")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Pharmacokinetic comparison of sustained- and immediate-release formulations of cilostazol after multiple oral doses in fed healthy male Korean volunteers
Authors Kim YH, Ghim J, Jung JA, Cho S, Choe S, Choi HY, Bae K , Lim H
Received 18 April 2015
Accepted for publication 19 May 2015
Published 9 July 2015 Volume 2015:9 Pages 3571—3577
DOI https://doi.org/10.2147/DDDT.S86845
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Yo Han Kim,1 Jong-Lyul Ghim,2 Jin Ah Jung,2 Sang-Heon Cho,3 Sangmin Choe,4 Hee Youn Choi,1 Kyun-Seop Bae,1 Hyeong-Seok Lim1
1Department of Clinical Pharmacology and Therapeutics, College of Medicine, University of Ulsan, Asan Medical Center, Seoul, 2Department of Clinical Pharmacology, Inje University, Busan Paik Hospital, Busan, 3Department of Clinical Pharmacology, Inha University Hospital, Inha University School of Medicine, Incheon, 4Clinical Trials Center, Pusan National University Hospital, Busan, Republic of Korea
Background: A new extended-release form of cilostazol has recently been developed. This study was conducted to compare the pharmacokinetic characteristics of sustained-release (SR) and immediate-release (IR) formulations of cilostazol after multiple oral doses in healthy male Korean volunteers.
Methods: This was an open-label, randomized, multiple-dose, crossover study conducted in 30 healthy Korean subjects. In each treatment period, subjects received oral doses of 200 mg SR formulation every 24 hours or 100 mg IR formulation every 12 hours for 5 consecutive days in a fed state, with a washout period of 9 days. The plasma concentrations of cilostazol and its metabolites were determined using a validated liquid chromatography-tandem mass spectrometry method. The area under the plasma concentration–time curve within a dosing interval (AUCT), the measured peak plasma concentration at steady state (Cmax,ss), and the time to reach Cmax,ss (tmax,ss) were analyzed using a noncompartmental method.
Results: A total of 24 healthy male subjects completed the study. The mean (standard deviation [SD]) AUCT (96–120 hours) values for SR and IR were 27,378.0 (10,301.6) ng·h/mL and 27,860.3 (7,152.3) ng·h/mL, respectively. The mean (SD) Cmax,ss values were 2,741.4 (836.0) ng/mL and 2,051.0 (433.2) ng/mL, respectively. The median tmax,ss values were 8.0 hours and 4.0 hours, respectively. The geometric mean ratios (90% confidence intervals) of the SR to IR formulations were 0.937 (0.863–1.017), 0.960 (0.883–1.043), and 0.935 (0.859–1.017) for AUCT and 0.644 (0.590–0.703), 0.586 (0.536–0.642), and 0.636 (0.577–0.702) for dose-normalized Cmax,ss of cilostazol, OPC-13015 (3,4-dehydro-cilostazol), and OPC-13213 (4'-trans-hydroxyl-cilostazol), respectively. All formulations were well tolerated.
Conclusion: At steady state, the AUCT of cilostazol SR 200 mg is comparable to that of cilostazol IR 100 mg twice a day in healthy male Korean subjects. Both formulations are well tolerated.
Keywords: cilostazol, bioavailability, sustained release, immediate release, pharmacokinetics, healthy subjects
Introduction
Cilostazol is a phosphodiesterase III inhibitor that inhibits platelet aggregation and vasodilation. It is approved for the treatment of intermittent claudication resulting from peripheral arterial disease.1,2 Recent studies have reported that cilostazol is also effective for the prevention of progression of symptomatic intracranial arterial stenosis and prevention of secondary cerebral infarction.3,4
The pharmacokinetics of cilostazol is linear at a dose range of 25–300 mg.5 A high-fat meal increases the rate and extent of absorption, with an approximately 90% increase in the peak plasma concentration (Cmax) and a 25% increase in the area under the curve (AUC).6 Approximately 95%–98% of cilostazol is bound to plasma proteins, mainly to albumins. After oral administration, cilostazol is extensively metabolized by cytochrome P450 isoenzymes, mainly by 3A4 and to a lesser extent by 2C19.7 Two major pharmacologically active metabolites of cilostazol are OPC-13015 (3,4-dehydro-cilostazol) and OPC-13213 (4′-trans-hydroxyl-cilostazol).8,9
Cilostazol has poor water solubility, and orally administered cilostazol is absorbed mainly in the upper gastrointestinal tract, with absorption reducing as the drug moves into the lower gastrointestinal tract.10 For this reason, cilostazol has been formulated as immediate-release (IR) tablets. Because these tablets must be taken twice daily, attempts have been made to develop a sustained-release (SR) cilostazol formulation. The SR formulation of cilostazol is prepared with a release-controlling polymer, which ensures a stable elution rate of cilostazol according to changes in pH. Thus, the SR formulation may have a similar efficacy to that of once-daily dosing frequency but with higher drug compliance. The objectives of this study were to assess the tolerability and pharmacokinetic (PK) properties of multiple oral doses of cilostazol SR and IR formulations in healthy male volunteers.
