")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Pharmacokinetics and Bioequivalence Evaluation of Two Montelukast Sodium Chewable Tablets in Healthy Chinese Volunteers Under Fasted and Fed Conditions
Authors Li W, Wang Y, Pei Y, Xia Y
Received 24 December 2020
Accepted for publication 23 February 2021
Published 9 March 2021 Volume 2021:15 Pages 1091—1099
DOI https://doi.org/10.2147/DDDT.S298355
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Weihong Li,1,* Yanrong Wang,1,* Yingzi Pei,2 Yue Xia2
1GCP Office of Cangzhou Central Hospital, Cangzhou, Hebei, 061000, People’s Republic of China; 2Research Center of Beijing Fuyuan Pharmaceutical Co., Ltd. (Formerly Beijing Wansheng Pharmaceutical Co., Ltd.), Beijing, 101113, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Weihong Li
GCP Office of Cangzhou Central Hospital, No. 16 on Xinhua Road, Cangzhou, Hebei, 061000, People’s Republic of China
Tel +86 18713731395
Fax +86 3172072825
Email [email protected]
Purpose: The aim of this study was to assess and compare the pharmacokinetic (PK) properties and bioequivalence of montelukast sodium chewable tablets prepared by two different manufacturers in healthy Chinese volunteers to obtain adequate PK evidence for the registration approval of the test formulation.
Patients and Methods: A randomized-sequence, single-dose, open-label, 2-period crossover study was conducted in fasted and fed healthy Chinese volunteers (Chinese Clinical Trials Registry identifier: CTR20182362). Eighteen subjects each were selected for a fasted study and a fed study. Eligible participants were randomly assigned in a 1:1 ratio to receive a single dose of the reference formulation or the test formulation, followed by a 5-day washout period and the administration of the alternate formulation. Plasma samples were collected over a 24-hour period following tablet administration and analyzed for montelukast contents by high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). The PK parameters, such as maximum serum concentration (Cmax), area under the curve (AUC) from t = 0 to the last quantifiable concentration (AUC0–t), AUC from t = 0 to infinity (AUC0–∞), half-life (t1⁄ 2), time to Cmax (Tmax), and terminal elimination rate constant (λz), were evaluated. The safety assessment included changes in vital signs (blood pressure, pulse, and temperature) or laboratory tests (hematology, blood biochemistry, hepatic function, and urinalysis) and the incidence of adverse events (AEs).
Results: The geometric mean ratios (GMRs) between the two formulations for the primary pharmacokinetic parameters (Cmax, AUC0– 24, and AUC 0–∞) and the corresponding 90% confidence intervals (Cis) were all within the range of 80.00– 125.00% for both the fasting and fed states. The safety profiles for both treatments were comparable.
Conclusion: The PK analysis revealed that the test and reference formulations of montelukast sodium chewable tablets were bioequivalent and well-tolerated by healthy Chinese subjects.
Keywords: montelukast sodium, bioequivalence, pharmacokinetic profile, HPLC-MS/MS, adverse events
Introduction
Montelukast sodium is a highly selective, type I, cysteinyl leukotriene receptor antagonist. Montelukast sodium has been shown to significantly improve the clinical symptoms of children with bronchial asthma by inhibiting the combination of cysteinyl leukotriene and its receptor, improving the inflammatory index of asthma and alleviating leukotriene-mediated bronchoconstriction, increased vascular permeability, and mucus secretion.1,2
The combination of montelukast sodium and inhaled glucocorticoid has demonstrated synergistic and substitutive effects: the dosage of inhaled glucocorticoid can gradually be reduced through the continued use of the combination, allowing for the avoidance and reduction of local and systemic side effects associated with glucocorticoid use.3–5
The montelukast sodium chewable tablet has been indicated as an effective drug for the prevention and long-term treatment of asthma in children aged 2–14 years. In February 1998, a montelukast sodium chewable tablet developed by Merck Sharp & Dohme Ltd. was first approved by the United States Food and Drug Administration (US FDA) under the trade name Singulair. Currently, this drug is listed in Japan, the United Kingdom, France, Italy, and other countries around the world as a first-line asthma control drug.6
The oral absorption of montelukast has been shown to be rapid and complete, with a time to maximum serum concentration (Tmax) of approximately 2 hours and an average bioavailability of 73%. More than 99% of montelukast sodium binds to plasma protein. The average apparent volume of distribution (Vd) of montelukast was reported as 8–11 L. Montelukast and its metabolites are almost completely excreted through bile.7–9 In vitro experiments have demonstrated that montelukast acts as an inhibitor of cytochrome P450 (CYP) 2C8 and as a substrate of CYP 2C8, 2C9, and 3A4.10–12
The montelukast sodium chewable tablet has been widely prescribed for nearly 20 years, and previous studies have been conducted to evaluate the pharmacokinetic (PK) parameters and tolerability of this drug.7,13–15 However, no studies have compared the bioequivalence of 5 mg montelukast sodium chewable tablets produced by two different manufactures under fasted and fed conditions in healthy Chinese volunteers. Thus, the present study was designed to assess the relative bioavailability of two 5 mg montelukast sodium chewable tablets (test and reference) under fasting and fed conditions in healthy Chinese adult volunteers to lay a foundation for the marketing of this drug formulation in China (Chinese Clinical Trials Registry identifier: CTR20182362).
