522.
Safety
Hepatic impairment trial
No severe, serious, significant, or other significant adverse events (according to ICH E3) occurred, and none of the subjects discontinued the trial due to an adverse event. Of the 32 treated subjects, adverse events of mild intensity occurred in two subjects from the moderate hepatic impairment group. Both subjects exhibited serum potassium concentrations that were slightly below the reference range at baseline (3.0 and 2.9 mmol·L−1, respectively) and which decreased to minima of 2.8 and 2.5 mmol·L−1, respectively, between 2 and 3 hours after olodaterol administration. Potassium chloride was administered 6 hours after olodaterol dosing, and the serum values normalized within 8 hours. Both adverse events were assessed as drug related. The time course of mean serum potassium concentrations is shown in Figure 3A. Mean baseline values were lowest in the moderate hepatic impairment group (3.86±0.55 mmol·L−1), due to the influence of the two subjects described earlier, followed by the mild hepatic impairment (4.19±0.33 mmol·L−1) and healthy subject groups (4.26±0.22 mmol·L−1). There were only minor fluctuations in mean potassium concentrations in any group (6% or less from baseline) over the further time course up to 8 hours.
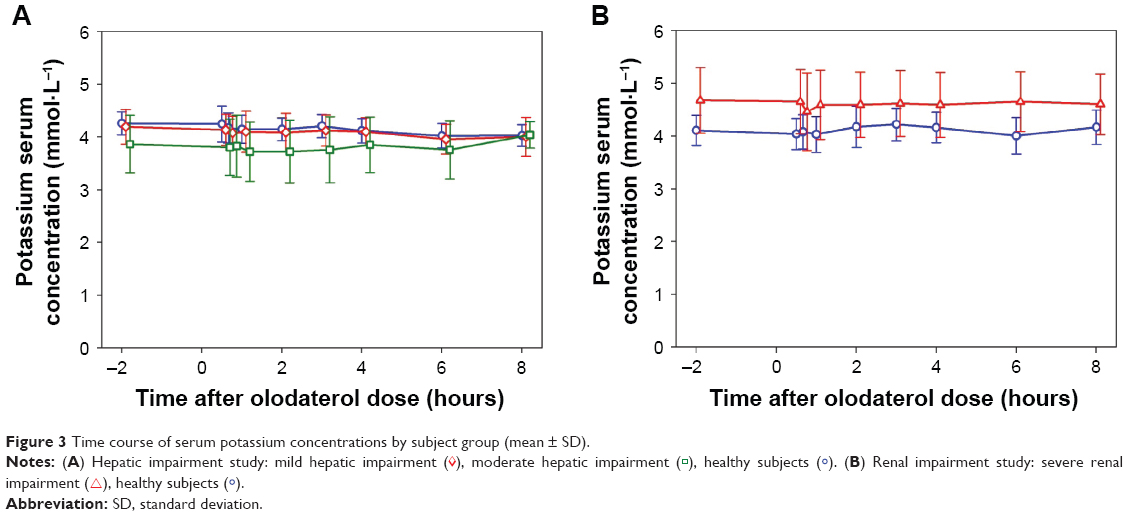 | Figure 3 Time course of serum potassium concentrations by subject group (mean ± SD). |
 ), moderate hepatic impairment (
), moderate hepatic impairment ( ), healthy subjects (
), healthy subjects ( ). (B) Renal impairment study: severe renal impairment (
). (B) Renal impairment study: severe renal impairment ( ), healthy subjects (
), healthy subjects (Renal impairment trial
Three of the 22 treated subjects experienced four adverse events, which were all in the healthy subject group (back pain, headache, and cough). None of the subjects with severe renal impairment experienced an adverse event. All adverse events were mild or moderate in intensity, and had resolved by the end of the trial. No adverse event was judged by the investigator as causally related to olodaterol, and no subject discontinued the trial prematurely due to the occurrence of an adverse event. The time course of mean serum potassium concentrations is shown in Figure 3B. Mean baseline values were 15% higher in the severely renally impaired group (4.67±0.62 mmol·L−1) than in the healthy subjects (4.11±0.29 mmol·L−1). Here also, over the further time course up to 8 hours, there were only minor fluctuations in mean potassium concentrations of 5% or less from baseline.
