")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 13
Pharmacogenomic Assessment of Patients with Colorectal Cancer and Potential Treatments
Authors Bruera G , Ricevuto E
Received 2 September 2020
Accepted for publication 30 October 2020
Published 16 November 2020 Volume 2020:13 Pages 601—617
DOI https://doi.org/10.2147/PGPM.S253586
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Gemma Bruera,1,2 Enrico Ricevuto1,2 On behalf of Oncology Network ASL1 Abruzzo
1Oncology Territorial Care, S. Salvatore Hospital, Oncology Network ASL1 Abruzzo, University of L’Aquila, L’Aquila, Italy; 2Department of Biotechnological and Applied Clinical Sciences, University of L’Aquila, L’Aquila, Italy
Correspondence: Gemma Bruera
Oncology Territorial Care, S. Salvatore Hospital, Oncology Network ASL1 Abruzzo, Department of Biotechnological and Applied Clinical Sciences, University of L’Aquila, Via Vetoio, L’Aquila 67100, Italy
Tel +39 0862368746
Fax +39 0862368308
Email [email protected]
Abstract: Evolving intensiveness of colorectal cancer (CRC) treatment, including chemotherapeutics and targeted agents associations, in adjuvant and metastatic CRC (MCRC) settings, increased overall survival (OS) with individual variability of toxicity. Pharmacogenomic guidelines recommended pre-treatment identification of at-risk patients suggesting dose adjustment of fluoropyrimidines based on dihydropyrimidine dehydrogenase (DPYD), and irinotecan on UDP glucuronosyl-transferase 1 family polypeptide A1 (UGT1A1) genetic variants, but they are poorly applied in clinical practice. This review highlighted clinically validated pharmacogenetic markers, to underline the need of their implementation in the multidisciplinary molecular board for individual CRC patients in clinical practice. Five clinically relevant DPYD variants with different prevalence impair enzymatic effectiveness and significantly increase toxicity: c.1236 G>A (c.1129– 5923 C>G, HapB3), 4.1– 4.8%; c.1679 T>G (DPYD*13), c.1905+1G>A (DPYD*2A), c.2846 A>T, c.2194 A>T (DPYD*6) 1% each. c.1679T>G and c.1905+1G>A are most deleterious on DPD effectiveness, moderately reduced in c.1236/HapB3 and c.2846A>T. Cumulatively, these variants explain approximately half of the estimated 10– 15% fluoropyrimidine-related gastrointestinal and hematological toxicities due to DPD. Prevalent UGT1A1 gene [TA]7TAA promoter allelic variant UGT1A1*28, characterized by an extra TA repeat, is associated with low transcriptional and reduced enzymatic effectiveness, decreased SN38 active irinotecan metabolite glucuronidation, vs wild-type UGT1A1*1 [A(TA)6TAA]. Homozygote UGT1A1*28 alleles patients are exposed to higher hematological and gastrointestinal toxicities, even more than heterozygote, at > 150 mg/m2 dose. Dose reduction is recommended for homozygote variant. Wild-type UGT1A1*28 alleles patients could tolerate increased doses, potentially affecting favorable outcomes. Implementation of up-front evaluation of the five validated DPYD variants and UGT1A1*28 in the multidisciplinary molecular tumor board, also including CRC genetic characterization, addresses potential treatments with fluoropyrimidines and irinotecan associations at proper doses and schedules, particularly for early CRC, MCRC patients fit for intensive regimens or unfit for conventional regimens, requiring treatment modulations, and also for patients who experience severe, unexpected toxicities. Integration of individual evaluation of toxicity syndromes (TS), specifically limiting TS (LTS), an innovative indicator of toxicity burden in individual patients, may be useful to better evaluate relationships between pharmacogenomic analyses with safety profiles and clinical outcomes.
Keywords: colorectal cancer, personalized treatments, pharmacogenomics
Introduction
Over the last 35 years, complexity of CRC medical treatment raised from single agent 5-fluorouracil (5-FU) as the only line of treatment in MCRC, with 30% of activity consisting of partial responses,1,2 giving a few months increase of overall survival (OS), to 5-FU, or newer antimetabolites, as single agents or associating with up to three different chemotherapeutic agents, also including an anti-vascular endothelial growth factor (VEGF) or anti-epidermal growth factor receptor (EGFR) targeted drug, as adjuvant and subsequent lines of MCRC treatment, gaining up to 80% efficacy,3–6 also consisting of complete responses, and justifying integration with secondary surgical resection of metastases,7 thus achieving >20% 5 years-OS. Chemotherapeutic drugs, including fluoropyrimidines, irinotecan, oxaliplatin, associated to targeted agents, antiangiogenic (bevacizumab, ziv-aflibercept, regorafenib, ramucirumab) or anti-EGFR (cetuximab, panitumumab), determined significant improvements of MCRC clinical outcomes, but correlated with a relevant increase of toxicity burden.
In clinical practice, the pathway addressing step-by-step proper treatment of each individual CRC patient consists of: evaluation of clinical parameters, such as age, comorbidities, performance status, to differentiate among patients fitting for intensive medical treatments or unfit, thus requiring modulated treatment strategies; evaluation of disease extension (primary tumor/nodes-limited; metastatic involvement, liver-/lung-limited or multiple metastatic sites); genetic characterization of cancer cells by KRAS/NRAS/BRAF genotyping, mismatch repair proteins (pMMR), and microsatellite instability (MSI), addressing among different drug options.8–17 Nowadays, RAS genotype identifies MCRC patients resistant to anti-EGFR treatments; BRAF V00E mutant patients could benefit from anti-BRAF (encorafenib)/cetuximab association in pretreated patients;18 deficient MMR, and/or MSI-high patients could benefit from immunotherapy.19
Evolving intensiveness of CRC medical treatment to achieve better clinical outcome is weighed by a wider spectrum of increased cumulative toxicities, with consistent individual variability, justifying on-treatment modulations of planned regimens.20 More, most MCRC patients are unfit for intensive regimens,16 thus requiring a priori individual modulations, including drug dose reductions and/or schedule modifications, reduction of number of associated drugs, and individual planning of different sequential treatment strategies.
Implementation of pharmacogenomic analyses in the multidisciplinary CRC management can increase the safety of different treatment options,21 determining cumulative adverse events of any grade in approximately 90% of patients at any disease stage with high individual variability, and can be unpredictable at clinical level. Pharmacogenomic analysis, based on the detection of inherited allelic variants, consisting of point mutations or single nucleotide polymorphisms (SNPs) differentially affecting Phase I and II enzymes function, specifically regarding ATP-binding cassette/solute carrier membrane transporters, proteins involved in DNA repair, folate pathway, and immune response, can identify at-risk patients and should be routinely implemented in tumor molecular board evaluations in clinical practice, to properly personalize chemotherapy and manage toxicity. Some genetic markers of toxicity were validated, specifically for fluoropyrimidines and irinotecan, and are under evaluation, not established to date for oxaliplatin-specific adverse events, such as peripheral neuropathy, nor for novel targeted agents. International scientific consortia, particularly the Clinical Pharmacogenomics Implementation Consortium and Dutch Pharmacogenetics Working Group,22 published pharmacogenomics guidelines, strongly recommending before treatment beginning the identification of CRC patients potentially at risk of toxicity, to increase safety, suggesting dose adjustment of fluoropyrimidines based on dihydropyrimidine dehydrogenase (DPYD), and of irinotecan based on UDP glucuronosyl transferase 1 family, polypeptide A1 (UGT1A1) genetic variants. In clinical practice, these recommendations are still poorly applied in the oncological settings. Nevertheless, chemotherapy-related toxicities prevalently remain unjustified.
This review highlights clinically validated pharmacogenetic markers to be implemented in the multidisciplinary pathway of proper selection of medical treatment in clinical practice for the individual CRC patient, also including CRC genetic characterization. Different retrospective studies investigated predictive/prognostic relevance of different pharmacogenomics biomarkers in CRC patients, related to the occurrence of different adverse events, prevalently gastrointestinal and hematological.23–25 To date, only some DPYD and UGT1A1 genetic variants demonstrated clinically relevant evidence and were validated for clinical application.
Fluoropyrimidines and Clinically Relevant DPYD Genetic Variants
Up to now, the fluoropyrimidine drugs class is a milestone in CRC treatment, in early and metastatic disease, and 5-FU and its pro-drug capecitabine remain the chemotherapy backbone of different associations. 5-FU, capecitabine, and tegafur can induce gastrointestinal and/or hematological limiting toxicities (LT), prevalently related to clearance deficiency. The most relevant rate-limiting enzyme involved in hepatic catabolism of about 85% of fluoropyrimidines administered dose is DPYD, codified by DPYD gene, frequently characterized by genetic variants justifying highly inter-patients variability in enzymatic effectiveness (about 8–21-fold).
A DPD deficiency familial linkage detected in a patient who experienced 5-FU-induced toxicity suggested potential biological pharmacogenetic variability in 5-FU metabolism.26 Molecular data suggested a correlation between DPYD gene allelic variants and DPD effectiveness deficiency, inducing fluoropyrimidine toxicity.
The metabolism of fluoropyrimidine involves enzymes with different intermediate metabolites, prevalently depending on the key enzyme DPD, that metabolize >80% administered 5-FU or capecitabine dose into 5-fluoro-5,6-dihydrouracil (5-FDHU).27 In the event of DPD inactivation or reduced effectiveness, the 5-FU amount for activation of anabolic pathway increases, inducing adverse effects.
DPYD is a gene characterized by a wide range of inter-individual genetic variants, with highly polymorphic features, also according to ethnicity. DPD deficiency is prevalently related to mutations within DPYD encoding genes, with more than 40 reported polymorphisms, point (introns and exons), splice-site, frame-shift nonsense mutations, deletions (exon skipping), functionally affecting splicing process, gene transcription, with complete inactivation or significant and different reduction of enzyme effectiveness. Different DPYD genetic variants alter mRNA splicing or the protein sequence, even if prevalently with not significantly impairment of enzyme effectiveness or inducing not clear functional effect. Four DPYD variants are of primary relevance due to their prevalence, impaired enzymatic effectiveness, and correlation with severe fluoropyrimidines-related adverse events (Table 1): c.1236 G>A (c.1129–5923 C>G, HapB3, rs75017182), c.1679 T>G (DPYD*13, p.I560S, rs55886062), c.1905+1G>A (DPYD IVS14+1G>A, DPYD*2A, rs3918290), c.2846 A>T (p.D949V, rs67376798). In Europeans, c.1129–5923C>G HapB3 is the most common DPYD variant with decreased function, with carrier frequencies 4.1–4.8%, followed by c.1905+1G>A (1–1.2%) and c.2846A>T (0.8–1.4%).