Patients and Methods
Formulations and Subject Selection
Montelukast sodium chewable tablets (5 mg/tablet; lot no.: 21,808,104; expiration date: July 2020), manufactured and provided by Beijing Fuyuan Pharmaceutical Co., Ltd. (formerly Beijing Wansheng Pharmaceutical Co., Ltd., Beijing, China) and montelukast sodium chewable tablets (5 mg/tablet; lot no.: R005764; expiration date: 3 October 2019) marketed under the brand name SINGULAIR®, manufactured by Merck Sharp & Dohme Ltd., were used as the test and reference formulations, respectively, for the assessment of bioequivalence.
Healthy Chinese male and female adults, aged 18 years and older and weighing at least 45 kg (female) or 50 kg (male), were eligible for inclusion in this study. Volunteers were assessed by a comprehensive medical examination to assess their health status, including a routine physical examination, measurement of vital signs (axillary temperature, blood pressure while seated at rest, heart rate, and pulse), medical history, laboratory tests (hematology, blood biochemistry, coagulation function, urinalysis, drug screening, hepatitis and human immunodeficiency virus [HIV] testing, syphilis specificity antibody, human chorionic gonadotropin [HCG] test, and alcohol breathalyzer test), 12-lead electrocardiography, and chest X-ray. Subjects with any history or evidence of the following were excluded: clinically relevant disease; acute or chronic disease; drug abuse in the past 2 years; smoking or alcohol abuse; allergic constitution; allergy to any ingredient in the montelukast sodium chewable tablets; and history of reacting to needle sticks or blood draws needle or blood sickness. Subjects who took any medications within 14 days before the first dosing, participated in any clinical studies within the past 3 months, experienced blood loss or blood donation of 200 mL or more, and female subjects who were pregnant, breastfeeding, or likely to become pregnant were also excluded.
All subjects were informed of the objectives, procedures, and risk-benefit analysis of the study by the clinical investigators, and each provided written informed consent, which was collected prior to study participation. Subjects were free to withdraw at any time.
Study Design and Treatment
This study was conducted at the Phase I Clinical Research Center of the Cangzhou Central Hospital between January 2019 and April 2019. The protocol was approved by the ethics committee of the Cangzhou Central Hospital (approval No. 2018-038-01, 2018-038-02). The study was performed in accordance with the Declaration of Helsinki,16 the International Conference on Harmonization Good Clinical Practice Guideline,17 and the Guideline for Good Clinical Practice recommended by the National Medical Products Administration (NMPA).18
This study was an open-label, single-dose, randomized, 2-formulation, 2-period crossover bioequivalence study, with a 5-day washout period between each administration. This study was comprised of two independent trials: a fasted trial and a fed trial. The subjects in each trial were assigned into Test/Reference (T/R) or Reference/Test (R/T) groups at a 1:1 ratio, according to a random number table generated by SAS statistical software, v9. 3.
Each subject received a single dose of the test or reference tablet (one 5 mg montelukast sodium chewable tablet), administered orally with 240 mL warm water under light-proof conditions. The subjects in the fasted trial performed an overnight fast of at least 10 hours, whereas the subjects in the fed trial received a standard, high-fat, and high-calorie (800–1000 kcal) breakfast (30.2% carbohydrate, 17.7% protein, and 52.1% fat) 30 minutes before drug administration. Additional water intake was withheld for 2 hours after treatment. A standardized meal was provided 4 hours and 10 hours after treatment. Alcoholic beverages, strenuous activity, and smoking were prohibited during the study.
Blood Sampling
For the fasted trial, blood samples (4 mL) were collected into coded, K2-EDTA anticoagulation tubes pre-dose (0 hour, baseline) and 0.25, 0.5, 1, 1.5, 2, 2.5, 3,3.5, 4, 4.5, 5, 6, 8, 10, 15, and 24 hours post-dose, followed by centrifugation at 2000 × g at 4°C for 10 min, within 1 hour of collection. The supernatant (plasma samples) was collected and stored at −70 ± 10°C within 2 hours of collection until used for analysis.
For the fed trial, blood samples (4 mL) were collected into coded, K2-EDTA anticoagulation tubes pre-dose (0 hours, baseline) and 0.5, 1, 2, 2.5, 3.3.5, 4, 4.5, 5, 5.5, 6, 7, 8, 10, 15, and 24 hours post-dose, followed by centrifugation at 2000 × g at 4°C for 10 min within 1 hour of collection. The supernatant (plasma samples) was collected and stored at −70 ± 10°C within 2 hours of collection until used for analysis.
All blood samples were light-proof during collection, processing, and storage.
Analytical Determinations
A high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) method was used to detect the plasma concentrations of montelukast using an API 4000 LC/MS/MS System. The method involved repeated solid-phase extraction steps and the quantitation of the target compounds, which was performed using a positive-ion mode and multiple reaction monitoring (MRM). The analytes were chromatographed using a Shimadzu LC-30AD and a Thermo Scientific Hypersil GOLD AQ column (30 × 2.1 mm; 3 µm). The mobile phase consisted of 0.1% formic acid in acetonitrile at a flow rate of 0.8 mL/min. The MS/MS API 4000 was used to analyze the mass. Analyst 1.6.3 was used for data acquisition and analysis.
The average deviation in accuracy for each sample concentration ranged from 96.5% to 97.7%, and the average coefficient of variation (CV%) was ≤3.2%. After 24 hours of treatment, at room temperature, under light-exposed or light-proof conditions, following four freeze-thaw cycles of drug-containing plasma, from −80 to −60°C, and 69 days of long-term storage at either −30 to −10°C or −80 to −60° C, the stability of montelukast was acceptable The linear range of montelukast was from 1 ng/mL to 500 ng/mL. The deviation accuracy of the standard curve, as determined by the lower limit of quantification (LLOQ), was −14.9% to 0.8%, and the deviation accuracy for samples with concentrations outside of the LLOQ was −4.4% to 2.6%.