Discussion
These two clinical trials systematically assessed the influence of mild or moderate hepatic impairment and of severe renal impairment on the pharmacokinetics and safety of single doses of inhaled olodaterol, compared to healthy control subjects who were matched closely to the organ function-impaired subjects with regard to sex, age, and body weight (Table 1). The pharmacokinetic parameters estimated for these four types of subjects (Tables 2 and 4) complement previous results that were obtained after single doses of 2–40 μg in COPD patients.8
In both trials, the Cmax of olodaterol at the selected doses was in the low pg·mL−1 range and rapidly decreased to levels below the assay LOQ of 2 pg·mL−1. Reliable estimation of terminal half-lives and the total exposure AUC0–∞ was thus not possible. As a consequence, in the majority of the study subjects, only the early systemic exposure as represented by the parameters Cmax and AUC0–4 could be assessed and statistically evaluated as primary end points for both trials, with dose normalization where appropriate.
In the hepatic impairment trial, subjects in both the moderate and mildly impaired groups exhibited primary pharmacokinetic parameters that were closely similar to those in the normal subjects (Table 2 [AUC0–4,norm and Cmax,norm] and Table 3 [geometric mean ratios and confidence intervals]). Although the geometric mean ratios (point estimators) were close to 100% (Table 3), the 90% confidence intervals were wider than the acceptance range of 80%–125% that would be applied in bioequivalence testing. Narrower confidence intervals could have been obtained by entering more subjects, but the sample sizes employed in this study (Table 2) were based on ethical and practical considerations and were judged adequate and consistent with the regulatory guidelines.9 The important finding is that systemic exposure to olodaterol was not increased as a result of reduced hepatic function. Evidently, the capacity for biotransformation of olodaterol in the liver by glucuronidation and oxidation was still adequate for the relatively low dose, even under conditions of moderate hepatic insufficiency.
Subjects with severe renal impairment showed slightly higher average AUC0–4 (35%) and Cmax (37%) than healthy subjects with normal renal function (Tables 3 and 4). The renal excretion of unchanged olodaterol accounts for less than 20% of its overall elimination.7,11 Therefore, even the observed 80% decrease of renal clearance in severe renal impairment is expected to translate into only a minor decrease of total clearance of olodaterol, and thus into only slightly increased systemic exposure. The magnitude of the observed difference in systemic exposure between the renally impaired subjects and the subjects with normal renal function may indicate that in addition to renal clearance, nonrenal clearance and/or the volume of distribution of olodaterol might be affected. An influence of renal impairment on nonrenal elimination is described in the literature for several other drugs, although the underlying mechanisms are incompletely understood.17 However, as evident from the confidence intervals of the Cmax and AUC0–4 ratios being wide and including unity, the sample size in the present trial was not sufficiently large to exclude overall between-subject variability as a cause for the observed differences in exposure between subjects with severe renal impairment and healthy subjects.
The currently recommended dose of olodaterol in COPD therapy is 5 μg once daily, but the twofold higher dose of 10 μg once daily was also shown to be well tolerated and safe when administered over 48 weeks in Phase III studies.4,5 Given the known linear pharmacokinetics of olodaterol, a moderate increase of systemic exposure, as observed in the renal impairment study, is thus not expected to give rise to safety concerns.
Olodaterol was well tolerated in both trials. In the hepatic impairment trial, the two adverse events consisted of decreased serum blood potassium observed in subjects with moderate hepatic impairment and resulted in a potassium decrease below the standard reference range. However, the predose potassium levels of those subjects were already low with regard to the reference range, which is a frequent clinical observation in patients with altered liver function.16 Both events were of mild intensity and assessed as drug related. The events were treated with concomitant medication and were recovered at the end of the trial. Such effects on serum potassium (temporary potassium shift) are a known class effect of β2-mimetic compounds.14,15,18 No adverse events were observed in healthy volunteers and subjects with mild hepatic impairment. No severe, serious, or significant adverse events were observed. In the renal impairment trial, olodaterol was equally well tolerated by subjects with severe renal impairment and healthy subjects. Adverse events were observed in healthy subjects only, and none was characterized by the investigator as related to treatment with olodaterol or involved serum potassium concentrations. Safety laboratory data and vital signs did not indicate any treatment-related untoward reactions.