Materials and methods
Subjects
Healthy male volunteers aged 19–55 years who were nonsmokers, had a body weight >50 kg, and were within 20% of their ideal body weight at screening (ideal body weight in kilograms = [height in centimeters − 100] ×0.9) were eligible for this study. Volunteers were considered to be in good health based on medical history, physical examinations, vital sign measurements (blood pressure, heart rate, and body temperature), 12-lead electrocardiograms (ECGs), clinical laboratory tests (hematology, blood chemistry, and urinalysis), serology (hepatitis B surface antigen, hepatitis C virus antibody, and HIV antibody), and urine drug screening (amphetamine, cocaine, opiate, barbiturate, and benzodiazepine).
Volunteers were excluded for the following reasons: exposure to any investigational drug or placebo within 60 days of the first study medication dose; any illness within 14 days of the first study medication dose; aspartate aminotransferase or alanine aminotransferase levels >1.25× the upper normal limit; total bilirubin level >1.5× the upper normal limit; a platelet count <170,000 or >360,000; prothrombin time (PT) or activated partial thromboplastin time (aPTT) >1.25× the upper normal limit; and bleeding time >8 minutes.
Study design
The study protocol was approved by the Korea Food and Drug Administration and the institutional review board of the Asan Medical Center (Seoul, Republic of Korea). The study was conducted at the Clinical Trials Center of Asan Medical Center from September 2008 to November 2008 and performed according to the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use–Good Clinical Practice (ICH-GCP) guidelines. All subjects provided written informed consent before screening tests. The subjects were also informed that they had the right to withdraw their consent at any time without penalty.
This study was a randomized, open-label, multiple-dose, crossover clinical trial. Subjects were randomly assigned to two groups; patients in the SR group were administered 200 mg SR formulation every 24 hours for 5 days, while those in the IR group were administered 100 mg IR formulation every 12 hours for 5 days. All treatments were given after consumption of a standard, high-fat breakfast of 900 Kcal to 1,000 Kcal. Following a 9-day washout period, subjects received alternate formulations.
For each treatment period, subjects were admitted in the Clinical Trials Center at Asan Medical Center from Day −1 through Day 7 (144 hours after dosing) and visited on Day 8 to assess drug tolerability and PK properties. The schedule for the second period was the same as in the first period. End-of-study visits were performed within 6–10 days after the last dose of treatment.
Tolerability
Tolerability was assessed throughout the study using vital sign measurements, 12-lead ECG recording, clinical laboratory tests (hematology, blood chemistry, and urinalysis), physical examinations, and monitoring of adverse events (AEs). Vital signs (sitting blood pressure and heart rate) were recorded at screening, at predose baseline, at 6 hours, 24 hours, 48 hours, 72 hours, 96 hours, 120 hours, 144 hours, and 168 hours after study drug administration, and at follow-up visit. ECG and clinical laboratory tests were performed at screening, predose baseline, and 7 days after each study drug administration. AEs were recorded in terms of symptoms and signs, duration, intensity, relationship to the study drug, action taken, outcome, and severity.