Safety Assessment
The safety assessment included monitoring changes in vital signs and laboratory test results, physical examination findings, and the incidence of adverse events (AEs). Vital signs, including temperature, blood pressure, and pulse, were measured at −1 d, 1 hour before administration, and 3 and 24 hours after each administration. Laboratory tests, including a routine blood test, urinalysis, liver function, renal function, blood glucose, and female β-HCG test, in addition to physical examination and 12-lead electrocardiogram, were performed at baseline and 24 hours after the second administration. Any AEs that occurred during the study were collected through medical observations and spontaneous reports by the subjects (both while in the hospital and at discharge).
The Medical Dictionary for Regulatory Activities, v20.0, was used to code all AEs to the preferred term and systemic organ class.
Pharmacokinetic and Statistical Analysis
All subjects who completed both Period 1 and Period 2 without any major protocol deviations were included in the PK analysis set. The primary endpoints were area under the curve, from t = 0 to infinity (AUC0-∞); AUC from t = 0 to the last quantifiable concentration (AUC0-t); and maximum serum concentration (Cmax). The secondary endpoints were time to Cmax (Tmax), half-life (t1/2), and terminal elimination rate constant (λz).
The PK parameters were calculated by Phoenix WinNonlin Software, v7.0, using the non-compartmental method, and individual plasma concentration–time curves were constructed. Cmax and Tmax were obtained from the acquired data. The linear trapezoidal rule was used to calculate AUC0-t. AUC0–∞ was calculated as the sum of AUC0–t and the extrapolated area, which was determined by dividing the last quantifiable concentration (Ct) by the slope of the terminal log-linear phase (Ke). The t1/2 value was calculated from the slope of the terminal log-linear phase, as 0.693/Ke. The relative bioavailability (F) of the tested formulation was calculated as follows: F = AUC0-t (test)/AUC0-t (reference) × 100%. According to the NMPA regulatory guidelines, a one-way analysis of variance (ANOVA) was used to analyze the logarithm (ln)-transformed PK parameters (AUC0–∞, AUC0-t, and Cmax) to assess the effects of treatment, sequence, period, and subjects nested in sequence. The Wilcoxon signed-rank test was used to compare Tmax values between groups. Statistical analysis was performed using the statistical software package SAS, V9.3 (SAS Institute Inc, Cary, North Carolina, license number 000063317058), using the General Linear Model (GLM) procedure. The two formulations were considered bioequivalent if the differences between the compared parameters were found to be insignificant (P>0.05) and if the 90% confidence interval (CI) for the geometric mean ratio (GMR) of AUC0-∞, AUC0-t, and Cmax fell within 80% and 125%.
Parametric 90% CIs for the GMR ratio between the two formulations (test–reference) were determined using the Schuirmann method.
Results
Subjects
A total of 156 healthy Chinese subjects were screened (see Figure 1). The fasting trial and the fed trial each enrolled 18 subjects (12 male/6 female and 11 male/7 female, respectively). No included subjects were excluded. Table 1 shows the demographic characteristics of all included subjects.
 |
Figure 1 Study design and disposition of subjects. |
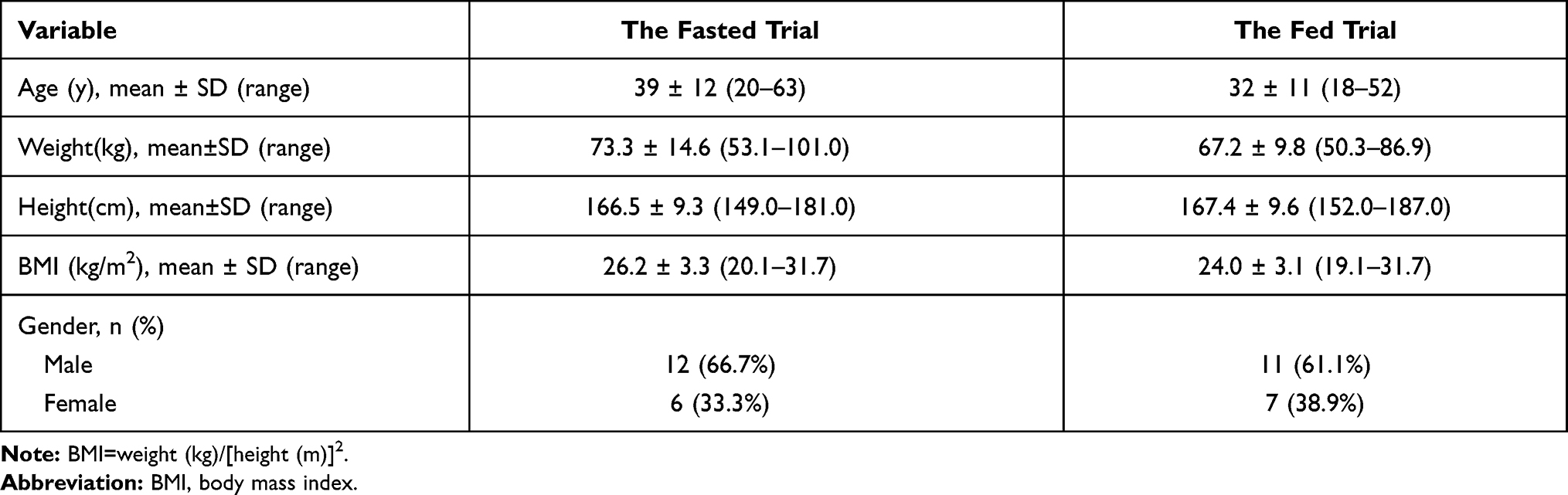 |
Table 1 Demographic Characteristics of Subjects in the Fasted Trial (n=18) and Fed Trial (n=18) |
Pharmacokinetic Properties
All 18 subjects in both the fasted and fed trials completed both periods and were included in the pharmacokinetic analysis.
