")
Back to Journals » International Journal of Nanomedicine » Volume 12
pH-triggered surface charge-switchable polymer micelles for the co-delivery of paclitaxel/disulfiram and overcoming multidrug resistance in cancer
Authors Huo Q, Zhu J, Niu Y, Shi H, Gong Y, Li Y, Song H, Liu Y
Received 20 June 2017
Accepted for publication 23 August 2017
Published 4 December 2017 Volume 2017:12 Pages 8631—8647
DOI https://doi.org/10.2147/IJN.S144452
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Dongwoo Khang
Qiang Huo,1,* Jianhua Zhu,1,2,* Yimin Niu,3 Huihui Shi,2 Yaxiang Gong,2 Yang Li,2 Huihui Song,4 Yang Liu2
1School of Pharmacy, Bengbu Medical College, Bengbu, 2School of Pharmacy, Nanjing Medical University, 3Department of Pharmacy, Zhongda Hospital, School of Medicine, Southeast University, Nanjing, 4Yangtze River Pharmaceutical Group, Taizhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: Multidrug resistance (MDR) remains a major challenge for providing effective chemotherapy for many cancer patients. To address this issue, we report an intelligent polymer-based drug co-delivery system which could enhance and accelerate cellular uptake and reverse MDR. The nanodrug delivery systems were constructed by encapsulating disulfiram (DSF), a P-glycoprotein (P-gp) inhibitor, into the hydrophobic core of poly(ethylene glycol)-block-poly(l-lysine) (PEG-b-PLL) block copolymer micelles, as well as 2,3-dimethylmaleic anhydride (DMA) and paclitaxel (PTX) were grafted on the side chain of l-lysine simultaneously. The surface charge of the drug-loaded micelles represents as negative in plasma (pH 7.4), which is helpful to prolong the circulation time, and in a weak acid environment of tumor tissue (pH 6.5–6.8) it can be reversed to positive, which is in favor of their entering into the cancer cells. In addition, the carrier could release DSF and PTX successively inside cells. The results of in vitro studies show that, compared to the control group, the DSF and PTX co-loaded micelles with charge reversal exhibits more effective cellular uptake and significantly increased cytotoxicity of PTX to MCF-7/ADR cells which may be due to the inhibitory effect of DSF on the efflux function of P-gp. Accordingly, such a smart pH-sensitive nanosystem, in our opinion, possesses significant potential to achieve combinational drug delivery and overcome drug resistance in cancer therapy.
Keywords: pH response, charge reversal, multidrug resistance, paclitaxel, disulfiram
Introduction
Multidrug resistance (MDR), where cells are instinctive or acquired, immensely limits the effectiveness of anticancer agents and remains one of the primary obstacles of chemotherapy in clinical practice.1–4 The mechanisms of MDR have been well studied, and one of the major mechanisms is the overexpression of P-glycoprotein (P-gp) in the tumor cell membrane.5,6 P-gp, a kind of adenosine triphosphate-binding cassette transporter, is able to remove many intracellular substrates, such as paclitaxel (PTX), doxorubicin (DOX), docetaxel, vincristine and etoposide into the tumor stroma in an adenosine triphosphate-dependent manner.7–9 This consequently leads to reducing the intracellular drug concentration and eventually limits its antitumor activity.10,11
Many strategies aiming at effective reversal of MDR have been investigated through the co-delivery of chemotherapeutic agents and P-gp inhibitor by nanoparticles.6,12–15 For example, Wang et al16 reported that polymeric micelles co-encapsulated with PTX and tacrolimus (a P-gp inhibitor) (P-F/M) could improve intracellular concentration of PTX and showed much stronger cytotoxicity against PTX-resistant human ovarian cancer A2780/PTX cells than PTX-loaded micelles. Unfortunately, the P-gp inhibitor actions in a concentration-dependent manner and the simultaneous release of P-gp inhibitor and cytotoxic agents may cause a lot of free chemotherapeutic agents being pumped out of the cells before the P-gp functions are restrained. Consequently, it is an attractive strategy to effectively overcome MDR with the P-gp inhibitor possibly releasing relatively quicker than the chemotherapy drug from the same carriers in the cells. Recently, Duan et al17 reported a pH-sensitive polymeric micelle system loaded with DOX and disulfiram (DSF; a P-gp inhibitor as well as apoptosis inducer) simultaneously, and they found that, because the DSF was released faster than DOX in the cells, the co-delivery system could obviously increase the intracellular accumulation of DOX and to demonstrate strong tumor suppression effects in vivo.
On the other hand, a class of stimuli-responsive nanostructures are recently developed and such systems could prolong circulation time in blood and enhance uptake by tumor cells through acidic tumor microenvironment-triggered surface charge switch based on acid-labile bonds.18–23 For example, Yuan et al24 prepared a charge-switchable nanoparticle (S-NP/DOX) based on zwitterionic polymers. The S-NP/DOX remained negative surface charge in the blood circulation system and switched to positive surface charge in response to the tumor acidity; the tumor-acidity-activated charge conversion could effectively increase the cellular uptake of the carriers and the intracellular concentrations of DOX and, in turn, resulted in enhancing tumor inhibitions. In addition, intracellular organelle microenvironments such as low pH in endo-/lysosomal compartments are also available for triggering drug release from drug-carrier conjugates which contain acid-cleavable linkages.25,26
In the aim to overcome the P-gp-mediated MDR, in this study we report novel surface charge-reversible polymer micelles which could cascade release PTX and DSF inside drug-resistant tumor cells (Scheme 1). The carriers were constituted with succinic anhydride-modified PTX (SA-PTX) and 2,3-dimethylmaleic anhydride (DMA)-conjugated poly(ethylene glycol)-b-poly(L-lysine). Besides, DSF, a hydrophobic drug, was encapsulated into the micelles. Such a smart drug delivery nanosystem was characterized as follows: 1) the micelles could ideally remain negative surface charge under blood circulation, while quickly changes to positive under the acidic tumor tissues owing to the hydrolysis of the DMA group.27 In addition, this change could increase the endocytosis rate of the micelles; 2) the internalization carriers could successively release two drugs, which means that DSF is quickly released accompanied with the deformation of micelles, and then copolymer–PTX prodrugs are released in a sustained manner in acidic organelles including endo-/lysosomes by pH-dependent forms;28 and 3) PTX, due to P-gp functions inhibited by DSF, the PTX could accumulate in cancer cells at a high concentration and effectively kill tumor cells. The drug co-delivery nanosystem adopted two strategies including enhanced cellular uptake and sequential drug release to combat cancer drug resistance and therefore had great potential in clinical applications.