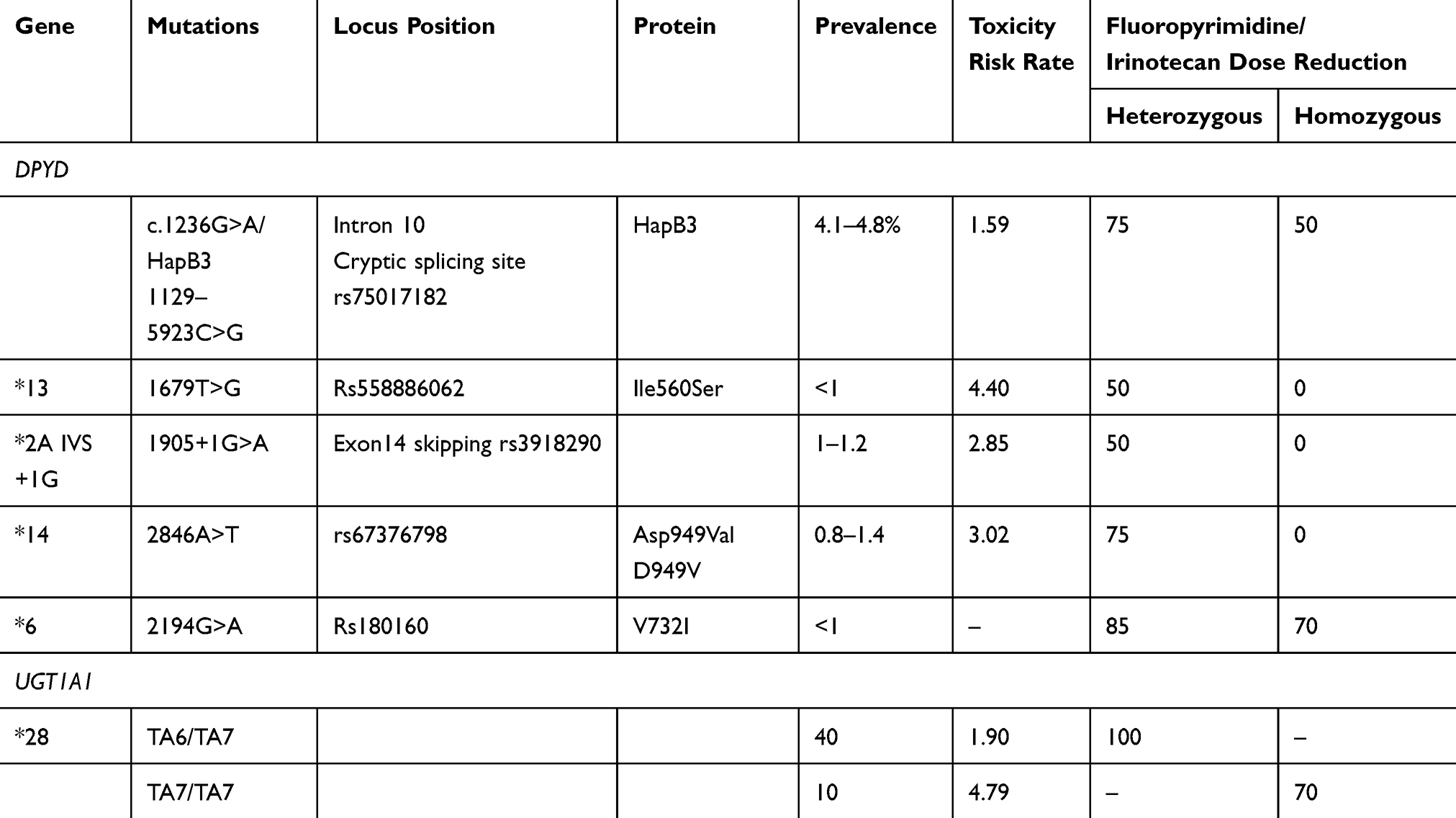 |
Table 1 DYPD and UGT1A1 Mutations with Pharmacogenetic Clinical Implications in Colorectal Cancer |
The c.1236 G>A (c.1129–5923 C>G, HapB3) variant in intron 10 inserts a cryptic splice site, determining partial production of a transcript with non-functional features. This variant in SNP is the determining DPYD haplotype (HapB3), spanning intron 5 to exon 11. The synonymous variant c.1236G>A (rs56038477) is in perfect linkage disequilibrium with c.1129–5923C>G.
The three other mutations were associated with more relevant fluoropyrimidine risk of toxicity:28 DPYD*2A, c.1905+1 G>A (DPYD:IVS14+1 G>A); DPYD*13, c.1679 T>G; c.2846 A>T. The most well-studied DPYD variant clearly associated with severe or even life-threatening toxicity, DPYD*2A, c.1905+1 G>A (DPYD:IVS14+1 G>A), localized at the exon 14 intron boundary, a splice site mutation, a purine transition at the first nucleotide of the intron 14, functionally consisting of a splicing mutation, determines entire exon 14 skipping and translation in a protein non-functional effectiveness.29 Thus, the heterozygous genotype consisting of mutated and wild-type alleles determines 50% reduction of its effectiveness; bi-allelic mutation or homozygous mutated genotype determines complete loss of function of enzymatic DPD effectiveness. DPYD*13, c.1679 T>G, and c.2846 A>T are missense mutations affecting the function of the protein, strongly related with fluoropyrimidine-induced adverse events: DPYD*13, c.1679 T>G induces the aminoacid change Ile560Ser in a DPD flavine binding domain; the non-synonymous variant c.2846 A>T, determining the Asp949Val aminoacid change near a DPD iron-sulfur motif. In a review reporting daily practice guidelines, omission or dose reductions of fluoropyrimidine drugs were recommended in homozygous and heterozygous carriers of these three variants, respectively. Thus, the most deleterious impact on DPD effectiveness is determined by DPYD*2A (c.1905+1G>A) and DPYD*13 (c.1679T>G); c.2846A>T and c.1129–5923C>G moderately reduced DPD effectiveness.
Among the four DPYD variants, DPYD c.2846A>T, DPYD*2A, and DPYD*13 are rarer with frequencies of 0.2–1.4% in Europeans; DPYD-HapB3 are reported with higher minor allele frequency of 4.8%. Considering the combined frequency of all four variants, 7% of Europeans harbor at least one of these DPYD variants with decreased function that cannot justify the reported estimation of 10–15% of fluoropyrimidine-induced toxicities related to DPD. Thus, DPYD analysis, by identification of poor metabolizers, may reduce toxicity occurrence, even if patients wild-type for the described genetic variants may experience severe toxicities.22
More than 30% of CRC patients receiving 5-FU and its pro-drug capecitabine report early-onset limiting or even life-threatening toxicity.
In a meta-analysis of eight studies including 7,365 patients, the four most relevant DPYD variants were clinically and significantly related to fluoropyrimidine-induced toxicity (Table 1):30 c.1236G>A/HapB3 adjusted RR=1.59 (CI=1.29–1.97, P<0.0001); DPYD*13 (c.1679T>G) RR=4.40 (CI=2.08–9.30, P<0.0001); DPYD*2A c.1905+1G>A RR=2.85 (CI=1.75–4.62, P<0.0001); and c.2846A>T RR=3.02 (CI=2.22–4.10, P<0.0001). Consistent associations were also reported for DPYD*13 (c.1679T>G) and c.1236G>A/HapB3, respectively, with: gastrointestinal adverse events, adjusted RR=5.72 (CI=1.40–23.33, P=0.015) and RR=2.04 (CI=1.49–2.78, P<0.0001); hematological adverse events, adjusted RR=9.76 (CI=3.03–31.48, P=0.00014), and RR=2.07 (CI=1.17–3.68, P=0.013). In a meta-analysis, also patients harboring DPYD*2A c.1905+1G>A were reported as at increased risk of overall adverse events, specifically hematological ones, diarrhea, and mucositis.31 A strong association was reported between DPYD c.2846A>T missense mutation and overall ≥G3 toxicity, and ≥G3 diarrhea. In prospective studies, an inverse linear relationship was described between odds ratio (OR) of DPYD*2A c.1905+1G>A and overall ≥G3 toxicity incidence, suggesting a relevant impact in patients with lower incidence of severe toxicity.
In the event of toxicity experienced after treatment, other variants, such as c.2194 G>A (DPYD*6, p.V732I, rs1801160), could be evaluated (European Medicine Agency Pharmacovigilance Risk Assessment Committee). The association between c.1601G>A (DPYD*5, p.S534N, rs1801159) and toxicity was not significant (adjusted RR=1.52, CI=0.86–2.70, P=0.15).30
Clinical relevance of the DPYD pharmacogenetic test to predict fluoropyrimidines-related toxicity was retrospectively investigated among 603 CRC patients treated with fluorouracil-based regimens.32 Patients were tested for eight DPYD polymorphisms, including DPYD*13 c.1679T>G,DPYD*2A c.1905+1G>A, DPYD c.2846A>T, DPYD*6 c.2194G>A, and four other variants (DPYD-rs2297595, DPYD-rs1801158, DPYD-rs1801159, DPYD-rs17376848) related with ≥G3 toxicity occurrence, within the first three treatment cycles. ≥G3 toxicity incidence in DPYD*2A c.1905+1G>A, DPYD c.2846A>T, DPYD*13 c.1679T>G heterozygous genotype carriers were 66.7%, 60.0%, and 50.0%, respectively. DPYD*2A c.1905+1G>A and DPYD c.2846A>T were significantly associated to ≥G3 toxicity (P=0.003, P=0.048, respectively); no significant correlations were reported with DPYD*13 c.1679T>G due to the low allelic frequency, even if death was reported after one treatment cycle in one out of two heterozygous patients (DPYD*2A c.1905+1G>A mutant). Among seven patients carrying one variant DPYD*2A c.1905+1G>A, DPYD*13 c.1679T>G, DPYD c.2846A>T allele, who did not develop ≥G3 toxicity, dose or schedule modification for moderate chronic toxicity was reported in 57% patients. No other DPYD polymorphism was associated with ≥G3 toxicity. In the daily practice, DPYD*2A c.1905+1G>A, DPYD*13 c.1679T>G, and DPYD c.2846A>T genotype analyses could prevent ≥G3 toxicity related to fluoropyrimidines and enhance compliance to treatment plan.
Among 1,545 stage III CRC patients enrolled in Pan-European Trials in an Alimentary Tract Cancer (PETACC)-8 Phase 3 trial, randomized to standard adjuvant fluorouracil, leucovorin, oxaliplatin (FOLFOX4), or cetuximab added to FOLFOX4 for 6 months, 25 DPYD variants were genotyped, and related with ≥G3 5-FU adverse events.33 Statistically significant correlations were found between DPYD c.2846 A>T and DPYD*6 c.2194 G>A variants (D949V and V732I) with increased incidence of ≥G3 5-FU adverse events: 18 out of 21 (85.7%, OR=6.3; CI =2.0–27.0, P<0.001) and 121 out of 199 (60.8%, OR=1.7, CI=1.3–2.4, P<0.001), respectively. The association with neutropenia ≥G3 was: DPYD c.2846 A>T OR=5.2 (CI =2.0–16.0); DPYD*6 c.2194 G>A, OR=1.8 (CI=1.3–2.4). Hematologic toxicities ≥G3 were associated with DPYD*6 c.2194 G>A variant, OR=1.9 (CI=1.4–2.6). The association between DPYD*6 c.2194 G>A variant and 5-FU-related ≥G3 side-effects, and overall hematological toxicities, was confirmed and validated in a population of 339 MCRC patients treated with FOLFOX4 in the Fédération Francophone de Cancérologie Digestive 2000–05 phase 3 trial.