The mean plasma concentration-time profiles for montelukast obtained following a single oral dose of the test and reference montelukast formulations under fasted and fed states are shown in Figures 2 and 3. The PK parameters for montelukast are shown in Table 2.
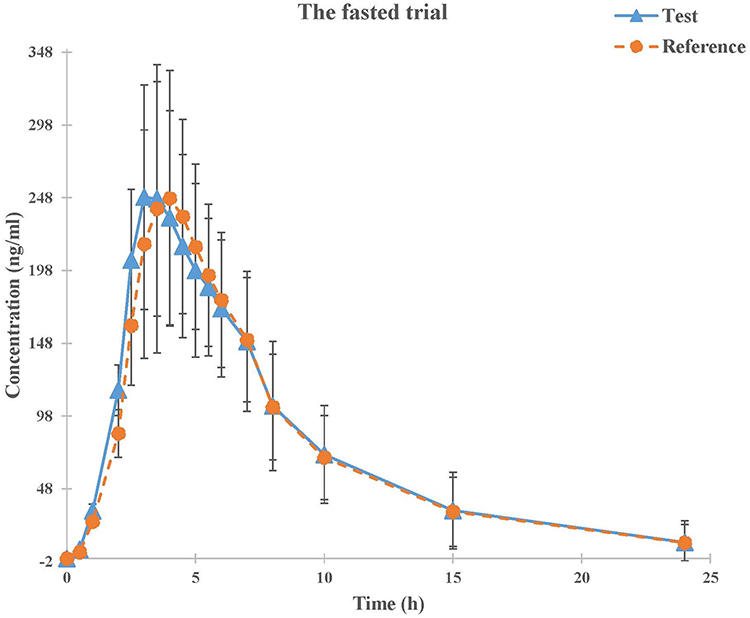 |
Figure 2 Mean plasma concentration-time profiles of test (n=18) and reference (n=18) formulations under fasted condition. Note: Data represent the mean value, and error bars represent the SD. |
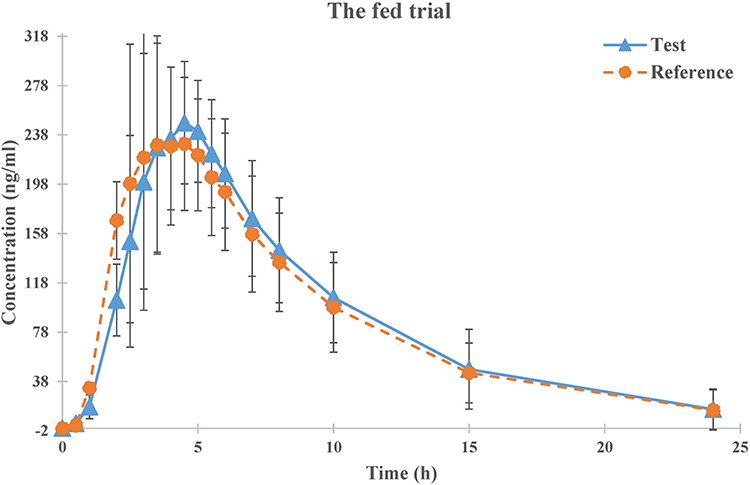 |
Figure 3 Mean plasma concentration-time profiles of test (n=18) and reference (n=18) formulations under fed condition. Note: Data represent the mean value, and error bars represent the SD. |
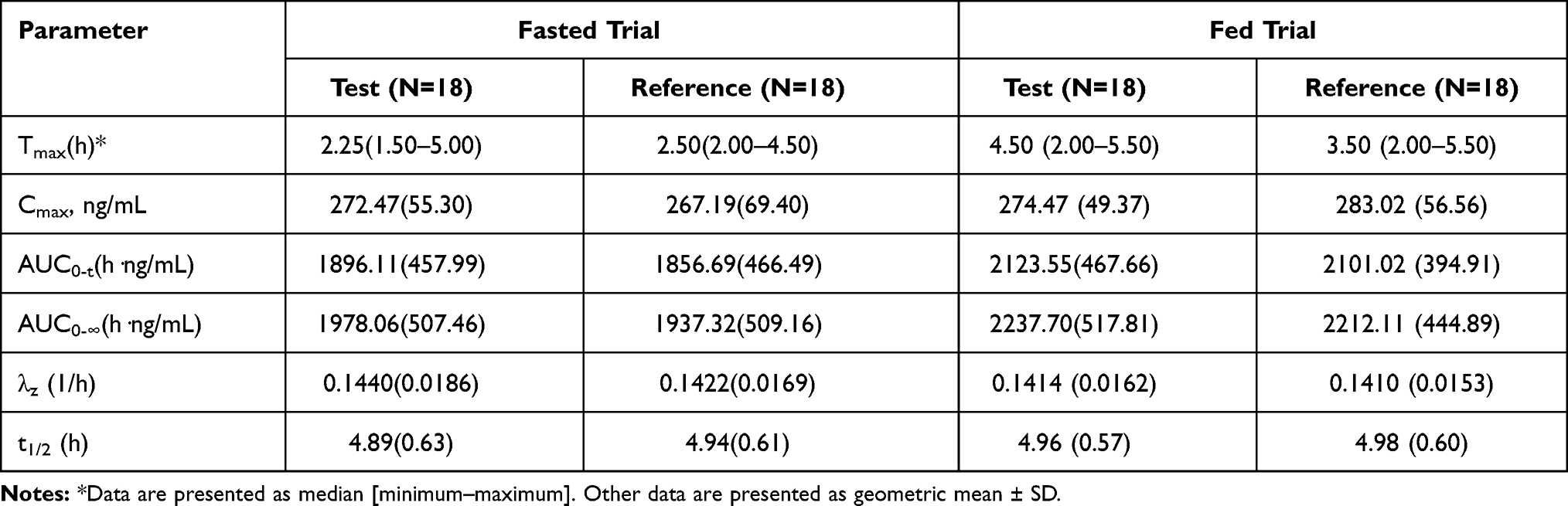 |
Table 2 PK Parameters of Montelukast After Administration of Test and Reference Formulations in Fasted or Fed State |
In the fasted trial, the comparison of primary PK parameters between the test and reference products for the bioequivalence evaluation showed that the GMR (90% CI) values for Cmax, AUC0–t, and AUC 0–∞ were 103.12% (96.88% to 109.76%), 102.24% (96.42% to 108.42%), and 102.16% (96.22% to 108.48%), respectively. All of the calculated 90% CIs were within the pre-defined equivalence margin of 80.00% to 125.00%. In