Sample collection and quantification
Blood samples were obtained from subjects administered the SR formulation immediately before and at 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 12 hours, 13 hours, 14 hours, 16 hours, 18 hours, 24 hours, 48 hours, 72 hours, 96 hours, 97 hours, 98 hours, 99 hours, 100 hours, 102 hours, 104 hours, 106 hours, 108 hours, 109 hours, 110 hours, 112 hours, 114 hours, 116 hours, 120 hours, 132 hours, 144 hours, and 168 hours after the first dose of the SR formulation. In subjects administered the IR formulation, blood was collected immediately before and at 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 10 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 97 hours, 98 hours, 99 hours, 100 hours, 102 hours, 104 hours, 106 hours, 108 hours, 112 hours, 120 hours, 132 hours, 144 hours, and 168 hours after the first dose of the IR formulation. After the first 1 mL of blood retained in cannula had been discarded, 6 mL of blood was drawn at each time point and collected in a sodium heparin-coated tube, followed by flushing with 1 mL of saline. Blood samples were immediately placed in an ice bath. Plasma was separated by centrifugation at 1,800× g for 10 minutes at 4°C and stored at −20°C until analysis.
A validated liquid chromatography-tandem mass spectrometry method to simultaneously measure cilostazol and its metabolites (OPC-13015 and OPC-13213) in human plasma was used on the basis of previous reports.11,12 Cilostazol and the internal standard, mosapride, were separated using a high-performance liquid chromatography system (Spark Holland, Emmen, The Netherlands) and detected by MS/MS (API 4000; Applied Biosystems/MDS Sciex, Toronto, Canada). The lower limit of quantification was 0.5 ng/mL, with the calibration curve ranging from 0.5 ng/mL to 2,000 ng/mL.
PK evaluations and statistical analysis
The plasma concentration–time profiles of cilostazol for each subject were analyzed by a noncompartmental method using WinNonlin® 6.1 (Pharsight Corporation, Mountain View, CA, USA). All analyses were made using actual times of sampling. The area under the curve for a dosing interval (AUCT) was calculated by the linear trapezoidal rule. The peak plasma concentration at steady state (Cmax,ss) and the time to reach Cmax,ss (tmax,ss) were determined from the observed values. The terminal elimination rate constant (λz) was estimated by linear regression of the terminal log-linear portion of the plasma concentration–time curves. The terminal elimination half-life (t1/2β) was calculated for each subject as ln(2)/λz.
All statistical analyses were conducted using SAS® 9.3 (SAS Institute, Cary, NC, USA) and WinNonlin 6.1 (Pharsight Corporation). Demographic data and PK results were summarized using descriptive statistics. For the comparison of PK characteristics between IR and SR formulations, the Cmax,ss and AUCT of each formulation were log-transformed and tested by a mixed-model analysis of variance. The mean differences and 90% confidence intervals (CIs) were back-transformed to obtain geometric mean ratios and CIs for those ratios. For the comparison of frequency of AEs between two formulations, P-value was obtained by a Fisher’s exact test.
Results
Study participants
A total of 30 healthy male subjects were enrolled, 24 of whom completed the study (Figure 1). Six volunteers withdrew from the study; five volunteers withdrew consent after experiencing AEs (headache), and one subject withdrew consent for other personal reasons. All subjects were included in the tolerability assessment, whereas only the subjects who completed the blood sampling as scheduled were included in the PK analysis. The demographic characteristics of the enrolled subjects are shown in Table 1.