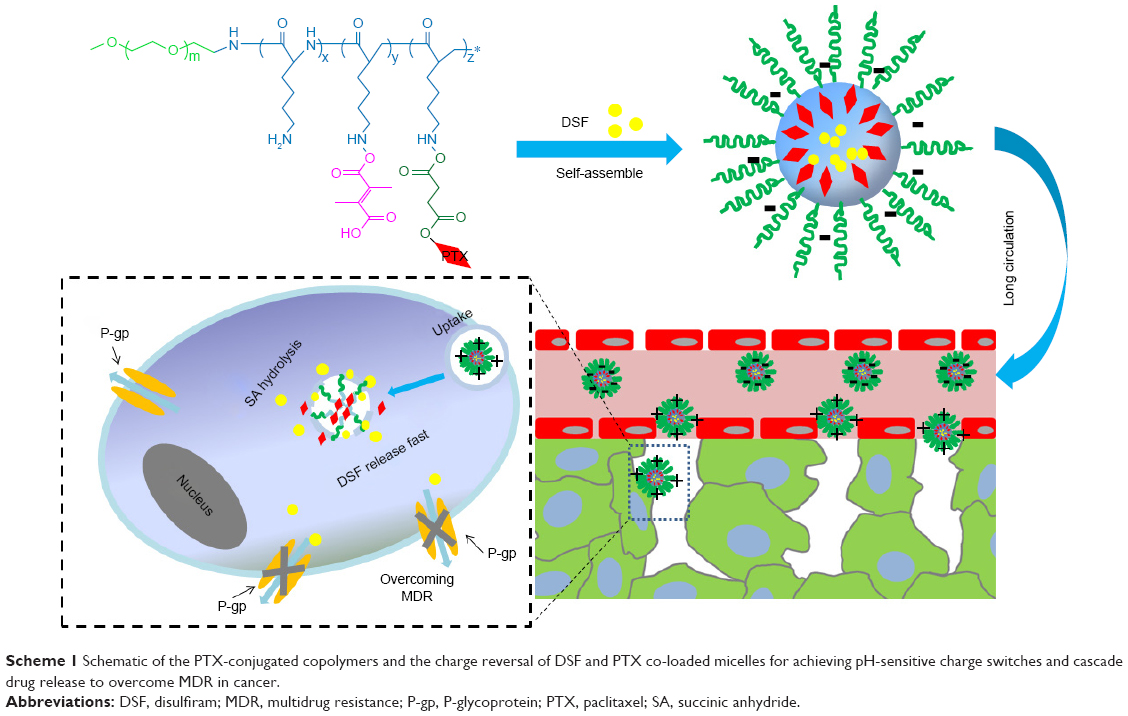 | Scheme 1 Schematic of the PTX-conjugated copolymers and the charge reversal of DSF and PTX co-loaded micelles for achieving pH-sensitive charge switches and cascade drug release to overcome MDR in cancer. |
Materials and methods
Materials
PTX was purchased from Beijing Huafeng United Technology Co., Ltd (Beijing, People’s Republic of China). DSF was obtained from Aladdin (Shanghai, People’s Republic of China). Succinic anhydride (SA) and DMA were purchased from Adamas Reagent Co., Ltd (Shanghai, People’s Republic of China) and used as received. N,N-dimethylformamide (DMF) was stored over calcium hydride (CaH2) and purified by vacuum distillation with CaH2. Amino-terminated methoxy poly(ethylene glycol) (PEG-NH2; molecular weight [MW] =5,000) was purchased from Shanghai Yare Biotech, Co., Ltd (Shanghai, People’s Republic of China) and used after dried under vacuum. ε-(Benzyloxycarbonyl)-L-lysine N-carboxyanhydride (Lys(Z)-NCA) were synthesized by the Fuchs-Farthing method using bis(trichloromethyl)carbonate (triphosgene) according to the literature.29 3-(4,5-Dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and Hoechst 33342 were purchased from Beyotime Institute of Biotechnology and used without further purification.
Characterization
1H nuclear magnetic resonance (NMR) spectra were performed on a Bruker Avance 500 spectrometer operating at 500 MHz by using D2O or dimethyl sulfoxide (DMSO)-d6 as the solvent depending on polymer solubility. The chemical shifts were calibrated against solvent signals. The morphology of nanoparticles (NPs) was observed using transmission electronic microscopy (TEM; Hitachi Ltd., Tokyo, Japan). The particle size and zeta potential were investigated using dynamic light scattering (DLS; Zs90; Malvern Instruments, Malvern, UK).
Synthesis of poly(ethylene glycol)-block-poly(L-lysine) (PEG-b-PLL) block copolymer
Methoxy PEG-b-PLL was synthesized by the ring-opening polymerization of Lys(Z)-NCA using PEG-NH2 as the macroinitiator and by the subsequent deprotection of benzyloxycarbonyl groups.30 Briefly, PEG-NH2 (1.50 g, 0.30 mM) was dissolved in 30 mL of dry DMF as the macroinitiator. Then, Lys(Z)-NCA (2.94 g, 9 mM) was dissolved in 40 mL of dry DMF and added to the PEG-NH2 solution under a dry argon atmosphere. The reaction mixture was stirred for 48 h at 40°C under a dry argon atmosphere, and then the solvent was evaporated under reduced pressure. The resulting product was precipitated into excess ice-cold diethyl ether to obtain methoxy poly(ethylene glycol)-block-poly-ε-(benzyloxycarbonyl)-L-lysine (PEG-b-PLLZ; yield 87.2%).