the fed trial, the comparison of primary PK parameters between the test and the reference formulations showed that the GMR (90% CI) values for Cmax, AUC0–t, and AUC 0–∞ were 97.36% (91.13% to 104.00%), 100.48% (95.81% to 105.39%), and 100.56% (95.41% to 105.99%), respectively. Similar to the fast trial, the corresponding 90% CIs for the fed trial were within the pre-defined equivalence margin of 80.00% to 125.00%. Tables 3 and 4 show the results of the variance analyses for Cmax, AUC0–t, and AUC0–∞ after logarithmic transformation and the average bioequivalence evaluations for the plasma PK parameters for montelukast administration under fasted or fed conditions.
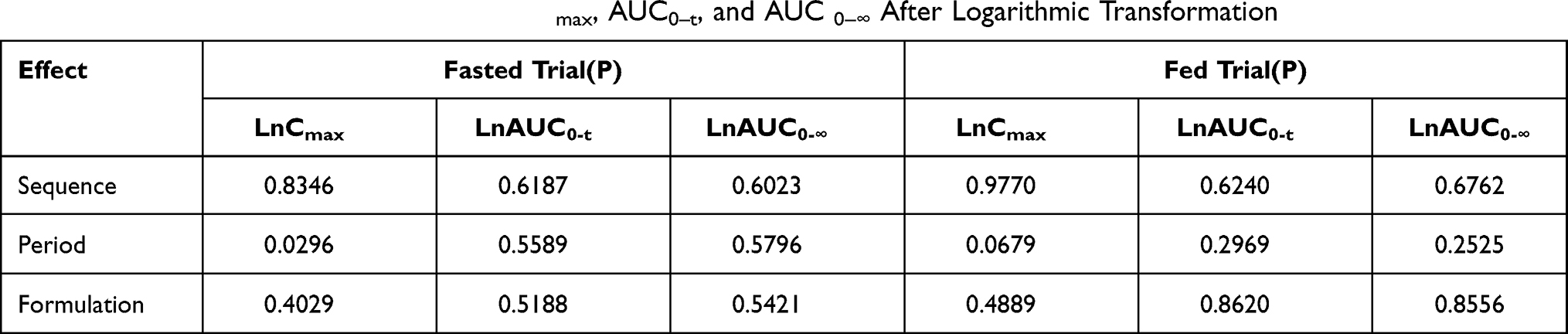 |
Table 3 The Results of Variance Analysis of Cmax, AUC0–t, and AUC 0–∞ After Logarithmic Transformation |
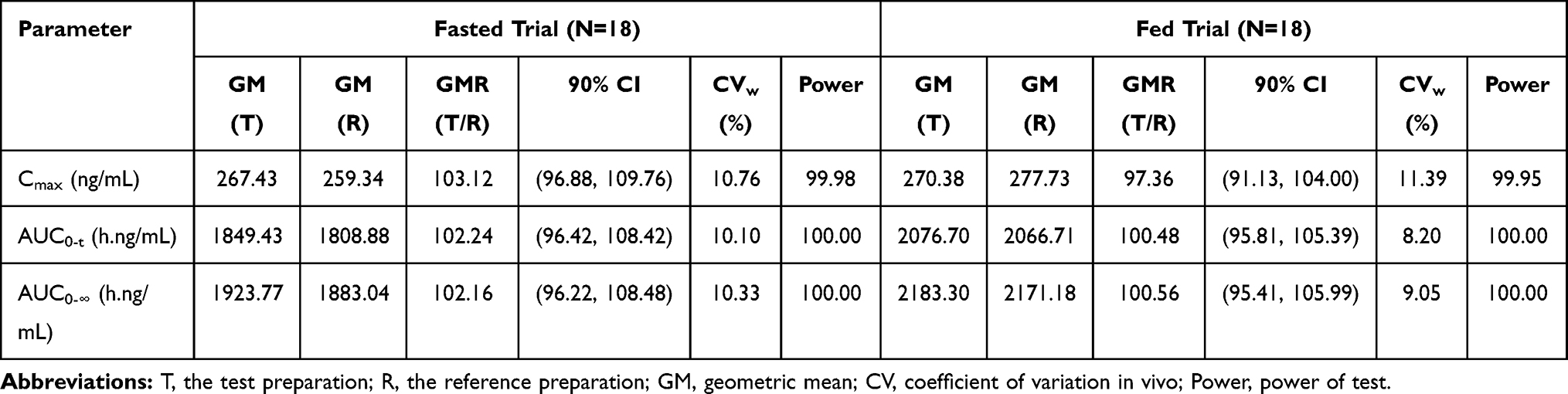 |
Table 4 The Results of Bioequivalence Evaluation for Plasma Pharmacokinetic Parameters of Montelukast Under Fasted or Fed Condition |
Safety Assessment
In the fasted trial, the incidence of AEs was 38.9%, with a total of 13 AEs recorded in 7 subjects. All of these AEs (100%) were identified through laboratory investigations and were considered mild, without requiring treatment. The incidence of AEs in the fasted trial is summarized in Table 5.