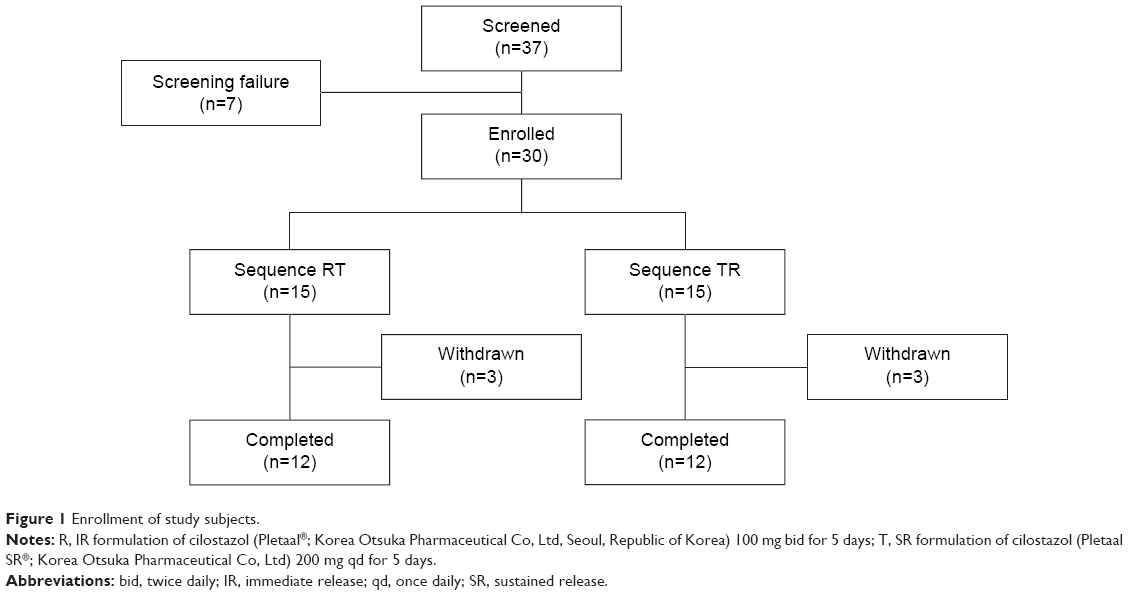 | Figure 1 Enrollment of study subjects. |
 | Table 1 Demographic characteristics of the study subjects |
PK analysis
The pharmacokinetics of cilostazol was analyzed in 24 subjects who had completed the entire treatment period. The mean plasma cilostazol, OPC13015, and OPC-13213 concentration–time profiles are shown in Figure 2. The PK characteristics of cilostazol in these subjects are summarized in Table 2. The 90% CIs for the geometric mean ratio of AUCT were within the comparative bioavailability range to assume bioequivalence of both cilostazol and its metabolites. The geometric mean ratios (90% CIs) of the SR to IR formulations were 0.937 (0.863–1.017), 0.960 (0.883–1.043), and 0.935 (0.859–1.017) for AUCT and 0.644 (0.590–0.703), 0.586 (0.536–0.642), and 0.636 (0.577–0.702) for dose-normalized Cmax,ss of cilostazol, OPC-13015, and OPC-13213, respectively (Table 3).