In an ancillary pharmacogenetic study of Italian TOSCA randomized trial including CRC patients treated with 3 or 6 months of FOLFOX-4 or XELOX adjuvant chemotherapy, 10 DPYD variants, including the three most relevant mutations (DPYD*2A c.1905+1G>A, DPYD*13 c.1679 T>G, DPYD c.2846 A>T), DPYD*6 c.2194 G>A, DPYD*5 c.1601G>A, and five different other variants (DPYD*4 rs1801158 G4A, *9A rs1801265 T4C, rs2297595 A4G, rs17376848 T4C, rs75017182 C4G) were retrospectively evaluated.28 Fluoropyrimidine-related adverse events were reported in 194 out of 508 evaluable patients (38.2%), more frequently in DPYD*6 c.2194 G>A carriers. Time to toxicity was significantly affected in patients harboring DPYD*6 c.2194 G>A, DPYD*2A c.1905+1 G>A, and rs2297595 GG genotype. Neutropenia was the prevalent adverse event (28.5%); diarrhea 6.5%. DPYD*6 c.2194 G>A, DPYD*2A c.1905+1 G>A variant alleles were significantly associated with time to neutropenia. Median time to toxicity was 7 months among homozygous genotype patients, significantly shortened (0.9–2.1 months) in homozygous DPYD*6 c.2194 G>A and rs2297595, and in DPYD*2A c.1905+1 G>A heterozygous genotypes. Shortened time to toxicity was reported in DPYD*6 c.2194 G>A. The early onset of toxicity underlines the relevance of enzymatic deficiency and could contribute to verify DPYD variants/DPD status in patients experiencing early-onset limiting fluoropyrimidine related toxicities after treatment administration.
In a multi-center toxicity evaluation conducted among 17 hospitals in the Netherlands, including 1,181 patients, the four most relevant DPYD mutations, DPYD*2A c.1905 G>A, DPYD*13 c.1679 T>G, c.2846 A>T, and c.1236 G>A/HAPB3 were prospectively genotyped prior to receive a treatment containing a fluoropyrimidine drug (capecitabine or fluorouracil alone or associated with other chemotherapeutic drugs or radiation therapy).34 Among 1,103 patients, 85 (8%) harbored heterozygous DPYD variant allele, 1,018 (92%) DPYD wild-type. Patients harboring heterozygous DPYD variant allele were treatment with up-front 25% (c.2846A>T and c.1236G>A) or 50% (DPYD*2A and c.1679T>G) dose reduction; patients with wild-type genotype were treated with standard dose. Overall, fluoropyrimidine-related limiting adverse events were prevalent in patients with DPYD variant (33 of 85 [39%] patients) compared with wild-type (231 of 1,018 [23%]; P=0.0013). DPYD genotype-based dose reductions improved safety: relative risk (RR) for severe fluoropyrimidine-related toxicity was 1.31 (95% CI=0.63–2.73) for dosing guided by genotype analyses compared with 2.87 (2.14–3.86) in the historical control for DPYD*2A carriers; no reported toxicity vs 4.30 (2.10–8.80) in DPYD*13 c.1679T>G patients, 2.00 (1.19–3.34) vs 3.11 (2.25–4.28) for c.2846A>T patients, 1.69 (1.18–2.42) vs 1.72 (1.22–2.42) for c.1236G>A ones. For DPYD*2A and DPYD*13 c.1679T>G carriers, an up-front dose reduction of 50% was useful. The authors suggested more investigation for c.1236G>A and c.2846A>T carriers and adequate dose modulation 50% vs 25%.
DPYD c.496A>G, c.1601G>A, c.1627A>G, c.1896T>C, and c.2194G>A variants were detected in a population of 982 patients who experienced ≥G2 gastrointestinal and/or ≥G3 hematological adverse events, and in a cohort of 272 patients not requiring reduction of drug dose, treatment delay, or discontinuation; while c.1905+1G>A and c.2846A>T were reported only in the cohort of patients experiencing LT.27 DPYD c.1679T>G and c.1236G>A/HapB3 were not detected. In an univariate analysis, DPYD*2A c.1905+1G>A, c.2846A>T, and c.2194G>A alleles were reported as significantly correlated with hematological and gastrointestinal adverse events (P<0.05); c.496A>G variant was associated with neutropenia (P=0.06). Specifically, DPYD*2A IVS14+1GA and AA genotypes were reported as significantly correlated with diarrhea (P=0.001), febrile neutropenia (P<0.0001), thrombocytopenia (P=0.012), and alopecia (P=0.007); c.2194GA/GG variants were associated with mucositis (P=0.053), leucopenia (P=0.003), and thrombocytopenia (P=0.049); c.2846AT/TT were reported as correlated with diarrhea (P=0.02). Thus, c.2194G>A was correlated with toxicities with clinical impact, as the c.1905+1G>A and c.2846A>T variants, and should be analyzed to prevent the risk of experiencing adverse events related to fluoropyrimidine administration.
In a population of 1,827 patients, 31 (1.7%) showed DPYD*2A SNP, 53 carried one additional SNP: 35 (66%) among 146 who developed severe toxicities, 18 (34%) out of 220 with no or mild toxicities (P<0.0001).29 DPYD*6 c.2194G>A was the prevalent SNP (12.5%), significantly correlated with severe neutropenia. In particular, neutropenia was reported in 50% of patients harboring c.2194G>A (23 out of 46) vs 21% of wild-type genotype (67 out of 320) (OR=3.75, CI=1.98–7.10; P<0.0001). DPYD c.2846A>T and DPYD*13 c.1679T>G were correlated to different adverse events: a significant correlation of c.2846A>T SNP was reported (P=0.0097), not statistically relevant for c.1679T>G due to the small population enrolled. DPYD c.2846A>T (1.37%, five out of 366 patients) was associated with neutropenia (P=0.0141) and other toxicity (P=0.0049), while DPYD*13 c.1679T>G (two out of 366, 0.55%) induced only gastrointestinal adverse events (P=0.0027). The event of toxicity was significantly different in patients with one of the three SNPs vs wild-type (median time to toxicity 5 vs 11 cycles; P<0.0001). In particular, patients harboring c.1679T>G variant vs wild-type reported very early occurrence of toxicity, 2 vs 8 cycles (P=0.02), as in c.2846A>T SNP, 1 vs 8 cycles (P<0.0001). Patients harboring DPYD*6 c.2194G>A variant showed an earlier toxicity onset, significantly different vs wild-type, 6 vs 10 cycles (P=0.0022). This could be related with a moderate reduction of about 15–20% of the enzymatic effectiveness of DPD, induced by this SNP, with a detrimental effect determining limiting toxicities by a cumulative effect evident in the advanced cycles, and not in CRC patients treated with fluoropyrimidine alone, but after exposure to poly-chemotherapy association. Thus, 50% dose reduction is recommended in DPYD*13 c.1679T>G genotype patients, as reported in Clinical Pharmacogenetics Implementation Consortium guidelines.
Among 828 patients who received fluoropyrimidine-based chemo-radiotherapy, occurrence of severe ≥G3 adverse events in patients harboring DPYD variant allele and receiving upfront reductions of fluoropyrimidine dose according to pharmacogenetic recommendations and patients who did not received specific reductions was compared with patients with DPYD wild-type genotype treated with standard dose.35 Patients harboring DPYD variant allele who received standard doses reported an increased risk of limiting gastrointestinal (adjusted OR=2.58, P=0.045) or hematological (adjusted OR=4.19, P=0.015) adverse events vs wild-type. Patients harboring DPYD variant allele treated with dose reductions reported comparable limiting gastrointestinal adverse events vs wild-type patients, but more, even if not statistically significant, limiting hematological adverse events. Hospitalizations for all patients harboring DPYD variant allele were comparable, not depending from dose modulation; mean duration of hospitalization was significantly shorter among patients treated with dose reduction (P=0.010).
Recently, Clinical Pharmacogenetics Implementation Consortium (CPIC), and the Royal Dutch Association for the Advancement of Pharmacy (DPWG) regulatory organizations tried to harmonize drug dosing adjustment according to genotype considering the gene activity score (GAS) model: based on the enzyme effectiveness, DPYD alleles are classified as completely non-functional (GAS=0), intermediate functional (GAS=0.5), or normally functional (GAS=1.0). The potential correlation between the 4-SNP panel of DPYD variants including DPYD*2A, DPYD*13, c.2846A>T, c.1236A>G-HapB3, strongly recommended by the current pharmacogenomic guidelines to prevent limiting toxicities induced by fluoropyrimidine, and the risk to experience dose-limiting toxicities was evaluated by stratifying patients according to DPYD GAS model.36 GAS 1.0, patients harboring one DPYD*2A or DPYD*13 allele, GAS 1.5, one c.2846A>T, or c.1236G>A-HapB3 allele. Non-carriers GAS 2.0. A population of 763 patients treated with fluoropyrimidine-based chemotherapy was retrospectively evaluated. Patients harboring at least one DPYD variant with decreased function in the 4-SNP panel reported a significant correlation with the risk of experience dose-limiting adverse events (≥G3 non-hematological or grade ≥G4 hematological toxicity), within the first three treatment cycles (OR=2.7, 95% CI=1.33–5.41), or during the entire treatment course (OR=2.7, 95% CI=1.42–5.04). The GAS model demonstrated to better evaluate the risk of experience dose-limiting acute (GAS=1.5, OR=1.80, and GAS=1.0, OR=10.12) and total adverse events (GAS=1.5, OR=2.08, and GAS=1, OR=7.09).
5-FU and capecitabine treatment in patients with low or absent DPD effectiveness is contraindicated by the Food and Drug Administration (FDA), no dose modulation is recommended for intermediate metabolizers, variably applied in clinical practice. Moreover, the recent publication of CRC European Society for Medical Oncology (ESMO) guidelines did not consider recommendations regarding pre-emptive DPYD evaluation. European Society for Medical Oncology guidelines suggest baseline pharmacogenomics evaluations as optional, strongly recommended before fluoropyrimidine re-introduction in patients who experienced severe toxicity.37 To prevent potential severe adverse events, particularly gastrointestinal, hematological, and hand–foot syndrome, the European Medicine Agency Pharmacovigilance Risk Assessment Committee recommended DPD evaluation for all patients candidate to receive a fluoropyrimidine-based regimen, specifically baseline evaluation of the following clinically validated mutations: c.1236 G>A (c.1129–5923 C>G, HapB3), c.1679 T>G (DPYD*13, p.I560S), c.1905+1G>A (DPYD*2A), c.2846 A>T (p.D949V).