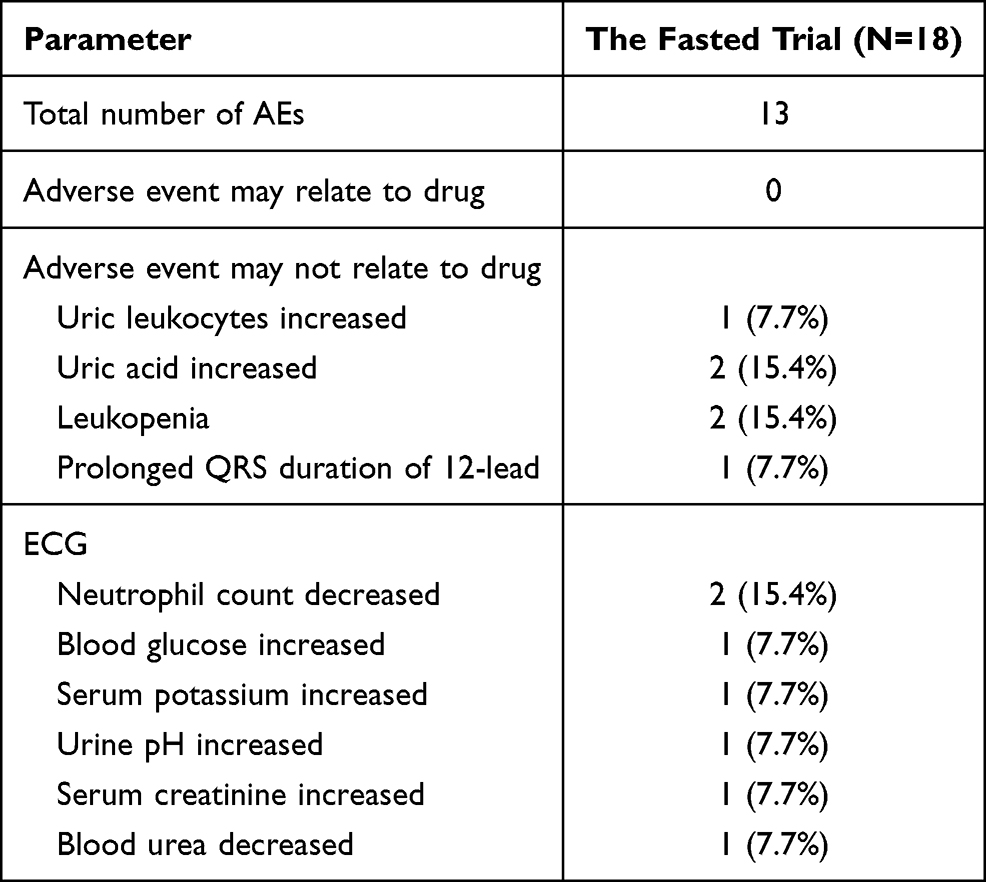 |
Table 5 AEs Profile in the Fasted Trial |
In the fed trial, no AEs were recorded. No serious AEs were reported in either trial.
Discussion
This study examined the PK properties and evaluated the bioequivalence between a test montelukast sodium chewable tablet and a reference montelukast sodium chewable tablet in healthy Chinese adult volunteers under fasted and fed conditions. For both the fasted and fed trials, all enrolled subjects completed the study, and no subjects dropped out or were excluded. All subjects were included in the pharmacokinetic analysis set and bioequivalence analysis set.