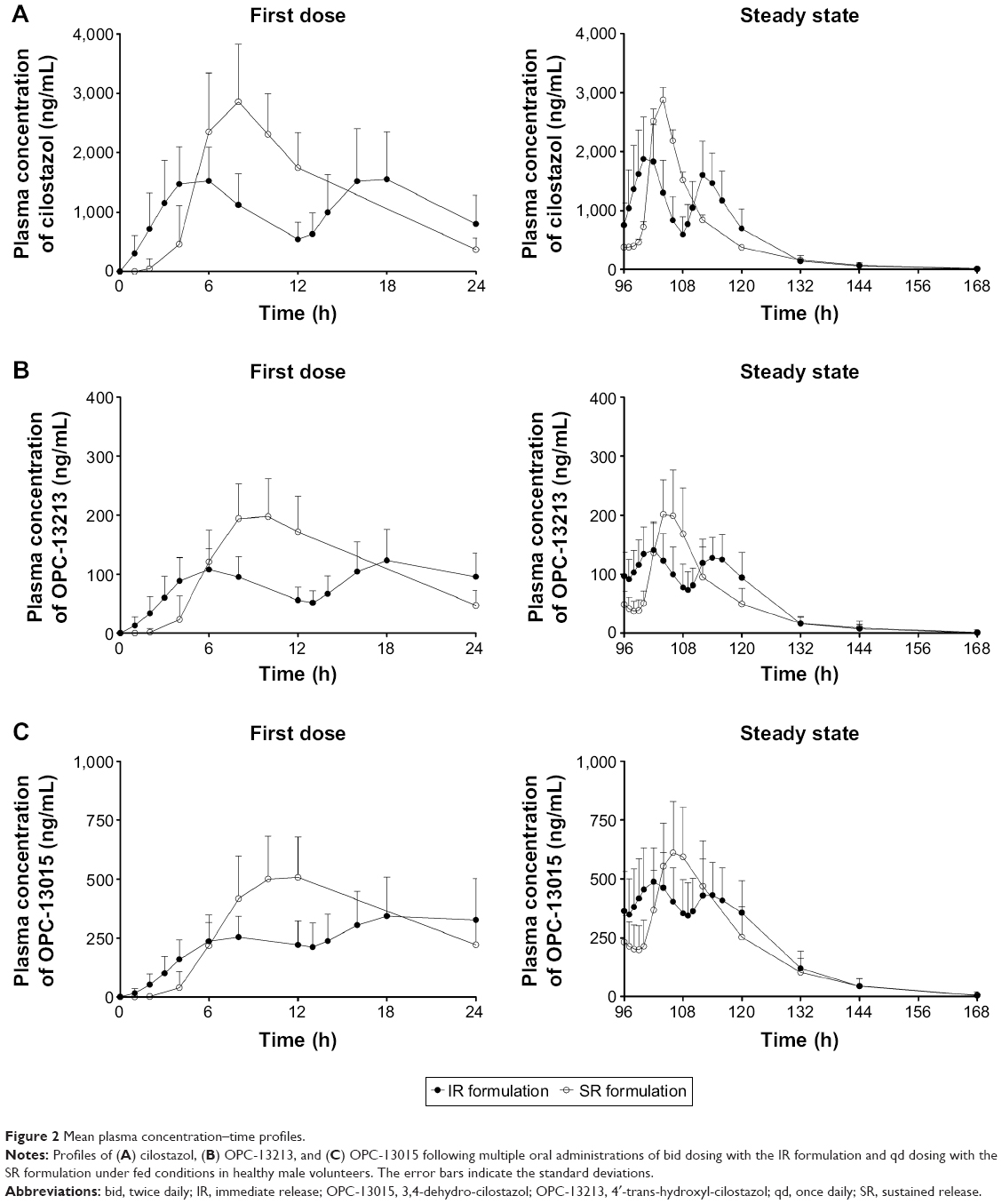 | Figure 2 Mean plasma concentration–time profiles. |
 | Table 2 Pharmacokinetic parameters of cilostazol, OPC-13015, and OPC-13213 after multiple oral administrations of bid dosing with the IR formulation and qd dosing with the SR formulation |
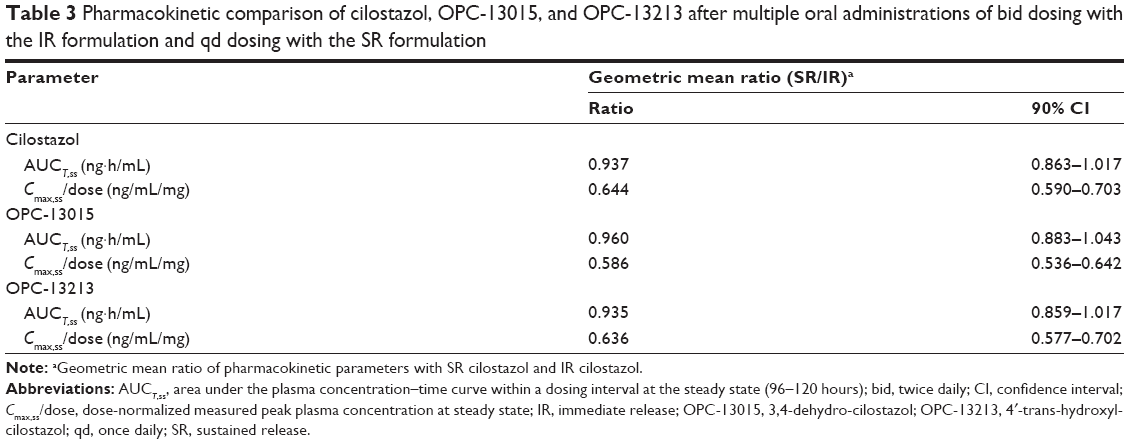 | Table 3 Pharmacokinetic comparison of cilostazol, OPC-13015, and OPC-13213 after multiple oral administrations of bid dosing with the IR formulation and qd dosing with the SR formulation |
Tolerability
No serious AEs or drug reactions occurred. Headache was the most frequently reported AE (in 26 of 26 subjects who received IR formulation and in 24 of 27 subjects who received SR formulation), followed by nausea, tachycardia, palpitation, and anorexia. All other AEs, including vomiting, diarrhea, upper respiratory tract infection, dizziness, paresthesia, herpes simplex infection, chest discomfort, blood bilirubin level increase, and hordeolum occurred in <3 subjects (Table 4).