PEG-b-PLL was obtained through the deprotection of Z groups on PEG-b-PLLZ. Typically, PEG-b-PLLZ (2.0 g) was dissolved in CF3COOH (20 mL). Then, 3 mL of HBr/acetic acid (33%) was added at 0°C. The reaction mixture was stirred for 30 min at 25°C, and then the reaction mixture was precipitated in excessive cold diethyl ether. The precipitation was dissolved in DMSO and further purified by dialyzed (molecular weight cutoff [MWCO] =3,500 Da) against distilled water. The product was obtained by lyophilization (yield 63.5%).
Synthesis of PEG-b-(PLL-g-PTX)
PTX was conjugated to PEG-b-PLL through an SA linker, as reported previously,28,31 and had some differences. Briefly, PTX (1.0 g, 1.17 mM) was dissolved in 80 mL of dry DMF. SA (0.176 g, 1.76 mM), 4-dimethylaminopyridine (DMAP; 0.143 mg, 1.17 mM) and triethylamine (TEA; 0.178 g, 1.76 mM) were added, and the reaction mixture was stirred at 40°C for 24 h. The solvent was evaporated under reduced pressure, and the product was washed with 0.01 M HCl three times and distilled water three times. After filtration and drying under vacuum for 24 h, a white solid of SA-PTX was obtained (yield 85%).
PTX-SA was conjugated to the amino groups of PEG-b-PLL to obtain the PEG-b-(PLL-g-PTX). In brief, in a 100 mL flame-dry flask, SA-PTX (0.2 g, 0.21 mM), 3-(ethyliminomethylideneamino)-N,N-dimethylpropan-1-amine·HCl (0.113 g, 0.63 mM) and N-Hydroxysuccinimide (0.073 g, 0.63 mM) were dissolved in 20 mL of dry DMF, and the reaction mixture was maintained under stirring for 2 h at 35°C to activate carboxyl group. Subsequently, PEG-b-PLL (2.0 g, 0.21 mM) and TEA (0.045 g, 0.45 mM) were dissolved in dry DMF and then added to the abovementioned solution. The reaction mixture was maintained at 35°C for 36 h. The whole solution was transferred to a dialysis bag (MWCO =3,500 Da) and dialyzed against DMF, and then deionized water. Finally, the product was obtained by lyophilization. PTX content in the PEG-b-(PLL-g-PTX) was determined by 1H NMR and high-performance liquid chromatography (HPLC) measurements.32 For the determination of PTX by HPLC method, the mobile phase consisted of water and acetonitrile (35:65); an Ultimate AQ-C18 column was used with a flow rate of 1 mL/min; the detection wavelength was 227 nm and the column temperature was maintained at 30°C. The PTX content was calculated according to the following formula:
|
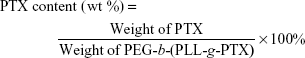
Synthesis of PEG-b-(PLL-g-PTX/DMA)
The charge-reversal polymers were prepared by grafting the amino residues of PLL blocks into β-carboxylicamide using DMA according to the previous literature.18 Briefly, PEG-b-(PLL-g-PTX) (0.20 g) and DMA (0.07 g, three equivalents of amino groups) were dissolved in 30 mL of deionized water, and the pH of the solution was kept at 8–9 by adding 1 M NaOH. After the reaction mixture was stirred for 12 h, the solution was dialyzed (MWCO =3,500 Da) against slightly basic water (pH 9–10) and a white solid was obtained by lyophilization. As a control, the no charge-reversal polymers, PEG-b-(PLL-g-PTX/SA), were synthesized through the reaction between PEG-b-(PLL-g-PTX) and SA by using the same method.
Preparation of the DSF and PTX co-loaded micelles
The DSF and PTX co-loaded micelles with charge reversal (abbreviated as DA-NPs) were fabricated by the nanoprecipitation method. Briefly, PEG-b-(PLL-g-PTX/DMA) (10 mg) and DSF (1 mg) were co-dissolved in 3 mL DMF stirred for 2 h, and then the mixture solution was added dropwise to 10 mL of pure water under vigorous stirring. The micelle solution was then dialyzed (MWCO =3,500 Da) against deionized water and filtered through a Millipore filter (pore size 0.45 μm) to remove unencapsulated DSF. The drug loading content (DLC) and drug loading efficiency (DLE) of DSF were measured by HPLC.
Moreover, the no charge-switchable micelles co-loaded with DSF and PTX were prepared as a control using the same protocol as DA-NPs, except that PEG-b-(PLL-g-PTX/DMA) polymer was replaced with PEG-b-(PLL-g-PTX/SA), and these micelles were abbreviated as SA-NPs.
The coumarin-6-labeled micelles were also prepared, and the method was similar to those of DA-NPs, except that DSF was replaced with coumarin-6.
Determination of the charge-reversal ability of DA-NPs
To study the surface charge characteristics of DA-NPs, the DA-NPs (0.1 mg·mL−1) were dispersed in PBS (pH 6.5 or 7.4, 0.01 M) and incubated at 37°C.33 Samples were taken at designated time intervals, and zeta potentials were measured.