Furthermore, proper evaluation of correlations between fluoropyrimidine and safety should take into account concomitant administered drugs and their potential pharmacological interaction with DPD, as well as previous experienced toxicities in the event of pre-treatment, to suggest adequate recommended dose reduction. Detection of specific pharmacogenomic biomarkers predictive of drug-induced adverse events, particularly in the adjuvant setting, remain a critical need, to avoid LT, potentially affecting treatment effectiveness, due to dose and/or schedule modifications or treatment discontinuation. The narrow therapeutic index may be particularly unfavorable in CRC patients potentially cured by surgery, and treated with fluoropyrimidines in the adjuvant setting. Suggested administered fluoropyrimidine doses should take into account DPYD genotype, and, specifically: for wild-type, 100% of standard dose; for heterozygous, 75% in c.1236 G>A, 50% in c.1679 T>G, c.1905+1G >A, c.2846 A>T, 85% in c.2194 G>A; for rare mutant homozygous, 50% in c.1236 A/A, 70% in c.2194 A/A, and should be avoided in c.1679 G/G, c.1905+1 A/A, and c.2846 T/T.
Irinotecan and Clinically Relevant UGT1A1 Genetic Variant
Irinotecan is a pro-drug, converted by carboxylesterases, ubiquitously distributed in the tissues, to its active metabolite SN-38, a topoisomerase I poison 100–1,000-fold more potent. Irinotecan could determine severe toxicities, prevalently gastrointestinal and hematological LT, primarily related to a clearance deficiency. Metabolism and drug excretion are primarily hepatic, less relevant renal.
Marked inter-patient variability has been reported for irinotecan-based regimens toxicity, due to highly variable levels of the active metabolite SN38 in the plasma and/or at the site of specific adverse event, as the bone marrow correlated with different factors. In particular, irinotecan conversion to SN38 by carboxylesterase enzymes and SN38 glucuronidation to inactive SN38 glucuronide (SN38G) by uridine diphosphate glucuronosyltransferase, UGT1A Phase II enzymes family, involved in bilirubin conjugation. Other transport and metabolic pathways are involved in irinotecan and SN38 disposition: SN-38 is subject to oxidation by cytochrome P450 family isoforms 3A4, 3A5, and adenosine triphosphate-binding cassette transporters; distribution qualities of SN38 compared with irinotecan may be relevant.
In the genome, the UGT1A enzymes family is a series of four invariant exons; transcribed product may be spliced to one of nine exons, defining specific substrate-binding domains. Due to the structural variability, the isoform UGT1A1 has the greatest affinity for SN-38, prevalently related to irinotecan catabolism, by conjugating the active metabolite SN-38 with glucuronic acid; UGT1A7 and UGT1A9 were recently reported as involved in the process.
UGT enzyme levels are prevalently regulated at transcriptional level, and the variability of the promoter structure affects the transcription rate. In the proximal promoter, a series of TA repeats vary from five to eight in length: the transcriptional effectiveness of the gene is more efficient with a lower number of repeats. The prevalent alleles have six and seven repeats. Ethnic and racial origin affect the allele frequency of these repeats: in a white population, about 50% [TA6/TA6], 40% [TA6/TA7], 10% [TA7/TA7] genotypes.
The prevalent allelic variant of promoter region of UGT1A1 gene [TA]7TAA, UGT1A1*28, rs8175347, characterized by an extra TA repeat in the promoter gene region [A(TA)7TAA] is associated with low transcriptional and reduced enzymatic effectiveness, decreased SN38 glucuronidation, vs wild-type UGT1A1*1 [A(TA)6TAA], that has six TA repeats. SN-38 is glucuronidated less efficiently in patients harboring homozygous UGT1A1*28 allele compared to patients who have one or two wild-type alleles, and therefore are exposed to higher SN-38 plasma concentrations after treatment.
Pharmacogenetic analyses aim to minimize toxicity and maximize treatment efficacy. In 2005, US FDA recommended to prospectively identify patients at higher risk for adverse effects induced by standard irinotecan doses, due to a genetic polymorphism in the gene encoding UGT1A1, inducing a lower capacity to metabolize and excrete SN-38, and a greater exposure to active drug after receiving standard drug dose.38 This warning was added to irinotecan package inserts:
Individuals who are homozygous for UGT1A1*28 allele are at increased risk for neutropenia following initiation of CAMPTOSAR treatment. A reduced initial dose should be considered for patients homozygous for UGT1A1*28 allele.
Also, the FDA approved the companion genetic test (Invader UGT1A1 Molecular Assay; Third Wave Technologies Inc, Madison, WI), evaluating genomic DNA isolated from peripheral blood, to identify patients homozygous harboring UGT1A1*28 allele, and recommended to consider a modulated initial irinotecan dose in these patients.
Major irinotecan-related adverse events in combination regimens are dose-limiting diarrhea and neutropenia, frequently and variably occurring together in the same patient. A relationship between UGT1A1*28 genotype and toxicity was demonstrated in patients treated with single-agent irinotecan administered every 3 weeks. The incidence of hematologic and gastrointestinal toxicities was increased in patients homozygous or heterozygous for *28 genotype; addiction of more drugs significantly increased the risk of experiencing adverse events. UGT1A1*28 homozygous genotype was clearly associated with neutropenia induced by the administration of irinotecan alone or in a combination regimen, only significant for diarrhea. Concomitant experience of both toxicities, that can be considered a major risk factor for early death as reported in trials proposing irinotecan addiction to 5-FU, may be correlated with a specific genotype. In a large cooperative group trial (N9741) including 520 patients, the preliminary genotyping evaluations confirmed the significant association between homozygous *28 and the risk of developing G4 neutropenia for irinotecan plus oxaliplatin arm (P=0.004), but not for weekly administration of irinotecan plus 5-FU (IFL; P=0.46). An increased risk of febrile neutropenia was associated with homozygous *28. No association was reported for the severity of diarrhea.
In a prospective study including 250 patients with metastatic disease treated with first-line irinotecan, 5-FU, leucovorin, prevalence of homozygous TA7/TA7, heterozygous TA6/TA7, wild-type TA6/TA6 genotype were 8.8%, 45.6%, 45.6%, respectively.39 TA6 and TA7 allele frequencies were 68.4% and 31.6%, respectively. UGT1A1*28 polymorphism correlated with increased risk of G3-4 hematologic adverse events (OR=8.63; CI=1.31–56.55), relevant for the first cycle alone. G3-4 neutropenia occurred in two out of 114 (1.7%) TA6/TA6, six out of 114 (5.3%) TA6/TA7, and three out of 22 (13.6%) TA7/TA7 patients. TA7 allele and G3-4 non-hematologic toxicity were not statistically significantly correlated. TA indel polymorphism did not affect diarrhea; G3 diarrhea was reported in three TA6/TA6 and three TA6/TA7 patients; no G4 diarrhea was reported in the first treatment cycle.
Hematologic and non-hematologic toxicities during the entire treatment course were not clearly associated with UGT1A1*28 polymorphism. Specifically, G3–4 neutropenia were reported in four out of 22 (18.2%) TA7/TA7, 20 out of 114 (17.5%) TA7/TA6, and 11 out of 114 (9.6%) TA6/TA6 patients. G3–4 diarrhea were reported in one out of 22 (4.5%) TA7/TA7, 14 out of 114 (12.3%) TA7/TA6, six out of 114 (5.3%) TA6/TA6 patients, respectively.
Among UGT1A1*28/*28 genotype patients, the association between irinotecan dose and the risk of G3-4 hematologic adverse events was assessed in a meta-analysis including 821 patients, treated with different regimens, including higher irinotecan doses (200–350 mg/m2), administered every 21 days, intermediate irinotecan dose (180 mg/m2), administered every 2 weeks, or lower doses with weekly administration (80–125 mg/m2); irinotecan was administered alone or in combination with other drugs.40 UGT1A1*28/*28 genotype was correlated with limiting hematologic adverse events in three out of 10 samples (P<0.05), associated with toxicity in two samples (P<0.1), and not associated in the other five.
The risk of developing irinotecan-related hematologic adverse events for patients harboring a UGT1A1*28/*28 genotype was related with administered dose. The risk of hematologic adverse events among patients with UGT1A1*28 and patients with UGT1A1*1/*1 or UGT1A1*1/*28 was significantly correlated with drug dose increase (P=0.028). The risk of develop toxicity was enhanced in patients with UGT1A1*28/*28 vs UGT1A1*1/*1 or UGT1A1*1/*28 genotype treated with moderate (150–250 mg/m2) (OR=3.22, CI=1.52–6.81; P=0.008) and high (>250 mg/m2) (OR=27.8, CI=4.0–195; P=0.005) irinotecan doses. Risk of toxicity was similar at lower irinotecan doses (OR=1.80, CI=0.37–8.84; P=0.41) (100–125 mg/m2), the therapeutic dose range commonly administered. At higher doses (>150 mg/m2), the risk of experience hematologic adverse event was strongly correlated with UGT1A1*28 polymorphism. At the other side, at lower doses (≤150 mg/m2), the risk of develop hematologic adverse events in patients harboring a UGT1A1*28/*28 genotype was not significantly different compared with patients with one or two wild-type alleles (UGT1A1*1/*28 or UGT1A1*1/*1, respectively).
UGT1A1*28/*28 genotype correlated with limiting diarrhea only in one sample (RR=3.40, CI=1.76–6.59; P=0.02), and the risk of develop diarrhea in patients harboring a UGT1A1*28/*28 genotype was not related with irinotecan dose. However, diarrhea rate in patients harboring one or two wild-type alleles inversely correlated with irinotecan dose.
Irinotecan maximum-tolerated dose was investigated in a dose-finding phase I trial enrolling patients with metastatic disease harboring UGT1A1*1/*1 and *1/*28 genotypes, undergoing first-line irinotecan plus infusional 5-FU/leucovorin (FOLFIRI).41 Patients with UGT1A1*28/*28 genotype were excluded; 59 with *1/*1 or *1/*28 genotype were eligible for dose titration. Irinotecan starting dose was 215 mg/m2 with biweekly administration for both subgroups, dose of infusional fluorouracil was fixed. At cycle 1, the prevalent limiting hematologic toxicity (24%) was G3–4 neutropenia; among non-hematologic toxicities, G3 diarrhea (7%), G3–4 asthenia (5%), G3 anorexia (3%). Along the entire treatment course, G3–4 neutropenia was 37% (7% G3–4 febrile neutropenia), and G3 diarrhea was 14%.
Irinotecan dose was escalated up to 370 mg/m2 in patients harboring *1/*28 genotype and up to 420 mg/m2 in patients harboring *1/*1 genotype. Dose-limiting toxicities were reported in two out of four patients with *1/*28 genotype at 370 mg/m2 and in two out of three patients with *1/*1 genotype at 420 mg/m2. No dose LT were reported in 10 patients with *1/*28 genotype treated at 310 mg/m2 and in 10 patients with *1/*1 genotype treated at 370 mg/m2. Thus, these levels represented the maximum tolerated doses for each group. The prevalent G3–4 adverse events were diarrhea and neutropenia.