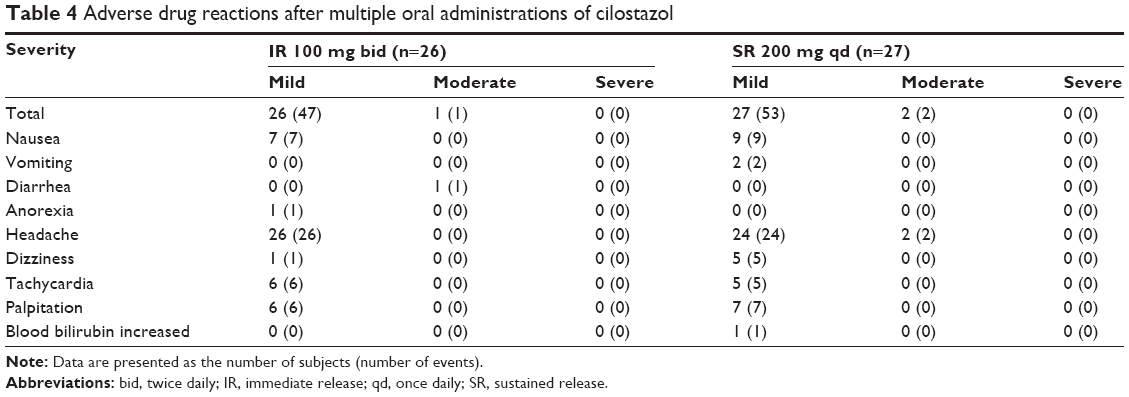 | Table 4 Adverse drug reactions after multiple oral administrations of cilostazol |
All AEs were mild or moderate (two patients experienced headaches following SR treatment and one patient had diarrhea in the IR treatment group). Most subjects who reported an AE recovered spontaneously within a few hours or days. The total incidence of AEs was not statistically different between IR and SR formulation groups (P-value =1.000). The total incidence of headache, which is a most common adverse drug reaction, was also not statistically different between IR and SR treatments (P-value =0.236). No clinically meaningful trends were found in vital signs, ECGs, clinical laboratory tests, and physical examinations.
Discussion
The findings of our current study demonstrate that the PK of cilostazol and its metabolites (OPC-13015 and OPC-13213) do not differ significantly among themselves. At steady state, the 90% CIs of the geometric mean ratios of AUCT (96–120 hours) for the SR and IR formulations were within the 0.8–1.25 range. As expected, the dose-normalized Cmax of cilostazol, OPC-13015, and OPC-13213 were 36%, 41%, and 36% lower, respectively, after administration of cilostazol SR compared to cilostazol IR (Table 3). The terminal half-lives of the IR and SR formulations were 7.0 hours and 11.8 hours, respectively, indicating that the SR formulation does indeed exert an extended release of cilostazol (Table 2). These PK properties enable the frequency of dosing to be reduced, which has been shown to improve patient adherence.13,14
The total incidence of AEs was not found to be different between the IR and SR formulations. The most common AE was headache, typically emerging about 4 hours after drug administration and resolving within 24 hours after last dose of drug. The headache-inducing effect of cilostazol in healthy volunteers was also reported in another study.15 The therapeutic focus of cilostazol is increasing cyclic adenosine monophosphate, which may play a role in the induction of headache. In a previous study,15 the median headache score peaked at 6–9 hours postdose, and the headaches were usually bilateral and pulsating; we found similar trends in our present study. Healthy volunteers may be more susceptible to headache than elderly patients, who comprise the demographic group that most frequently requires cilostazol treatment.16
Our present study was conducted in healthy male volunteers, meaning that our results on the PK profile of cilostazol are limited to this demographic group. However, a previous study7 has reported that the PK characteristics of cilostazol are not affected by age or sex, and the pharmacodynamic effects were shown in another report to be well correlated with the PK profile.17 Thus, the SR formulation would be expected to provide similar effects in patients compared to the IR formulation. Overall, our current analysis indicates that the SR formulation of cilostazol has a PK profile comparable to that of the IR formulation in terms of the extent of absorption. Both formulations are also well tolerated.