The plasma protein binding of olodaterol was unaltered by either hepatic impairment or renal impairment compared to normal subjects. The pharmacologically active olodaterol metabolite SOM 1522 was not detectable in the plasma of any subject in either trial. Therefore, there is no indication that this metabolite could reach pharmacologically relevant systemic exposure in hepatic or renal impairment.
Conclusion
In conclusion, single oral doses of olodaterol were safe and well tolerated in the trial subjects. Impaired hepatic or renal function did not translate into a clinically relevant change of systemic exposure to olodaterol compared to normal subjects.
Acknowledgments
The authors would like to acknowledge Dr Holger Fuchs for performance of the plasma protein-binding experiments. Dr Paul Tanswell provided editorial support with funding from Boehringer Ingelheim.
Disclosure
Christina Kunz, Doreen Luedtke, Anna Unseld, Alan Hamilton, Martina Wein and Stephan Formella are employees of Boehringer Ingelheim. Atef Halabi was the principal investigator at CRS Clinical Research Services Kiel GmbH, Kiel, Germany, where the studies, sponsored by Boehringer Ingelheim, were conducted. The authors report no other conflicts of interest in this work.
References
Gibb A, Yang LP. Olodaterol: first global approval. Drugs. 2013;73:1841–1846. | ||
Bouyssou T, Casarosa P, Naline E, et al. Pharmacological characterization of olodaterol, a novel inhaled β2-adrenoceptor agonist exerting a 24-hour-long duration of action in preclinical models. J Pharmacol Exp Ther. 2010;334:53–62. | ||
Casarosa P, Kollak I, Kiechle T, et al. Functional and biochemical rationales for the 24-hour-long duration of action of olodaterol. J Pharmacol Exp Ther. 2011;337:600–609. | ||
Ferguson GT, Feldman GJ, Hofbauer P, et al. Efficacy and safety of olodaterol once daily delivered via Respimat in patients with GOLD 2–4 COPD: results from two replicate 48-week studies. Int J Chron Obstruct Pulmon Dis. 2014;9:629–645. | ||
Koch A, Pizzichini E, Hamilton A, et al. Lung function efficacy and symptomatic benefit of olodaterol once daily delivered via Respimat versus placebo and formoterol twice daily in patients with GOLD 2–4 COPD: results from two replicate 48-week studies. Int J Chron Obstruct Pulmon Dis. 2014;9:697–714. | ||
Hodder R, Price D. Patient preferences for inhaler devices in chronic obstructive pulmonary disease: experience with Respimat Soft Mist inhaler. Int J Chron Obstruct Pulmon Dis. 2009;4:381–390. | ||