In a meta-analysis of 16 trials conducted in Caucasian population, including different irinotecan combination regimens and doses in CRC, UGT1A1*28/*28 genotype significantly correlated with more than 4-fold (OR=4.79, CI=3.28–7.01; P<0.00001) and 3-fold (OR=3.44, CI=2.45–4.82; P<0.00001) increased risk of develop neutropenia vs wild-type and patients with at least one UGT1A1*1 allele, respectively (Table 1).42 UGT1A1*1/*28 genotype showed an OR of 1.90 (CI=1.44–2.51; P<0.00001) for increased risk of experience neutropenia. An increased risk of irinotecan-related neutropenia was reported in patients with heterozygote or homozygote UGT1A1*28 allele, regardless of the administrated dose and association of fluoropyrimidine.
The risk of diarrhea was 2-fold increased in patients with UGT1A1*28/*28 genotype (OR=1.84, CI=1.24–2.72; P=0.002), with irinotecan high dose (OR=2.37, CI=1.39–4.04; P=0.002) or in the event of fluoropyrimidine association (OR=1.78, CI=1.16–2.75; P=0.009). UGT1A1*28 homozygous variant showed a stronger association with toxicity, also reported for UGT1A1*28 heterozygous variant.
Thus, pre-emptive UGT1A1*28 polymorphism genotyping for CRC patients can contribute to personalize treatments, reducing irinotecan-related adverse events.
To prevent potentially severe adverse events, UGT1A1 pharmacogenetic analysis is recommended before irinotecan treatment to avoid LT, or after irinotecan exposure in the event of G3–4 gastrointestinal and/or G4 hematological, or any unexpected severe toxicity. To properly evaluate potential correlation between irinotecan and induced toxicity, concomitant administered treatments and their potential pharmacological interaction with UGT1A1, as well as previously experienced toxicities, should take into consideration, to recommend dose modifications, specifically: 100% of standard dose for wild-type 6/6, and heterozygote 6/7; 70% in mutant homozygote 7/7.
For UGT1A1*28 homozygous CRC patients, the US Food and Drug Administration (FDA) recommend initial irinotecan dose modulation, with dose reduction not better specified. The European Medicines Agency (EMA) recommends reduction from 80 to 60 mg/m2 of irinotecan starting dose in UGT1A1*28 homozygous patients.
Potential Correlations Between DPYD and UGT1A1 Activity and Efficacy
Some studies proposed tumoral DPD effectiveness as pharmacogenomic markers of response, correlated with 5-FU efficacy.
SNPs in DPYD were not significantly associated with OS or PFS among 568 MCRC patients treated with first line capecitabine, oxaliplatin, bevacizumab ± cetuximab in the CAIRO2 trial.43
In a prospective study including 250 previously untreated MCRC patients treated with irinotecan, 5-FU, leucovorin,39 TA7/TA7 patients (OR=0.32; CI=0.12–0.86) compared with TA6/TA6 reported higher response rate, potentially related to different pharmacokinetics, specifically higher biliary index and lower glucuronidation ratio, associated with TA7/TA7 genotype, suggesting functional relevance of the polymorphism. Patients harboring the TA7/TA7 (HR=0.52; CI=0.31–0.90) and TA6/TA7 (HR=0.73; CI=0.55–0.98) variant allele significantly showed different time to progression, vs wild type patients: 316, 239, and 226 days, respectively. Patients with TA7 variant allele vs TA6/TA6A showed a non-significant survival advantage: HR=0.81 (95% CI=0.45–1.44) for TA7/TA7, HR=0.84 (95% CI=0.58–1.21) for TA6/TA7, respectively; median OS=686, 669, and 613 days for TA7/TA7, TA6/TA7, and TA6/TA6 patients, respectively.
In the dose-finding phase I trial enrolling MCRC patients harboring UGT1A1*1/*1 and *1/*28 genotypes, treated with first-line irinotecan added to infusional 5-FU/leucovorin (FOLFIRI),41 ORR was higher in patients with *28 allele or treated at doses higher than the maximum tolerated doses; time to progression was not different between higher and lower than maximum tolerated doses.
In a meta-analysis, in Caucasian CRC patients who had received irinotecan, OS and PFS were not significantly affected by the presence of ≥1 UGT1A1*28 alleles (homozygous, heterozygous, or wild-type).44
Other UGT1A1 variants include the UGT1A1*6 (rs4148323, 211G>A) polymorphism, prevalent in the Asian populations, considered as a predictor for irinotecan-induced adverse events in this geographic area. UGT1A1*6/*6 genotype correlated with an approximately 70% reduction of UGT1A1 effectiveness. UGT1A1*6 correlated with the risk of irinotecan-related neutropenia, and was reported as a significant correlate with limiting diarrhea.45
UGT1A9*22 genotype carriers were more at risk for diarrhea due to higher enzyme expression and SN-38 glucuronidation.46 Other UGT1A9 variants, such as UGT1A9*3 and UGT1A9*5, are rare in Caucasians and therefore were not reported as significantly affecting irinotecan pharmacokinetics and pharmacodynamics in this specific population.47
Patients with UGT1A7*346 and UGT1A7*4 polymorphisms48 showed lower enzyme activity and reduced SN-38 conjugation: an increased risk of irinotecan-induced toxicity was reported in UGT1A7*3/*3 carriers.46,49
Irinotecan and SN-38 represent substrates of ABC transporters, therefore irinotecan pharmacokinetics and related toxicities may be affected by polymorphisms in ABC.50 Increased SN-38 plasma concentrations and/or decreased absolute neutrophil counts were associated with ABCC1 SNPs rs6498588 and rs17501331.51 Increased risk of early toxicity, reduced objective response, and shorter OS were reported in patients harboring ABCB1 SNP (rs1045642).52
Also, enhanced SN-38 plasma concentrations and major risk of neutropenia was reported for SLCO1B1*5 (rs4149056), combined with UGT1A1*28 variant alleles.53
Other Pharmacogenomic Biomarkers
This review aimed to focus on recommended biomarkers with enhanced evidence to be applied in clinical practice and add relevant information to guide potential treatments and contribute to better management of CRC patients.
Other potential pharmacogenomics biomarkers were reported requiring more evidence for recommendation, both for fluoropyrimidines and irinotecan, and also for oxaliplatin.
In the ancillary pharmacogenetic evaluation of Italian TOSCA randomized study enrolling CRC patients treated with FOLFOX-4 or XELOX adjuvant chemotherapy, for 3 or 6 months, 17 polymorphisms in 11 genes (specifically, TS, MTHFR, ERCC1, XRCC1, XRCC3, XPD, GSTT1, GSTP1, GSTM1, ABCC1, ABCC2) were analyzed, and none of them showed a clinically relevant association with G3-4 fluoropyrimidines and oxaliplatin-related (G2-4 for neurotoxicity) toxicity.54
TYMS is the key intracellular 5-FU target. A polymorphic tandem repeat sequence in the 5ʹ-untranslated region (5ʹUTR) into TYMS Sequence Enhancer Region (TSER) defines different haplotypes (TSER*2 up to TSER*9). Many studies evaluated the associations between TSER genotype (predominantly TSER*2 and *3) and treatment response.55 Allele harboring the triple tandem repeat (3R) has increased TYMS expression compared with those harboring the double repeat (2R). In MCRC, low TYMS levels correlated with more favorable treatment outcome.56 TYMS 3R/3R vs TYMS 3R/2R genotype carriers have a 1.6-fold increased risk of toxicity (43% of patients treated with 5-FU). Only 3% of patients with TYMS 3/3 genotype developed G3–4 toxicity.56
An important predictor of response to 5-FU is methylenetetrahydrofolate reductase, encoded by MTHFR gene. Several SNPs influence MTHFR activity; and MTHFR A222V was significantly associated with 5-FU response, increased risk of adverse events, and OS in stage III and IV CRC patients, even if conflicting results were reported.
The ABCB1 gene is part of the drug transporter gene family. A significantly higher risk of diarrhea was reported in patients harboring ABCB1 I1145I treated with 5-FU,57 while lower risk of hand-and-foot syndrome was reported for patients harboring the same genotype and treated with capecitabine.58 Among 239 CRC patients treated with capecitabine, a significant correlation was reported between ABCB1*1 haplotype [1236C, 2677G (893Ala), and 3435C] and limiting overall adverse events.57
Exon 5 GSTP1 313A>G polymorphisms (GSTP1*B, Ile105Val, rs1695) were correlated with inhibition of enzyme effectiveness, cancer resistance or toxicity, specifically increased risk of neutropenia,54 and neurotoxicity,59 while homozygous mutant patients reported a lower risk of neurotoxicity and tumor progression vs wild-type.
The Excision Repair Cross-Complementing group (ERCC also named XPD) and the X-Ray Cross-Complementing group (XRCC) are genes involved in DNA adducts repair. ERCC2 encode DNA Helicase; resistance to platinum drugs is related with nuclear protein suppression. A significant correlation between A/A (Lys/Lys) genotype at codon 751 and favorable OS in MCRC treated with 5-FU and platinum was reported.60 ERCC2 variants Lys751Gln and Asp312Gln were correlated with favorable outcomes in patients treated with oxaliplatin-based chemotherapy. Heterozygous genotype for Lys751Gln correlated with increased risk of relapse.61 Lower response, unfavorable PFS, and OS in Caucasians were reported in the CC and the heterozygous AC.
Arg399Gln germline variant of the gene encoding XRCC1 was associated with lower risk of toxicity: patients harboring Gln mutant allele (heterozygous plus homozygous) reported a 5.2-fold increased risk of resistance to 5-FU/oxaliplatin therapy.62 Carriers of both AA (for ERCC2) and GG (for XRCC1) may show enhanced DNA repair ability, reducing the effect of nucleoside analogs drugs, inducing drug resistance and unfavorable outcomes.
Cost-Effectiveness Analyses
Moreover, among 550 CRC patients who received fluoropyrimidine-based chemotherapy, a cost analysis was performed on the experienced adverse events. Retrospective genotype of DPYD*2A, DPYD*13, DPYD c.2846A>T, DPYD-HapB3, and UGT1A1*28 did not affect patients’ treatments.63 DPYD and UGT1A1*28 variants were correlated with the costs to manage adverse events during fluoropyrimidine-based chemotherapy: patients with at least one DPYD variant experienced higher costs (€2,972; 95% CI=€2,456–€3,505) compared with wild-type (€825; 95% CI=€785-€864) (P<0.0001), and showed an increased risk of adverse events requiring hospitalization (odds ratio=4.14; 95% CI=1.87–9.14). In patients treated with fluoropyrimidine plus irinotecan, the incremental cost between patients harboring DPYD variant and UGT1A1*28/*28 and wild-type was €2,975.