Acknowledgments
The study was funded by Korea Otsuka Pharmaceutical Co, Ltd (Seoul, Republic of Korea), the manufacturer of cilostazol IR and SR formulations. The sponsor’s financial support did not include bonuses for the success of the present study or for the recruitment of subjects.
Disclosure
The authors report no conflicts of interest in this work.
References
Dawson DL, Cutler BS, Meissner MH, Strandness DE Jr. Cilostazol has beneficial effects in treatment of intermittent claudication: results from a multicenter, randomized, prospective, double-blind trial. Circulation. 1998;98(7):678–686. | ||
Pearce L, Ghosh J, Counsell A, Serracino-Inglott F. Cilostazol and peripheral arterial disease. Expert Opin Pharmacother. 2008;9(15):2683–2690. | ||
Kwon SU, Cho YJ, Koo JS, et al. Cilostazol prevents the progression of the symptomatic intracranial arterial stenosis: the multicenter double-blind placebo-controlled trial of cilostazol in symptomatic intracranial arterial stenosis. Stroke. 2005;36(4):782–786. | ||
Gotoh F, Tohgi H, Hirai S, et al. Cilostazol stroke prevention study: a placebo-controlled double-blind trial for secondary prevention of cerebral infarction. J Stroke Cerebrovasc Dis. 2000;9(4):147–157. | ||
Niki T, Mori H. Phase I study of cilostazol. Safety evaluation at increasing single doses in healthy volunteers. Arzneimittelforschung. 1985;35(7A):1173–1185. | ||
Bramer SL, Forbes WP. Relative bioavailability and effects of a high fat meal on single dose cilostazol pharmacokinetics. Clin Pharmacokinet. 1999;37(suppl 2):13–23. | ||
Suri A, Forbes WP, Bramer SL. Pharmacokinetics of multiple-dose oral cilostazol in middle-age and elderly men and women. J Clin Pharmacol. 1998;38(2):144–150. | ||
Bramer SL, Forbes WP, Mallikaarjun S. Cilostazol pharmacokinetics after single and multiple oral doses in healthy males and patients with intermittent claudication resulting from peripheral arterial disease. Clin Pharmacokinet. 1999;37(suppl 2):1–11. | ||
Hiratsuka M, Hinai Y, Sasaki T, et al. Characterization of human cytochrome p450 enzymes involved in the metabolism of cilostazol. Drug Metab Dispos. 2007;35(10):1730–1732. | ||
Shimizu T, Osumi T, Niimi K, Nakagawa K. Physico-chemical properties and stability of cilostazol. Arzneimittelforschung. 1985;35(7A):1117–1123. | ||
Nirogi RV, Kandikere VN, Shukla M, et al. Simultaneous quantification of cilostazol and its primary metabolite 3,4-dehydrocilostazol in human plasma by rapid liquid chromatography/tandem mass spectrometry. Anal Bioanal Chem. 2006;384(3):780–790. | ||
Fu CJ, Tata PN, Okada K, Akiyama H, Bramer SL. Simultaneous quantitative determination of cilostazol and its metabolites in human plasma by high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl. 1999;728(2):251–262. | ||
Malinowski H, Marroum P, Uppoor VR, et al. Draft guidance for industry extended-release solid oral dosage forms. Development, evaluation and application of in vitro-in vivo correlations. Adv Exp Med Biol. 1997;423:269–288. | ||
Srivastava K, Arora A, Kataria A, Cappelleri JC, Sadosky A, Peterson AM. Impact of reducing dosing frequency on adherence to oral therapies: a literature review and meta-analysis. Patient Prefer Adherence. 2013;7:419–434. | ||
Birk S, Kruuse C, Petersen KA, Tfelt-Hansen P, Olesen J. The headache-inducing effect of cilostazol in human volunteers. Cephalalgia. 2006;26(11):1304–1309. | ||
Pratt CM. Analysis of the cilostazol safety database. Am J Cardiol. 2001;87(12A):28D–33D. | ||
Woo SK, Kang WK, Kwon KI. Pharmacokinetic and pharmacodynamic modeling of the antiplatelet and cardiovascular effects of cilostazol in healthy humans. Clin Pharmacol Ther. 2002;71(4):246–252. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.