The analysis, which was based on Cmax, AUC0-t, and AUC0-∞, revealed that the 2 formulations of montelukast were similar in terms of PK properties in healthy Chinese subjects. The 90% CIs for the GMR values of Cmax, AUC0–t, and AUC0-∞ were all within the 80.00% to 125.00% range, which represents the standard bioequivalence acceptance range according to NMPA guidelines.19
In the fasted trial, subject FA-15 (FA-S069) presented with nitrite-positive during the routine urinalysis during the screening period, which was abnormal, had clinical significance, did not meet the inclusion criteria, and was not detected in time to exclude the volunteer from the study. Therefore, the subject was included and allowed to complete the trial. Because this was a minor protocol violation, we conducted a sensitivity analysis in which we excluded this subject. The analysis results are shown in Table 6 and indicated that after excluding subject FA-15 (FA-S069) to perform the sensitivity analysis, the 90% CIs for the GMR values of Cmax, AUC0–t, and AUC0-∞ remained between 80.00% and 125.00%. These results are consistent with those (shown in Table 7) using the same reference formulation previously reported.15
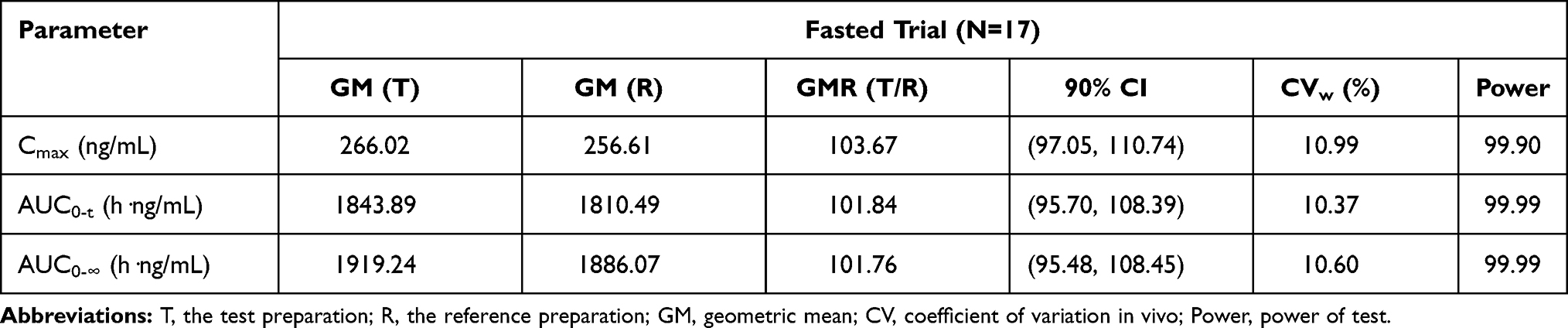 |
Table 6 The Results of Bioequivalence Evaluation for Plasma Pharmacokinetic Parameters of Montelukast Under Fasted Condition (Sensitivity Analysis) |
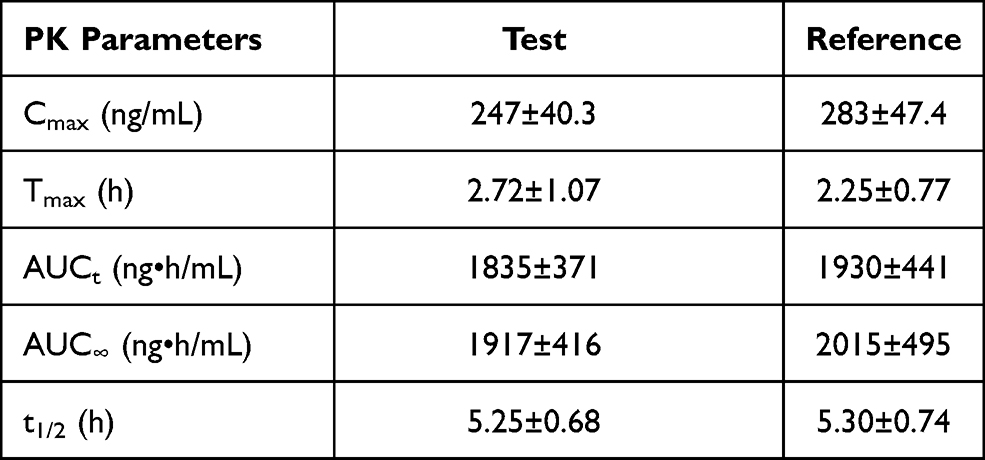 |
Table 7 PK Parameters of Montelukast After Administration of Test and Reference Formulations in Healthy Korean Adult Male Volunteers Under Fasted State (n=32) |
Moreover, the Wilcoxon test comparing Tmax values between groups indicated no significant difference between the test and reference montelukast formulations, regardless of whether they were administered under fasting or fed conditions, as shown in Table 8.
 |
Table 8 Wilcoxon Signed Rank Test Results of Tmax (h) |
Additionally, we compared the PK data between the fasting and fed trial and found that food influenced the AUC0-t, AUC0-∞, and Tmax values for both the test and reference montelukast formulations. When montelukast was administered after a standard high-fat, high-calorie breakfast, approximately 13% increases in the AUC0-t and AUC0-∞ values were observed for both the test and reference montelukast formulations, with approximately 100% and 40% increases in Tmax observed for the test and reference formulations, respectively. Thus, food appeared to slow the rate of absorption and increase the absorption extent of montelukast.
In this study, no serious AEs were observed, and all observed AEs were mild. Thus, both the test and the reference formulations were well-tolerated among our study population.
Conclusion
After the oral administration of 5 mg of either the test or reference preparation under both fasted and fed conditions, no subjects withdrew from the study due to AEs, and no serious AEs occurred, which indicated that both formulations were well-tolerated and their safety profiles were acceptable The PK analysis showed that the primary PK parameters (Cmax, AUC0–t, and AUC0–∞) were within the pre-defined equivalence margin of 80.00% to 125.00% and met the regulatory criteria established by the People’s Republic of China, regardless of whether it was administered under fasted or fed conditions. Therefore, the test and reference products were determined to be bioequivalent.