Among 571 Italian patients who were treated with fluoropyrimidines-based therapy, the DPYD gene was retrospectively analyzed to analyze the cost sustained to manage toxicity guided by DPYD and clinical benefit, based on quality adjusted life years (QALYs).64 Effectiveness was measured as OS from chemotherapy, data regarding safety, efficacy, and resource utilization to manage specific adverse events, were used to measure costs to treat drug-related toxicity. Fluoropyrimidines treatment guided by DPYD genotype was reported as a cost-saving option for the Italian healthcare system. Patients without any DPYD c.2846A>T, DPYD*2A, DPYD*13, and DPYD-HapB3 variant alleles (group A) had more QALYs and favorable outcomes, in terms of survival months from the beginning of treatment, vs patients harboring one of the DPYD c.2846A>T, DPYD*2A, DPYD*13, and DPYDHapB3 risk variant alleles (group B), treated with fluoropyrimidine-containing regimen. The mean cost to manage fluoropyrimidine-induced toxicity in group A was estimated at €1,010, compared with the average cost of group B (€3,712). These incremental costs were mainly related with more frequent limiting adverse events in group B vs group A, specifically G4 diarrhea and febrile neutropenia. DPYD extensive metabolizers (528 patients) reported enhanced effectiveness and lower cost, compared with DPYD intermediate and poor metabolizers (43 patients) with mean QALYs 4.18 (95% CI=3.16–5.55) vs 3.02 (95% CI=1.94–4.25), representing a cost-saving option. The cost of DPYD extensive vs intermediate/poor metabolizers was significantly lower (P<0.01); some differences in survival between the two groups were reported (P>0.05).
In a prospective clinical trial proposing upfront fluoropyrimidine dose adjustment based on DYPD genotype, the mean treatment cost per individual was significantly lower in patients analyzed for DYPD*2A variant before drug administration (€2,772), compared with patients treated with standard dose (€2,817), showing a cost-effective rationale.65
Discussion
The evolving intensiveness of CRC medical treatment to increase clinical outcome is weighed by a wider spectrum of enhanced cumulative toxicities, with consistent individual variability, justifying on-treatment modulations of planned regimens in clinical practice.20 More, most MCRC patients are unfit for intensive treatment strategies,16 thus requiring a priori individually modulated approaches, consisting of drugs dose reductions and/or schedule modifications, regimens associating with reduced number of drugs, and individual planning of different sequential treatment strategies.
Chemotherapeutic drugs administered as a single agent or in multiple association in CRC patients are frequently associated with severe toxicities, that compromise planned dose and schedule, and treatment efficacy. Detection of genetic variants in genes involved in drugs metabolism could predict the safety profile and enable us to tailor proper individualized treatment strategies. Different studies demonstrated that genetic variants of different enzymes involved in fluoropyrimidine and irinotecan metabolism increased the risk of adverse events. Clinically relevant data involve DPD for fluoropyrimidine, and UGT1A1 for irinotecan, through its metabolite SN-38.66
A reduced DPD effectiveness could result in an increased 5-FU half-life, inducing a higher risk of adverse events occurrence.67 Although prospective identification of known DPYD variants determining a consistent reduction of DPD effectiveness is validated to prevent severe, treatment-related adverse events, this clinical approach is not yet routinely implemented in clinical practice. Prospective detection of DPYD variants, guiding upfront personalization of fluorouracil dose, could translate into a relevant clinical benefit in patient care by reducing toxicity, and maintaining the treatment plan and efficacy. To date, three DPYD variants showed highest evidence to predict 5-FU-induced severe toxicity: DPYD*2A c.1905+1G>A, DPYD*13 c.1679T>G, DPYD c.2846A>T, each reported with approximately 1.0% prevalence in the Caucasian population.68 Different studies demonstrated that at least four most relevant DPYD genetic variants should be conventionally evaluated, also including DPYD c.1236 G>A (c.1129–5923 C>G, HapB3), the most prevalent DPYD variant. Patients carrying c.IVS14+1G>A DPYD variant develop severe toxicities when treated with fluoropyrimidine, with different reported frequencies (5.5–29%). Recently, further studies showed that DPYD*6 c.2194G>A should also be evaluated due to significant association with clinically-relevant adverse events. Overall, approximately 8% of CRC patients are heterozygous DPYD variant allele carriers, mostly determined by these five mutations significantly determining fluoropyrimidines-related toxicity, justifying different dose reductions.
Allelic variant UGT1A1*28 is associated with reduced irinotecan metabolism, and hematological and gastrointestinal toxicities. The risk to experience adverse events seems to be dose-dependent, generally not reported for doses <150 mg/m2. Reduced Irinotecan dose is recommended in patient carriers of homozygote UGT1A1*28 variant. Moreover, some data suggest that patients carrying wild-type UGT1A1 allele could tolerate increased irinotecan doses, potentially affecting more favorable clinical outcome.
Moreover, coexisting diarrhea and neutropenia represent major related irinotecan adverse events, and in this event proper and careful doses adjustments are required. Thus, oncologists should better define different and specific genotype-phenotype implications, to properly personalize treatments in clinical practice.
Pharmacogenomic analyses should be performed when chemotherapy treatment is planned, according to specific clinical settings, particularly to avoid LT in elderly and frail patients with comorbidities. These evaluations should be recommended for all early CRC potentially cured by surgery, and suitable for adjuvant treatment, and particularly in specific settings such as in the high-risk stage II early CRC, when the proposal of treatment can be considered optional for the patient, adding limited advantage in terms of clinical outcomes. The usefulness of these baseline analyses should be properly considered in the metastatic setting for patients potentially suitable for intensive first line treatment associations, with potentially increased toxicities determined by more drug combinations, particularly for more intensive triplet chemotherapy plus targeted agent regimens, and specifically related to fluoropyrimidines and irinotecan associations (Table 2).
 |
Table 2 Implementation of Pharmacogenetic Analysis in Colorectal Cancer in Clinical Practice |
Moreover, pharmacogenomic analyses should be performed after treatment in the event of limiting G3–4 gastrointestinal and/or G4 hematological toxicity or in the event of any unexpected toxicity, to adequately modulate treatment, to reduce LT, maintain adequate dose and schedule, and avoid affecting the clinical effectiveness of the administered treatment.
Specific pharmacogenetic analyses will help to properly relate genotype variants with adverse events to verify: lack of toxicity in patients with wild-type genotype; the occurrence and the spectrum of adverse events in patients with variant genotypes treated with standard- 5-FU and/or irinotecan doses, even if about 50% experience severe toxicity. To this aim, prospective studies including larger patient populations will provide an accurate risk evaluation.
Recently, pharmacogenomic analyses was even more evaluated among patients treated with intensive regimens, associating fluorouracil, irinotecan, oxaliplatin plus targeted agent, administered according to different schedules, underlying their contribution to adequately manage treatments combining different drugs potentially affecting the same hematological and gastrointestinal safety profile. DPYD and UGT1A1 variants were evaluated in 87% MCRC patients enrolled in TRIBE trial randomized to FOLFIRI vs FOLFOXIRI plus bevacizumab.69
Detected DPYD variants were heterozygous DPYD*2A c.1905+1G>A and DPYD c.2846 A>T 1.1%, respectively; no DPYD*13 c.1679T>G variant was reported. UGT1A1 wild-type, heterozygote *1/*28, and homozygote *28/*28 were 33.5%, 57.6%, and 8.9%, respectively. Concomitant DPYD and UGT1A1*28 minor variants were detected in seven patients. Genotyped patients with any ≥G3 adverse events were 51%; ≥G3 gastrointestinal 23%, and hematological 38%. The most frequent adverse events were neutropenia (37%), diarrhea (15%), febrile neutropenia (8%), and stomatitis (7%). DPYD c.1905+1G/A and DPYD c.2846A/T variants were significantly associated with ≥G3 hematological adverse events and stomatitis; and UGT1A1*28 variant with ≥G3 hematological toxicities, particularly neutropenia. Genotype/phenotype relations between DPYD variants and at least one ≥G3 adverse event during treatment occurred in eight out of 10 (80%) DPYD c.1905+1G/A or DPYD c.2846A/T carriers; seven out of 10 patients (70%) with a DPYD variant allele had a ≥G3 adverse event within the first four cycles of therapy, compared to 39% with wild-type genotype (P=0.055). The most frequent adverse events were neutropenia (70%), febrile neutropenia (20%), stomatitis (40%), diarrhea (20%), and thrombocytopenia (10%). DPYD c.1905+1G/A and DPYD c.2846A/T were significantly associated with ≥G3 stomatitis (OR=9.69, respectively); and DPYD c.1905+1G/A with ≥G3 thrombocytopenia (OR=21.50). DPYD c.1905+1G/A and DPYD c.2846A/T confirmed the association with ≥G3 stomatitis in multivariate analysis (OR=17.32 and 14.11, respectively), also confirmed for DPYD c.1905+1G/A with thrombocytopenia (OR=62.81); DPYD c.1905+1G/A was significantly associated with higher risk of anemia (OR=41.26); DPYD c.1905+1G/A or DPYD c.2846A/T variants with increased risk of ≥G3 overall hematological adverse events (OR=3.88), neutropenia (OR=4.12), thrombocytopenia (OR=9.42), and stomatitis (OR=10.33), compared to wild-type genotypes, and increased overall gastrointestinal adverse events (OR=4.59).
Patients with UGT1A1*1/*28 heterozygote or homozygote genotype showed ≥G3 overall adverse events of 54% and 62%, respectively; UGT1A1*28/*28 homozygote patients showed more frequent ≥G3 adverse events within the first four cycles of treatment (56.4%), compared to heterozygote (43.0%) and wild-type genotypes (29.5%) (P=0.002). UGT1A1 variants and neutropenia were significantly associated, higher for homozygote (OR=3.75) than heterozygote (OR=1.66), compared with wild-type.
Patients bearing DPYD c.1905+1G/A or DPYD c.2846A/T and/or UGT1A1*28/*28 genotypes showed a significantly higher risk of ≥G3 overall toxicity (OR=1.89), overall hematological (OR=2.79), stomatitis (OR=3.32), neutropenia (OR=2.98), and febrile neutropenia (OR=2.78), compared to patients with wild-type genotypes.
Genetic variants of DPYD and UGT1A1 genotype modify fluoropyrimidines and irinotecan safety profile, and justify inter-patient variability of toxicity.70 In phase II trials evaluating the COI regimen (capecitabine, oxaliplatin, irinotecan) plus bevacizumab or cetuximab,71 DPYD c.496 A>G and c.1896 T>C were independently and significantly associated with severe toxicity and treatment modulations; UGT1A1*28 heterozygote variant was associated (P=0.054),71 also with increased risk of severe neutropenia.70–72
Moreover, in the last few years, in order to better evaluate, at the clinical level, the burden of toxicities in the individual CRC patient, we proposed the consideration of individual toxicity syndrome (TS), and specifically limiting TS (LTS), consisting of at least a LT associated or not to other limiting or G2 toxicities,5,13,20 defined as single site-LTS (LTS-ss), characterized only by the LT, from multiple sites-LTS (LTS-ms), characterized by ≥2 LTs or a LT associated to other, at least G2, non-limiting toxicities. Recently, we added the evaluation of LTS as an indicator of the individual toxicity in the phase II trial of MCRC RAS wild-type patients treated with FIr-C/FOx-C intensive treatment regimen, adding cetuximab to triplet chemotherapy;14 more, we used LTS as the parameter to which relate 5-FU/irinotecan-related pharmacogenomic analyses, including DPYD and UGT1A1 genetic variants to predict gastrointestinal toxicity in individual patients,14 evaluated on-treatment in patients who experienced LTS at first dose level, and before starting treatment at recommended dose level. LTS were observed in 65.5% (19 out of 29), and 83% young-elderly patients (five out of six), mostly LTS-ms 59%, consisting of LT associated to other at least G2 non-limiting toxicities (34%) or ≥2 LTs (24%). Pharmacogenomic analyses including DPYD*2A c1905+1 G>A and A166G, UGT1A1*28 genetic variants were related with LTS. The prevalence of pharmacogenomic alterations were: DPYD genetic variants, two out of 13 (15%), none out of eight patients with LTS, and all in five patients without LTS; UGT1A1*28, six out of 12 patients (50%), five out of seven patients with LTS (71%), and one out of five without LTS (20%), respectively. Upfront detection of UGT1A1*28 variants may be useful to select patients fit for intensive treatments including anti-EGFR associated with triplet chemotherapy. DPYD and UGT1A1 wild-type genotype may justify increased tolerability at the recommended doses. Evaluation of TS, specifically LTS, may address selection of MCRC patients suitable for intensive regimens adding triplet chemotherapy and anti-EGFR drug, or to modulate intensive triplet chemotherapy-based regimens. Furthermore, implementation of a pharmacogenetic profile, including DPYD and UGT1A1, of the individual patient, particularly treated with intensive regimens associating different drugs, may also be related to individual TS, specifically LTS, to better evaluate the relationship between individual genetic identity and the occurrence of TS.