Characterization of the stability of DA-NPs and SA-NPs
The DA-NPs and SA-NPs were gently mixed with RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) at pH 7.4 or 6.5. The mean diameters of DA-NPs and SA-NPs at different periods of incubation time were measured.24
Protein adsorption of the DA-NPs and SA-NPs
Bovine serum albumin (BSA) was used as a model protein to investigate the protein adsorption on the different micelles. Typically, DA-NPs and SA-NPs with the final concentration of 0.57 mg/mL were incubated with the solution of BSA (0.5 mg/mL) dissolved in PBS with different pH values (pH 6.5 or 7.4), respectively. After incubation at 37°C for 2 h, each sample was withdrawn and centrifuged at 10,000 rpm for 15 min to precipitate the protein adsorbed on DA-NPs or SA-NPs. In addition, the concentration of free BSA in supernatant was determined by the commercial BCA Protein Assay kits.18
In vitro pH-responsive release profiles of PTX and DSF
The release behaviors of PTX and DSF from DA-NPs were measured by ultrafiltration centrifugation method in PBS (0.2 M, pH 7.4) or HAc-NaAc buffer (0.04 M, pH 5.0) containing 0.5% (w/v) Tween-80 at 37°C.34 Briefly, DA-NPs were dispersed into 4 mL media, and every sample was shaken at 100 rpm and 37°C. At predetermined intervals, the solution was transferred to Centriprep centrifugal filter units (MWCO =3,000 Da) and then centrifuged at 4,000 rpm for 10 min. The ultrafiltrate was collected, and the amount of the released PTX and DSF was measured by HPLC.
Cell culture
MCF-7 was obtained from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, People’s Republic of China) and cultured in RPMI 1640 medium containing 10% FBS, 100 U/mL penicillin G sodium and 100 μg/mL streptomycin sulfate at 37°C and 5% CO2 in humidified incubator. MCF-7/ADR was obtained from Central South University (Changsha, People’s Republic of China) and maintained in media containing DOX at a concentration of 1 μg/mL as reported previously.12,35 All cell experiments were performed according to protocols evaluated and approved by the ethics committee of Nanjing Medical University.
PTX accumulation assay
MCF-7 and MCF-7/ADR cells were seeded in 24-well plates (5×104 cells/well) and then treated with PTX, PTX + DSF, DA-NPs and SA-NPs for 1, 2 and 4 h at 37°C. The concentrations of PTX and DSF were 5 and 1 μg/mL, respectively.17 After incubation, the cells were washed twice with cold PBS. Subsequently, 200 μL of cell lysis buffer (1% of TritonX-100) was added and incubated for 30 min. After that, the cell lysate (100 μL) was mixed with acetonitrile (200 μL) by ultrasonication for drug extraction followed by centrifugation at 6,000 rpm for 10 min, the supernatant was collected and the concentration of PTX was measured by HPLC as described earlier. All the determination of PTX content was normalized to protein concentrations of cell lysate.
Fluorescence microscopy (FLM) observation
MCF-7/ADR cells were seeded into six-well plates at a density of 1×105 cells/well and incubated for 24 h. Subsequently, cells were incubated with coumarin-6-loaded DA-NPs or SA-NPs at pH 7.4 or 6.5 for 2 h at 37°C. After that, the cells were washed with precooled PBS and fixed with 4% formaldehyde for 15 min, and then the cell nuclei were stained by 10 mM Hoechst 33342 for 10 min. Afterward, the cells were analyzed by using FLM (Leica Microsystems, Wetzlar, Germany).
Rhodamine 123 (Rh123) efflux assay
Rh123, as a P-gp substrate fluorescent dye, is an index of assaying the transport activity of P-gp.12,35 The transport activity of P-gp was first studied in MCF-7/ADR and MCF-7 cells via fluorescence spectrophotometer. Briefly, MCF-7/ADR or MCF-7 cells were seeded in 24-well plates at 5×104 cells/well. After overnight attachment, the cells were incubated with Rh123 at different concentrations (5, 10 or 20 μg/mL) at 37°C for 30 min. Subsequently, the free Rh123 was removed with PBS for washing three times, and then the cells were maintained in dye-free medium for another 2 h. At the end of the incubation period, the cells were lysed and centrifuged. The concentration of Rh123 in the supernatant was measured by fluorescence spectrophotometer (F-4600; Hitachi Ltd.). All the determination of Rh123 content was normalized to protein concentrations of cell lysate.
To study DSF inhibition of P-gp efflux function, MCF-7/ADR cells were seeded in 24-well plates and treated with verapamil, taxol, taxol + DSF, DA-NPs and SA-NPs overnight, followed by incubation with 10 μg/mL of Rh123 for another 30 min at 37°C. The other steps were the same as mentioned earlier.
In vitro cytotoxicity
The MTT assay was used to examine the vitro cytotoxicity of different drug-loaded NPs against MCF-7 and MCF-7/ADR cells. Briefly, the cells were seeded in 96-well plates at a seeding density of 1×104 cells/well. After 24 h of incubation, the medium was replaced with a 0.2 mL of fresh medium containing PTX, DSF, PTX + DSF, DA-NPs and SA-NPs at various drug concentrations (mass ratio of PTX to DSF was fixed at 5:1) for 48 h at pH 7.4. Afterward, 20 μL of MTT (5 mg/mL in PBS) was added to each well, and the plates further were incubated for 4 h in the dark. Then, the solution was removed, and 200 μL of DMSO was added to each well. Finally, the absorbance was detected at 490 nm by using a microplate reader (Thermo Multiskan MK3; Thermo Fisher Scientific, Waltham, MA, USA).
Statistical analysis
All experimental values were expressed as mean ± SD, n=3 or 5. All data were analyzed using the one-way ANOVA analysis. A p-value of <0.05 was considered to be statistically significant.
Results and discussion
Preparation and characterization of PEG-b-(PLL-g-PTX/DMA)
The detailed synthesis protocol of PEG-b-(PLL-g-PTX/DMA) is shown in Figure S1. The preparation of PEG-b-(PLL-g-PTX/DMA) mainly contained three steps: first, PEG-b-PLL copolymers and PTX-SA were synthesized; then, PTX-SA was conjugated to the side groups of PEG-b-PLL through esterification reaction; and finally, DMA was grafted on the residue amino groups of PEG-b-PLL through amidation reaction.