Thus, the 5 mg montelukast sodium chewable tablets manufactured by Beijing Fuyuan Pharmaceutical Co., Ltd. (formerly Beijing Wansheng Pharmaceutical Co., Ltd.) would represent an affordable, acceptable, and beneficial alternative for use in Chinese patients to the currently available reference formulation.
Data Sharing Statement
Individual deidentified participant data will not be shared. All available data have been included in this manuscript. No other study-related documents will be made available.
Acknowledgments
We thank all of the healthy subjects for their participation in this study and the sponsor, Beijing Fuyuan Pharmaceutical Co., Ltd. (formerly Beijing Wansheng Pharmaceutical Co., Ltd.), for their financial support.
Disclosure
Yingzi Pei and Xia Yue are affiliated with Beijing Fuyuan Pharmaceutical Co., Ltd. (formerly Beijing Wansheng Pharmaceutical Co., Ltd.). The authors report no other conflicts of interest in this work.
References
1. Ellis KL, Fan LT, Leung AKC. Clinical effectiveness and safety of montelukast in asthma. What are the conclusions from clinical trials and meta-analyses? Drug Des Devel Ther. 2014;2014:839–850. doi:10.2147/DDDT.S39100
2. Zubairi ABS, Salahuddin N, Khawaja A, et al. A randomized, double-blind, placebo-controlled trial of oral montelukast in acute asthma exacerbation. BMC Pulm Med. 2013;13(1):1–7. doi:10.1186/1471-2466-13-20
3. Yang DZ, Liang J, Zhang F, Yao HB, Shu Y. Clinical effect of montelukast sodium combined with inhaled corticosteroids in the treatment of osas children. Medicine. 2017;96(19):e6628. doi:10.1097/MD.0000000000006628
4. Joos S, Miksch A, Szecsenyi J, et al. Montelukast as add-on therapy to inhaled corticosteroids in the treatment of mild to moderate asthma: a systematic review. Thorax. 2008;63(5):453–462. doi:10.1136/thx.2007.081596
5. Keith PK, Koch C, Djandji M, et al. Montelukast as add-on therapy with inhaled corticosteroids alone or inhaled corticosteroids and long-acting beta-2-agonists in the management of patients diagnosed with asthma and concurrent allergic rhinitis (the RADAR trial). Can Respir J. 2016;16(Suppl A):17A. doi:10.1155/2009/145071
6. Montella S, Maglione M, Stefano SD, et al. Update on leukotriene receptor antagonists in preschool children wheezing disorders. Ital J Pediatr. 2012;38(1):29. doi:10.1186/1824-7288-38-29
7. Naser ZA, Murad A, David W, et al. Investigation of the bioequivalence of montelukast chewable tablets after a single oral administration using a validated LC-MS/MS method. Drug Des Devel Ther. 2015;9:5315. doi:10.2147/DDDT.S87938
8. Kim S, Kim KJW
9. Moon SJ, Yu KS, Jung J, Kim Y, Kim MG. Comparative pharmacokinetics of a montelukast/levocetirizine fixed-dose combination chewable tablet versus individual administration of montelukast and levocetirizine after a single oral administration in healthy Korean male subjects. Int J Clin Pharmacol Ther. 2020;58(6):354–362. doi:10.5414/CP203709
10. Dennison J, Puri A, Warrington S, et al. Amenamevir: studies of potential CYP2C8‐ and CYP2B6‐mediated pharmacokinetic interactions with montelukast and bupropion in healthy volunteers. Clin Pharmacol Drug Dev. 2018;7(8):860–870. doi:10.1002/cpdd.578
11. Cardoso JDO, Oliveira RV, Lu JBL, et al. In vitro metabolism of montelukast by cytochrome P450s (CYPs) and UDP-glucuronosyltransferases (UGTs). Drug Metab Dispos. 2015;43(12):1905–1916. doi:10.1124/dmd.115.065763
12. Karonen T, Neuvonen PJ, Backman JT. CYP2C8 but not CYP3A4 is important in the pharmacokinetics of montelukast. Br J Clin Pharmacol. 2012;73(2):257–267. doi:10.1111/j.1365-2125.2011.04086.x
13. Haarman MG, Van Hunsel F, De Vries TW. Adverse drug reactions of montelukast in children and adults. Pharmacol Res Perspect. 2017;5(5):e00341. doi:10.1002/prp2.341
14. Arnold DH, Bowman N, Reiss TF, Hartert TV, Seger DL. Adverse events are rare after single-dose montelukast exposures in children. Clin Toxicol (Phila). 2018;56(1):25–29. doi:10.1080/15563650.2017.1337123
15. Kim HT, Song YK, Lee SD, Park Y, Kim CK. Relative bioavailability of two 5-mg montelukast sodium chewable tablets: a single dose, randomized, open-label, 2-period crossover comparison in healthy Korean adult male volunteers. Arzneimittelforschung. 2012;62(3):123–127. doi:10.1055/s-0031-1298004
16. World Medical Association. Declaration of Helsinki. Available from: https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/.
17. The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Guideline for good clinical practice. Available from: https://www.ich.org/products/guidelines/efficacy/efficacy-single/article/integrated-addendum.good-clinical-practice.html.
18. National Medical Products Administration. Guideline for good clinical practice. Available from: http://www.nmpa.gov.cn/WS04/CL2101/329583.html.
19. National Medical Products Administration. Center for Drug Evaluation. Guideline for bioavailability and bioequivalence studies of generic drug products. Available from: http://www.nmpa.gov.cn/WS04/CL2093/331454.html.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.