In conclusion, implementation of up-front evaluation of the five validated DPYD and UGT1A1*28 variants in the multidisciplinary molecular tumor board, also including CRC genetic characterization, addresses potential treatments with fluoropyrimidines and irinotecan associations at proper doses and schedules, particularly for early CRC, MCRC patients fit for intensive regimens or unfit for conventional regimens requiring treatment modulations, and also for patients who experienced severe, unexpected toxicities. Integration of individual evaluation of toxicity syndromes (TS), specifically limiting TS (LTS), as an innovative indicator of toxicity burden in individual patients, may be useful to better evaluate relationships between pharmacogenomic analyses with safety profile and clinical outcome.
Abbreviations
anti-EGFR, anti-epidermal growth factor receptor; anti-VEGF, anti-vascular endothelial growth factor; CRC, colorectal cancer; DPYD, dihydropyrimidine dehydrogenase; LT, limiting toxicity; LTS, limiting toxicities syndromes; LTS-ms, limiting toxicity syndrome multiple sites; LTS-ss, limiting toxicity syndrome single site; MCRC, metastatic colorectal cancer; OS, overall survival; SNP, Single Nucleotide Polymorphisms; UGT1A1, UDP glucuronosyl transferase 1 family, polypeptide A1; 5-FU, 5-fluorouracil; 5-FUDR, 5-fluorouracil degradation rate.
Funding
There is no funding to report.
Disclosure
The authors declare that there are no conflicts of interest.
References
1. Machover D, Goldschmidt E, Chollet P, et al. Treatment of advanced colorectal and gastric adenocarcinomas with 5-fluorouracil and high-dose folinic acid. J Clin Oncol. 1986;4(5):685–696. doi:10.1200/JCO.1986.4.5.685
2. Barone C, Astone A, Garufi C, et al. High-dose folinic acid (HDFA) combined with 5-fluorouracil (5-FU) in first line chemotherapy of advanced large bowel cancer. Eur J Cancer Clin Oncol. 1987;23(9):1303–1306. doi:10.1016/0277-5379(87)90112-X
3. Loupakis F, Cremolini C, Masi G, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med. 2014;371(17):1609–1618. doi:10.1056/NEJMoa1403108
4. Cremolini C, Loupakis F, Antoniotti C, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015;16(13):1306–1315. doi:10.1016/S1470-2045(15)00122-9
5. Bruera G, Santomaggio A, Cannita K, et al. “Poker” association of weekly alternating 5-fluorouracil, irinotecan, bevacizumab and oxaliplatin (FIr-B/FOx) in first line treatment of metastatic colorectal cancer: a phase II study. BMC Cancer. 2010;10(1):567. doi:10.1186/1471-2407-10-567
6. Bruera G, Ricevuto E. Intensive chemotherapy of metastatic colorectal cancer: weighing between safety and clinical efficacy. Evaluation of Masi G, Loupakis F, Salvatore L, et al. Bevacizumab with FOLFOXIRI (irinotecan, oxaliplatin, fluorouracil, and folinate) as first-line treatment for metastatic colorectal cancer: a Phase 2 trial. Lancet Oncol 2010;11:845–52. Expert Opin Biol Ther. 2011;11(6):821–824.
7. Bruera G, Cannita K, Giuliante F, et al. Effectiveness of liver metastasectomies in metastatic colorectal cancer (MCRC) patients treated with triplet chemotherapy plus bevacizumab (FIr-B/FOx). Clin Colorectal Cancer. 2012;11(2):119–126.
8. Bruera G, Russo A, Galvano A, Rizzo S, Ricevuto E. Clinical parameters to guide decision-making in elderly metastatic colorectal cancer patients treated with intensive cytotoxic and anti-angiogenic therapy. Oncotarget. 2017;8(23):37875–37883. doi:10.18632/oncotarget.14333
9. Ficorella C, Bruera G, Cannita K, et al. Triplet chemotherapy in patients with metastatic colorectal cancer: toward the best way to safely administer a highly active regimen in clinical practice. Clin Colorectal Cancer. 2012;11(4):229–237.
10. Bruera G, Cannita K, Di Giacomo D, et al. Prognostic value of KRAS genotype in metastatic colorectal cancer (MCRC) patients treated with intensive triplet chemotherapy plus bevacizumab (FIr-B/FOx) according to extension of metastatic disease. BMC. Medicine. 2012;10(1):135.
11. Bruera G, Cannita K, Di Giacomo D, et al. Worse prognosis of KRAS c.35 G > A mutant metastatic colorectal cancer (MCRC) patients treated with intensive triplet chemotherapy plus bevacizumab (FIr-B/FOx). BMC Med. 2013;11(1):59. doi:10.1186/1741-7015-11-59
12. Bruera G, Cannita K, Tessitore A, et al. The prevalent KRAS exon 2 c.35 G > A mutation in metastatic colorectal cancer patients: a biomarker of worse prognosis and potential benefit of bevacizumab-containing intensive regimens? Crit Rev Oncol Hematol. 2015;93(3):190–202. doi:10.1016/j.critrevonc.2014.10.004
13. Bruera G, Cannita K, Giordano AV, Vicentini R, Ficorella C, Ricevuto E. Effectiveness and safety of intensive triplet chemotherapy plus bevacizumab, FIr-B/FOx, in young-elderly metastatic colorectal cancer (MCRC) patients. Biomed Res Int. 2013;2013:143273. doi:10.1155/2013/143273
14. Bruera G, Massacese S, Pepe F, et al. Intensive first-line FIr-C/FOx-C triplet chemotherapy plus cetuximab in RAS wild-type metastatic colorectal cancer patients: preliminary phase II data and prediction of individual limiting toxicity syndromes by pharmacogenomic biomarkers. Ther Adv Med Oncol. 2019;11:1–13. doi:10.1177/1758835919846421
15. Bruera G, Pepe F, Malapelle U, et al. KRAS, NRAS and BRAF mutations detected by next generation sequencing, and differential clinical outcome in metastatic colorectal cancer (MCRC) patients treated with first line FIr-B/FOx adding bevacizumab (BEV) to triplet chemotherapy. Oncotarget. 2018;9(41):26279–26290. doi:10.18632/oncotarget.25180
16. Bruera G, Cannita K, Giordano AV, Vicentini R, Ficorella C, Ricevuto E. Prognostic relevance of KRAS genotype in metastatic colorectal cancer patients unfit for FIr-B/FOx intensive regimen. Int J Oncol. 2014;44(6):1820–1830. doi:10.3892/ijo.2014.2369
17. Bruera G, Cannita K, Giordano AV, Vicentini R, Ficorella C, Ricevuto E. Differential prognosis of metastatic colorectal cancer patients post-progression to first-line triplet chemotherapy plus bevacizumab, FIr-B/FOx, according to second-line treatment and KRAS genotype. Int J Oncol. 2014;44(1):17–26. doi:10.3892/ijo.2013.2179
18. Van Cutsem E, Huijberts S, Grothey A. Binimetinib encorafenib, and cetuximab triplet therapy for patients with BRAF V600E-mutant metastatic colorectal cancer: safety lead-in results from the phase III BEACON colorectal cancer study. J Clin Oncol. 2019;37(17):1460–1469. doi:10.1200/JCO.18.02459
19. Geva R, Jäger D, Hara H, et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol. 2020;38(1):11–19. doi:10.1200/JCO.19.02107
20. Bruera G, Ricevuto E. Toxicity-syndromes, patient-related indicator of toxicity burden induced by intensive triplet chemotherapy-based regimens in gastrointestinal cancers with metastatic disease. Front Oncol. 2020;10:172.
21. Cecchin E, De Mattia E, Ecca F, Toffoli G. Host genetic profiling to increase drug safety in colorectal cancer from discovery to implementation. Drug Resist Updat. 2018;39:18–40. doi:10.1016/j.drup.2018.07.001
22. Amstutz U, Henricks LM, Offer SM, et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin Pharmacol Ther. 2018;103(2):210–216. doi:10.1002/cpt.911
23. Di Francia R, De Lucia L, Di Paolo M, et al. Rational selection of predictive pharmacogenomics test for the fluoropyrimidine/oxaliplatin based therapy. Eur Rev Med Pharmacol Sci. 2015;19(22):4443–4454.
24. Ab Mutalib NS, Md Yusof NF, Abdul SN, Jamal R. Pharmacogenomics DNA biomarkers in colorectal cancer: current update. Front Pharmacol. 2017;8:736. doi:10.3389/fphar.2017.00736
25. de Man FM, Goey AKL, van Schaik RHN, Mathijssen RHJ, Bins S. Individualization of irinotecan treatment: a review of pharmacokinetics, pharmacodynamics, and pharmacogenetics. Clin Pharmacokinet. 2018;57(10):1229–1254.