In this study, the PEG-b-PLL was synthesized by the ring-opening polymerization. The 1H NMR spectra of PEG-b-PLLZ and PEG-b-PLL demonstrated that PEG-b-PLL was synthesized successfully. All the peaks in the spectra were consistent with the literature (Figure 1).30,36 The degree of polymerization (DP) of PLLZ was calculated by comparing the signal intensities of PLLZ methylene protons (–CH2–CH2–CH2–CH2–NH) with methylene protons of PEG (–CH2–CH2–O–), and the value was 15. After deprotection, the typical peaks of carbobenzoxy groups at δ 5.00 and 7.17–7.35 ppm disappeared in PEG-b-PLL (Figure 1B), which indicated that the deprotection was complete. The DP of PLL in PEG-b-PLLZ was calculated to be 15 by using the same method as for PEG-b-PLLZ.
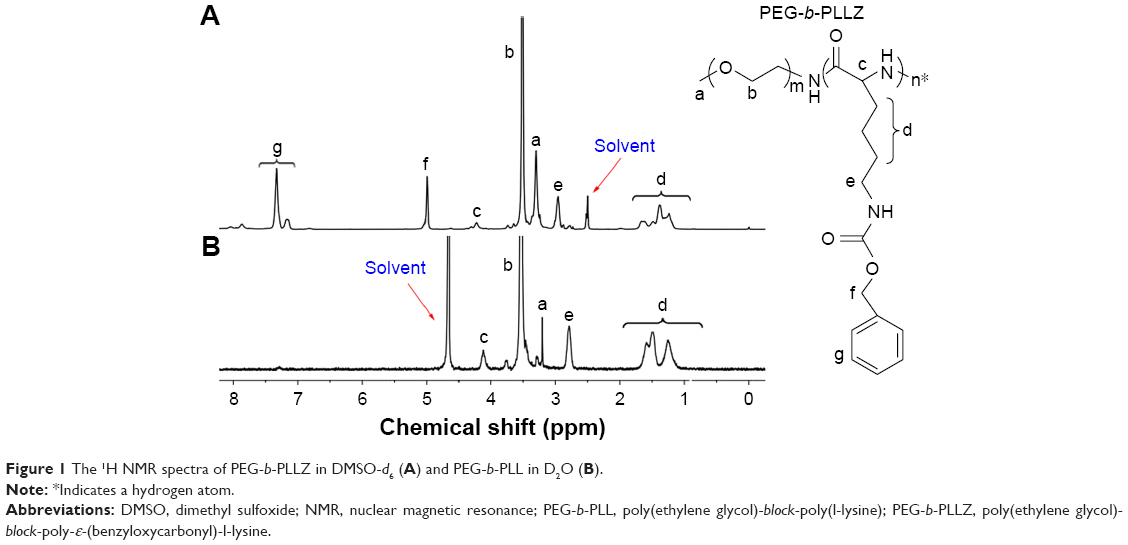 | Figure 1 The 1H NMR spectra of PEG-b-PLLZ in DMSO-d6 (A) and PEG-b-PLL in D2O (B). |
PTX, a cytotoxic drug, was then conjugated to PEG-b-PLL through SA as a linker. 1H NMR spectrum confirmed that PTX-SA was successfully prepared and the signals at δ 2.75 and 2.92 ppm were the methylene peaks of SA.37 Moreover, the 2′-CH proton peak on the 1H NMR spectra was shifted from 4.67 ppm (free PTX) to 5.90 ppm (conjugated PTX; Figure 2).38,39
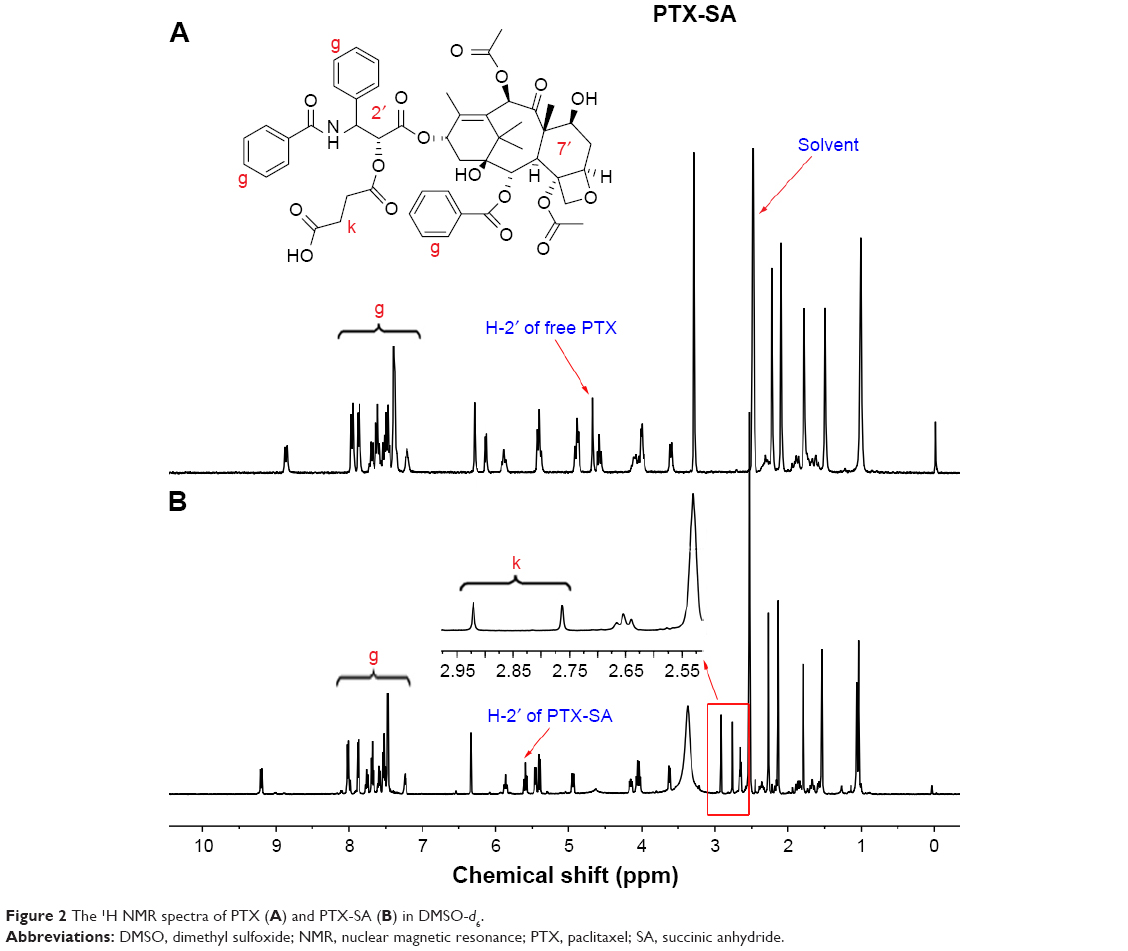 | Figure 2 The 1H NMR spectra of PTX (A) and PTX-SA (B) in DMSO-d6. |