Striverdi Respimat (olodaterol) inhalation spray for oral inhalation [prescribing information]. 2014. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/203108s000lbl.pdf. Accessed October 25, 2014. | ||
van Noord JA, Smeets JJ, Drenth BM, et al. 24-Hour bronchodilation following a single dose of the novel β2-agonist olodaterol in COPD. Pulm Pharmacol Ther. 2011;24:666–672. | ||
US Food and Drug Administration. Guidance for Industry: Pharmacokinetics in Patients with Impaired Hepatic Function – Study Design, Data Analysis, and Impact on Dosing and Labeling. Rockville (MD): FDA; 2003. | ||
US Food and Drug Administration. Guidance for Industry: Pharmacokinetics in Patients with Impaired Renal Function – Study Design, Data Analysis, and Impact on Dosing and Labeling. Rockville (MD): FDA; 1998. | ||
Borghardt JM, Weber B, Staab A, Kunz C, Formella S, Kloft C. Investigating pulmonary and systemic pharmacokinetics of inhaled olodaterol in healthy volunteers using a population pharmacokinetic approach. Br J Clin Pharmacol. Epub 2015 Sep 8. | ||
Antonelli Incalzi R, Fuso L, De Rosa M, et al. Co-morbidity contributes to predict mortality of patients with chronic obstructive pulmonary disease. Eur Respir J. 1997;10:2794–2800. | ||
Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–649. | ||
Adamus WS. Pharmacodynamic methods for investigating antiasthma drugs in healthy volunteers. Methods Find Exp Clin Pharmacol. 1998; 20:139–145. | ||
Vincent HH, Boomsma F, Man in’t Veld AJ, Derkx FH, Wenting GJ, Schalekamp MA. Effects of selective and nonselective β-agonists on plasma potassium and norepinephrine. J Cardiovasc Pharmacol. 1984;6:107–114. | ||
Tietz NW, Prüden EL, Siggaard-Andersen O. Electrolytes. In: Burtis CA, Ashwood ER, editors. Tietz Textbook of Clinical Chemistry. 2nd ed. Philadelphia: WB Saunders; 1994:1354–1374. | ||
Zhang Y, Zhang L, Abraham S, et al. Assessment of the impact of renal impairment on systemic exposure of new molecular entities: evaluation of recent new drug applications. Clin Pharmacol Ther. 2009;85:305–311. | ||
Elliott MJ, Ronksley PE, Clase CM, Ahmed SB, Hemmelgarn BR. Management of patients with acute hyperkalemia. CMAJ. 2010;182:1631–1635. |
Supplementary materials
Sensitivity analysis
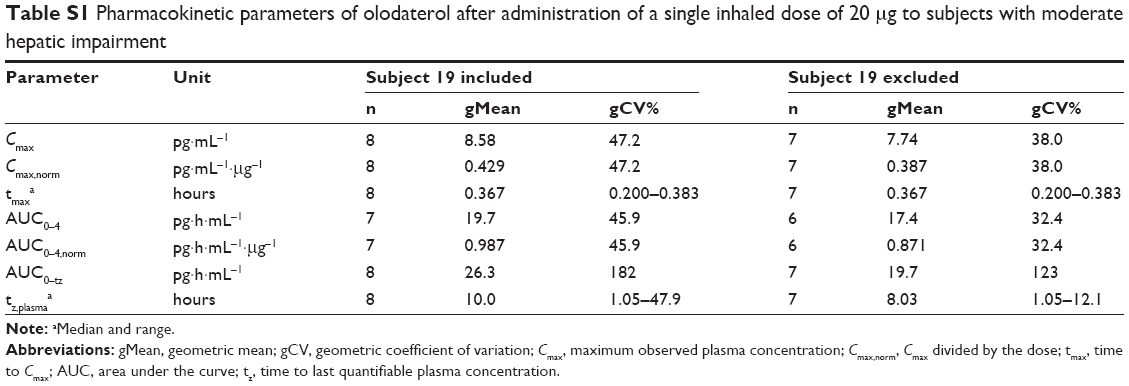 | Table S1 Pharmacokinetic parameters of olodaterol after administration of a single inhaled dose of 20 μg to subjects with moderate hepatic impairment |
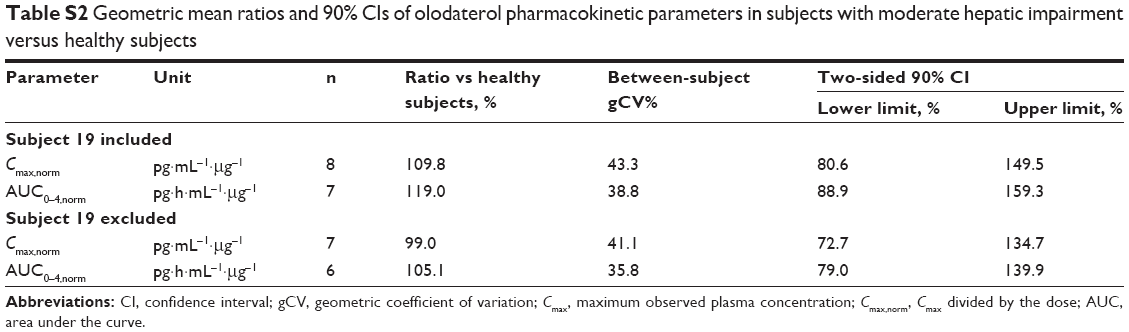 | Table S2 Geometric mean ratios and 90% CIs of olodaterol pharmacokinetic parameters in subjects with moderate hepatic impairment versus healthy subjects |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.