26. Mattison LK, Soong R, Diasio RB. Implications of dihydropyrimidine dehydrogenase on 5-fluorouracil pharmacogenetics and pharmacogenomics. Pharmacogenomics. 2002;3(4):485–492. doi:10.1517/14622416.3.4.485
27. Del Re M, Cinieri S, Michelucci A, et al. DPYD*6 plays an important role in fluoropyrimidine toxicity in addition to DPYD*2A and c.2846A>T: a comprehensive analysis in 1254 patients. Pharmacogenomics J. 2019;19(6):556–563. doi:10.1038/s41397-019-0077-1
28. Ruzzo A, Graziano F, Galli F, et al. Dihydropyrimidine dehydrogenase pharmacogenetics for predicting fluoropyrimidine-related toxicity in the randomised, phase III adjuvant TOSCA trial in high-risk colon cancer patients. Br J Cancer. 2017;117:1269–1277. doi:10.1038/bjc.2017.289
29. Iachetta F, Bonelli C, Romagnani A, et al. The clinical relevance of multiple DPYD polymorphisms on patients candidate for fluoropyrimidine based-chemotherapy. An Italian case-control study. Br J Cancer. 2019;120(8):834–839. doi:10.1038/s41416-019-0423-8
30. Meulendijks D, Henricks LM, Sonke GS, et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015;16(16):1639–1650. doi:10.1016/S1470-2045(15)00286-7
31. Terrazzino S, Cargnin S, Del Re M, Danesi R, Canonico PL, Genazzani AA. DPYD IVS14+1G>A and 2846A>T genotyping for the prediction of severe fluoropyrimidine-related toxicity: a meta-analysis. Pharmacogenomics. 2013;14(11):1255–1272. doi:10.2217/pgs.13.116
32. Toffoli G, Giodini L, Buonadonna A, et al. Clinical validity of a DPYD -based pharmacogenetic test to predict severe toxicity to fluoropyrimidines. Int J Cancer. 2015;137(12):2971–2980. doi:10.1002/ijc.29654
33. Boige V, Vincent M, Alexandre P, et al. DPYD genotyping to predict adverse events following treatment with fluorouracil-based adjuvant chemotherapy in patients with stage III colon cancer: a secondary analysis of the PETACC-8 randomized clinical trial. JAMA Oncol. 2016;2(5):655–662. doi:10.1001/jamaoncol.2015.5392
34. Henricks LM, Lunenburg CATC, de Man FM, et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol. 2018;19(11):1459–1467. doi:10.1016/S1470-2045(18)30686-7
35. Lunenburg CATC, Henricks LM, Dreussi E, et al. Standard fluoropyrimidine dosages in chemoradiation therapy result in an increased risk of severe toxicity in DPYD variant allele carriers. Eur J Cancer. 2018;104:210–218. doi:10.1016/j.ejca.2018.07.138
36. Dalle Fratte C, Polesel J, Roncato R, et al. DPYD gene activity score predicts dose-limiting toxicity in fluoropyrimidine-treated colorectal cancer patients. J Mol Clin Med. 2018;1(3):143–149.
37. Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27(8):1386–1422. doi:10.1093/annonc/mdw235
38. O’Dwyer PJ, Catalano RB. Uridine diphosphate glucuronosyltransferase (UGT) 1A1 and irinotecan: practical pharmacogenomics arrives in cancer therapy. J Clin Oncol. 2006;24(28):4534–4538. doi:10.1200/JCO.2006.07.3031
39. Toffoli G, Cecchin E, Corona G, et al. The role of UGT1A1*28 polymorphism in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J Clin Oncol. 2006;24(19):3061–3068. doi:10.1200/JCO.2005.05.5400
40. Hoskins JM, Goldberg RM, Qu P, Ibrahim JG, McLeod HL. UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J Natl Cancer Inst. 2007;99(17):1290–1295. doi:10.1093/jnci/djm115
41. Toffoli G, Cecchin E, Gasparini G, et al. Genotype-driven phase I study of irinotecan administered in combination with fluorouracil/leucovorin in patients with metastatic colorectal cancer. J Clin Oncol. 2009;28(5):866–871. doi:10.1200/JCO.2009.23.6125
42. Liu X, Cheng D, Kuang Q, Liu G, Xu W. Association of UGT1A1*28 polymorphisms with irinotecan induced toxicities in colorectal cancer: a meta-analysis in Caucasians. Pharmacogenomics J. 2014;14(2):120–129. doi:10.1038/tpj.2013.10
43. Deenen MJ, Tol J, Burylo AM, et al. Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer. Clin Cancer Res. 2011;17(10):3455–3468. doi:10.1158/1078-0432.CCR-10-2209
44. Dias MM, Pignon JP, Karapetis CS, et al. The effect of the UGT1A1*28 allele on survival after irinotecan-based chemotherapy: a collaborative meta-analysis. Pharmacogenomics J. 2014;14(5):424–431.
45. Cheng L, Li M, Hu J, et al. UGT1A1*6 polymorphisms are correlated with irinotecan-induced toxicity: a system review and meta-analysis in Asians. Cancer Chemother Pharmacol. 2014;73(3):551–560. doi:10.1007/s00280-014-2382-3
46. Inoue K, Sonobe M, Kawamura Y, et al. Polymorphisms of the UDP-glucuronosyl transferase 1A genes are associated with adverse events in cancer patients receiving irinotecan-based chemotherapy. Tohoku J Exp Med. 2013;229(2):107–114. doi:10.1620/tjem.229.107
47. Paoluzzi L, Singh AS, Price DK, et al. Influence of genetic variants in UGT1A1 and UGT1A9 on the in vivo glucuronidation of SN-38. J Clin Pharmacol. 2004;44(8):854–860. doi:10.1177/0091270004267159
48. Gagne JF, Montminy V, Belanger P, Journault K, Gaucher G, Guillemette C. Common human UGT1A polymorphisms and the altered metabolism of irinotecan active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38). Mol Pharmacol. 2002;62(3):608–617. doi:10.1124/mol.62.3.608
49. Cecchin E, Innocenti F, D’Andrea M, et al. Predictive role of the UGT1A1, UGT1A7, and UGT1A9 genetic variants and their haplotypes on the outcome of metastatic colorectal cancer patients treated with fluorouracil, leucovorin, and irinotecan. J Clin Oncol. 2009;27(15):2457–2465. doi:10.1200/JCO.2008.19.0314
50. Innocenti F, Kroetz DL, Schuetz E, et al. Comprehensive pharmacogenetic analysis of irinotecan neutropenia and pharmacokinetics. J Clin Oncol. 2009;27(16):2604–2614. doi:10.1200/JCO.2008.20.6300
51. Li M, Seiser EL, Baldwin RM, et al. ABC transporter polymorphisms are associated with irinotecan pharmacokinetics and neutropenia. Pharmacogenomics J. 2018;18(1):35–42. doi:10.1038/tpj.2016.75
52. Glimelius B, Garmo H, Berglund A, et al. Prediction of irinotecan and 5-fluorouracil toxicity and response in patients with advanced colorectal cancer. Pharmacogenomics J. 2011;11(1):61–71. doi:10.1038/tpj.2010.10
53. Teft WA, Welch S, Lenehan J, et al. OATP1B1 and tumour OATP1B3 modulate exposure, toxicity, and survival after irinotecan-based chemotherapy. Br J Cancer. 2015;112(5):857–865.
54. Ruzzo A, Graziano F, Galli F, et al. Genetic markers for toxicity of adjuvant oxaliplatin and fluoropyrimidines in the phase III TOSCA trial in high-risk colon cancer patients. Sci Rep. 2014;4(1):6828. doi:10.1038/srep06828
55. Liersch T, Langer C, Ghadimi BM, et al. Lymph node status and TS gene expression are prognostic markers in stage II/III rectal cancer after neoadjuvant fluorouracil-based chemoradiotherapy. J Clin Oncol. 2006;24(25):4062–4068. doi:10.1200/JCO.2005.04.2739
56. Lecomte T, Ferraz JM, Zinzindohoue F, et al. Thymidylate synthase gene polymorphism predicts toxicity in colorectal cancer patients receiving 5-fluorouracil-based chemotherapy. Clin Cancer Res. 2004;10(17):5880–5888. doi:10.1158/1078-0432.CCR-04-0169
57. García-González X, Cortejoso L, García MI, et al. Variants in CDA and ABCB1 are predictors of capecitabine-related adverse reactions in colorectal cancer. Oncotarget. 2015;6(8):6422–6430. doi:10.18632/oncotarget.3289
58. Gonzalez-Haba E, García MI, Cortejoso L, et al. ABCB1 gene polymorphisms are associated with adverse reactions in fluoropyrimidine-treated colorectal cancer patients. Pharmacogenomics. 2010;11(12):1715–1723. doi:10.2217/pgs.10.159
59. De Monaco A, Faioli D, Di Paolo M, et al. Pharmacogenomics markers for prediction response and toxicity in cancer therapy. WCRJ. 2014;1:e276.
60. Park DJ, Zhang W, Stoehlmacher J, et al. ERCC1 gene polymorphism as a predictor for clinical outcome in advanced colorectal cancer patients treated with platinum-based chemotherapy. Clin Adv Hematol Oncol. 2003;1:162–166.
61. Yin M, Yan M, Martinez-Balobrea E, et al. ERCC1 and ERCC2 polymorphisms predict clinical outcomes of oxaliplatin-based chemotherapies in gastric and colorectal cancer: a systemic review and meta-analysis. Clin Cancer Res. 2011;17(6):1632–1640. doi:10.1158/1078-0432.CCR-10-2169
62. Stoehlmacher J, Ghaderi V, Iobal S, et al. A polymorphism of the XRCC1 gene predicts for response to platinum based treatment in advanced colorectal cancer. Anticancer Res. 2001;213075–213079.
63. Toffoli G, Innocenti F, Polesel J, et al. The genotype for DPYD risk variants in patients with colorectal cancer and the related toxicity management costs in clinical practice. Clin Pharmacol Ther. 2019;105(4):994–1002. doi:10.1002/cpt.1257
64. Fragoulakis V, Roncato R, Dalle Fratte C, et al. Estimating the effectiveness of DPYD genotyping in italian individuals suffering from cancer based on the cost of chemotherapy-induced toxicity. Am J Hum Genet. 2019;104(6):1158–1168. doi:10.1016/j.ajhg.2019.04.017
65. Deenen MJ, Meulendijks D, Cats A, et al. Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy: a safety and cost analysis. J Clin Oncol. 2016;34(3):227–234. doi:10.1200/JCO.2015.63.1325
66. Cortejoso L, López-Fernández LA. Pharmacogenetic markers of toxicity for chemotherapy in colorectal cancer patients. Pharmacogenomics. 2012;13(10):1173–1191.
67. Amstutz U, Froehlich TK, Largiadèr CR. Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5-fluorouracil toxicity. Pharmacogenomics. 2011;12(9):1321–1336. doi:10.2217/pgs.11.72
68. Lunenburg CATC, Henricks LM, Guchelaar HJ, et al. Prospective DPYD genotyping to reduce the risk of fluoropyrimidine-induced severe toxicity: ready for prime time. Eur J Cancer. 2016;54:40–48. doi:10.1016/j.ejca.2015.11.008
69. Cremolini C, Del Re M, Antoniotti C, et al. DPYD and UGT1A1 genotyping to predict adverse events during first-line FOLFIRI or FOLFOXIRI plus bevacizumab in metastatic colorectal cancer. Oncotarget. 2018;9(8):7859–7866. doi:10.18632/oncotarget.23559
70. Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP working group: can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan? Genet Med. 2009;11(1):15–20. doi:10.1097/GIM.0b013e31818efd9d
71. Falvella FS, Cheli S, Martinetti A, et al. DPD and UGT1A1 deficiency in colorectal cancer patients receiving triplet chemotherapy with fluoropyrimidines, oxaliplatin and irinotecan. Br J Clin Pharmacol. 2015;80(3):581–588. doi:10.1111/bcp.12631
72. Dias MM, McKinnon RA, Sorich MJ. Impact of the UGT1A1*28 allele on response to irinotecan: a systematic review and meta-analysis. Pharmacogenomics. 2012;13(8):889–899. doi:10.2217/pgs.12.68
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.