PTX-SA was conjugated to the side chains of the amino groups in PEG-b-PLL through a typical amidation reaction to obtain an ester bond containing polymer PEG-b-(PLL-g-PTX). As shown in Figure 3, PTX-SA was successfully conjugated to PEG-b-PLL, which was certified by the typical signals of the phenyl proton (δ 7.28–8.0 ppm) of PTX that appeared in the 1H NMR spectra of PEG-b-(PLL-g-PTX). Subsequently, the mass fraction of PTX to PEG-b-(PLL-g-PTX) was 12.8% by calculating the peak area of the 1H NMR spectrum (Figure 3)32 and was 11.3% by HPLC detection through complete acid hydrolysis of SA. Finally, DMA was modified on the PEG-b-(PLL-g-PTX) to obtain PEG-b-(PLL-g-PTX/DMA), and the characteristic peaks of DMA were shown in the 1H NMR spectrum (Figure 4).
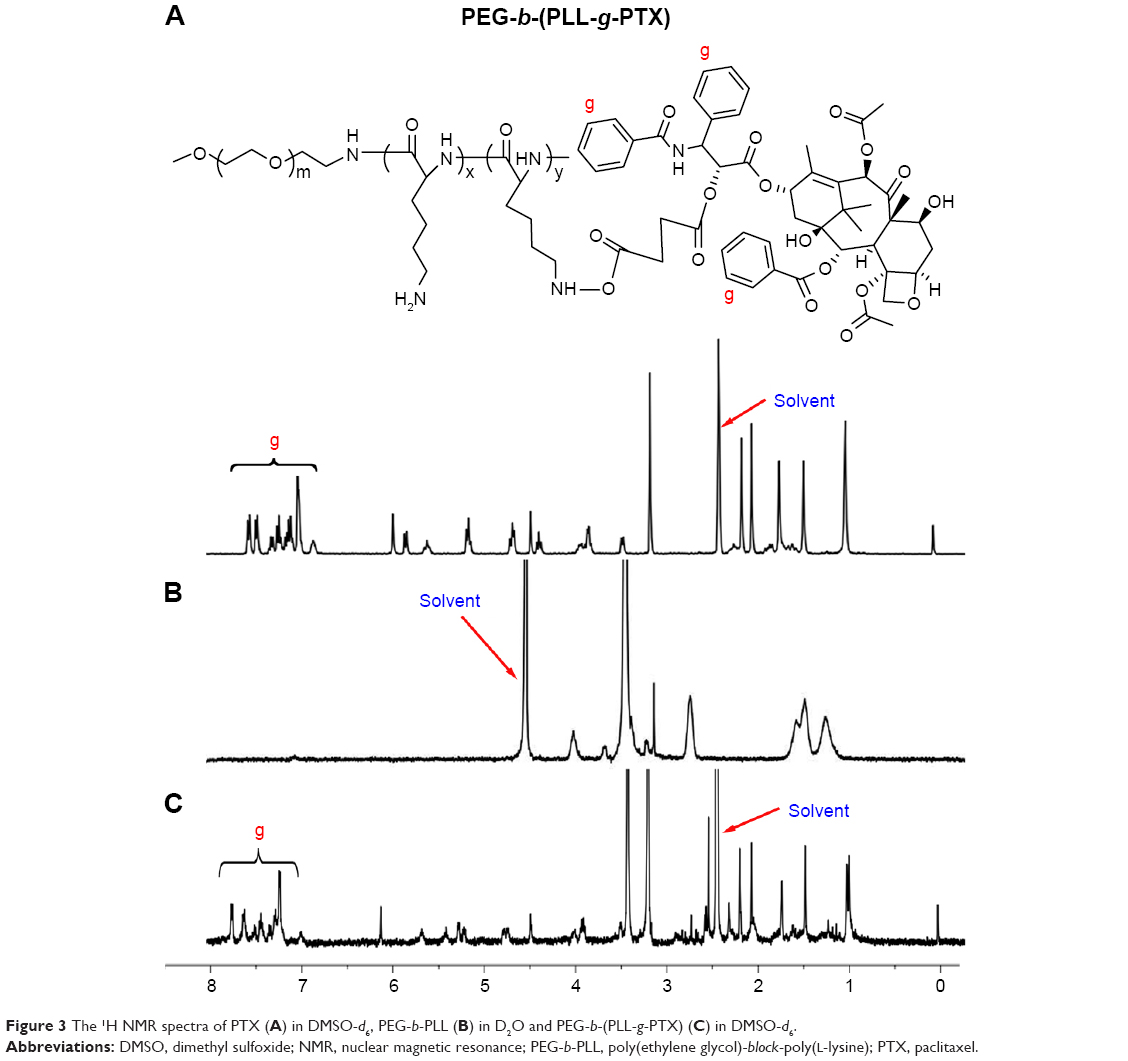 | Figure 3 The 1H NMR spectra of PTX (A) in DMSO-d6, PEG-b-PLL (B) in D2O and PEG-b-(PLL-g-PTX) (C) in DMSO-d6. |
 | Figure 4 The 1H NMR spectra of PEG-b-(PLL-g-PTX/DMA) in DMSO-d6. |
The characteristics of DA-NPs
The well-defined amphiphilic PTX-conjugated copolymers could form micelles (DA-NPs or SA-NPs) by a self-assembly process in aqueous solution. The morphology and size of DA-NPs and SA-NPs were investigated by TEM and DLS, respectively. As shown in Figure 5, the size of DA-NPs and SA-NPs was 129.8±8.5 and 118.6±2.1 nm (n=3) with a narrow distribution, respectively. The TEM images showed that both micelles had good monodispersity and were nearly in spherical shape with diameter about 100 nm, which were consistent with the results determined by DLS.
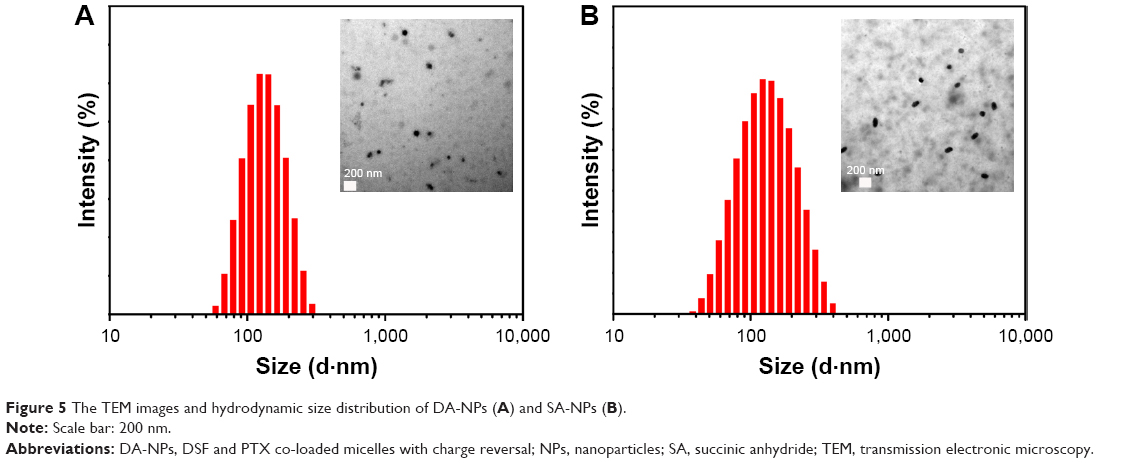 | Figure 5 The TEM images and hydrodynamic size distribution of DA-NPs (A) and SA-NPs (B). |
Surface charge conversion behavior of DA-NPs
Recently, nanocarriers with the characteristics of pH-dependent charge conversion which could enhance tumor cell adhesion and trigger drug release were developed for drug delivery. DA-NPs or SA-NPs were dispersed in the PBS solution with different pH values (6.5 and 7.4), respectively, and the zeta potential was monitored. As shown in Figure 6A, the zeta potential of DA-NPs changed rapidly with time at pH 6.5, and the corresponding values were increased from −13.3 to 6.5 mV after 15 min and further increased to 10.5 mV after 1 h, which could be attributed to the gradual hydrolysis of DMA groups under acidic conditions. However, the zeta potential of DA-NPs was increased slowly at pH 7.4 and still maintained a negative charge (−1.5±0.5 mV) within 1 h, which suggested that the DMA groups of DA-NPs remained relatively stable at physiological pH. These phenomena were also in good consistent with previous report.19,40,41 In sharp contrast, the zeta potential of SA-NPs, which were set as a negative control, did not exhibit significant change, whether at pH 6.5 (12.3±1.7 mV) or at pH 7.4 (12.4±1.3 mV), in 2 h, which indicated that SA groups possessed higher stability compared to the DMA group (Figure 6B). Overall, these results indicated that DA-NPs were capable of changing surface charge on receiving a pH signal, which would promote its adsorption on tumor cells and have a higher probability of entering the cells.
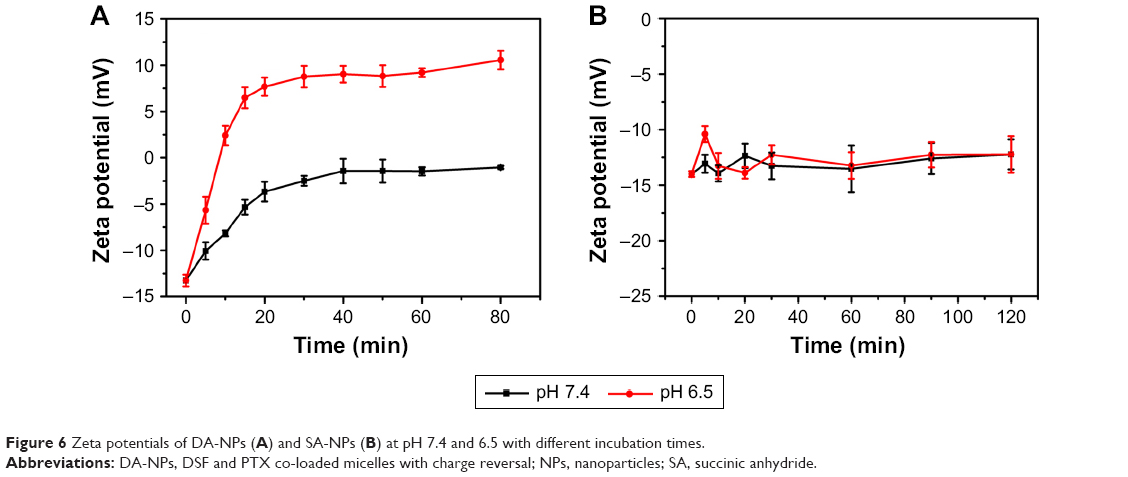 | Figure 6 Zeta potentials of DA-NPs (A) and SA-NPs (B) at pH 7.4 and 6.5 with different incubation times. |
In vitro stability study and protein adsorption
To study the stability of DA-NPs and SA-NPs, cell cultured with 10% FBS was used to simulate blood microenvironment (pH 7.4) and tumor tissues at slightly acidic conditions (pH 6.5). As shown in Figure 7A, after 24 h of incubation at pH 7.4, the particle size of DA-NPs was maintained at 138±8.5 nm, indicating that they are relatively stable at blood circulation. However, the size of DA-NPs drastically changed from 138 to 1,000 nm after 24 h of incubation at pH 6.5, whcih maybe because the negative surface charge changes to positive charge resulting in abundant protein were adsorbed on the surface of NPs. As a control, there was almost no change in the size of SA-NPs, whether at pH 7.4 or at pH 6.5. These phenomena were also in good consistent with previous reports.24
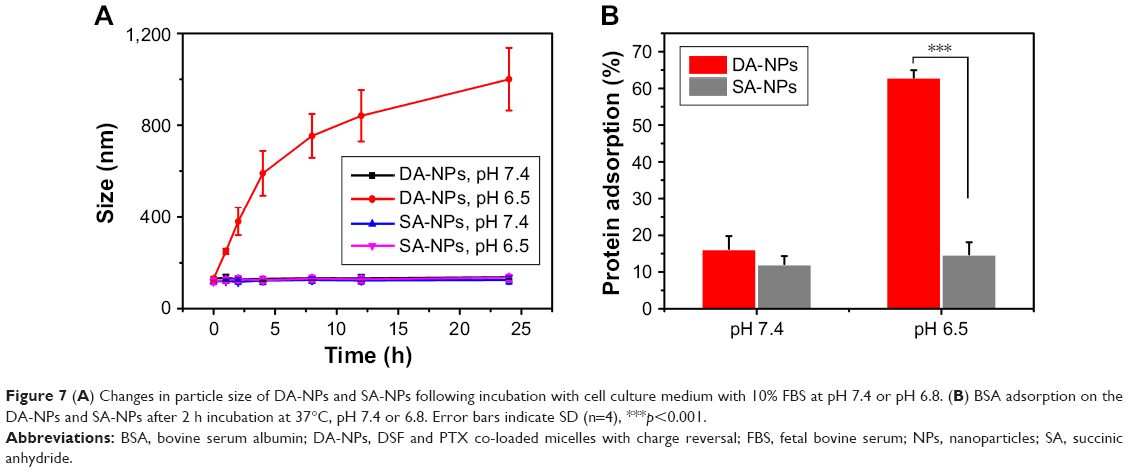 | Figure 7 (A) Changes in particle size of DA-NPs and SA-NPs following incubation with cell culture medium with 10% FBS at pH 7.4 or pH 6.8. (B) BSA adsorption on the DA-NPs and SA-NPs after 2 h incubation at 37°C, pH 7.4 or 6.8. Error bars indicate SD (n=4), ***p<0.001. |
Reduced nonspecific protein adsorption of the drug-loaded NPs is an important indicator for the prolonged blood circulation.19,42 We hypothesized that the surface charge reversal of DA-NPs may be advantageous to the reduction in nonspecific protein adsorption to NPs during circulation. To test this hypothesis, BSA was set as a model of plasma protein to simulate the interaction between micelles and protein. As shown in Figure 7B, when the pH values decreased to 6.5, DA-NPs with positive charge strongly interacted with the protein and adsorbed more than 60% protein within 2 h. In contrast, SA-NPs showed negligible protein adsorption under similar conditions. However, under the physiological pH conditions (pH 7.4), both DA-NPs and SA-NPs exhibited minimal protein adsorption (less than 20%) after 2 h incubation. These results were in good agreement with the pH-dependent charge-switchable and size-changeable behaviors of DA-NPs at pH 6.5.
Drug release
As previously described, DA-NPs were expected to have the ability of temporal release of DSF and PTX, which was aimed that DSF could inhibit the function of P-gp first and then the release of PTX was activated by a pH change in the intracellular endosomal compartments in a sustained manner. To confirm this property, the release kinetic behavior of each drug from DA-NPs was measured. PBS (pH 7.4) and HAc-NaAc buffer (pH 5.0) were used to simulate the environment of plasma and endosome, respectively. The DLC of PTX and DSF in DA-NPs was 10.73%±0.8% and 1.97%±0.5% and in SA-NPs were 11.42%±0.7% and 2.15%±0.5% (n=3), respectively, as measured by HPLC method (Table 1). As shown in Figures 8A and S2, 72.5% and 44.3% of DSF were released from DA-NPs within 12 h after incubation at pH 5.0 and 7.4, respectively, which was convenient to inhibit P-gp quickly. In contrast, trace amounts of PTX were released from DA-NPs at pH 7.4 within 12 h (Figure 8B), which maybe that the ester bond between PTX and SA was still stable under such conditions. However, the release amount of PTX from DA-NPs was modestly increased at pH 5.0 in a sustained manner up to 72 h and released 53.6%. These results suggested that DA-NPs could minimize the amount of premature drug release during the blood circulation, and release cargoes in the acidic intracellular compartments.
 | Table 1 The drug loaded in DA-NPs and SA-NPs (n=3) |
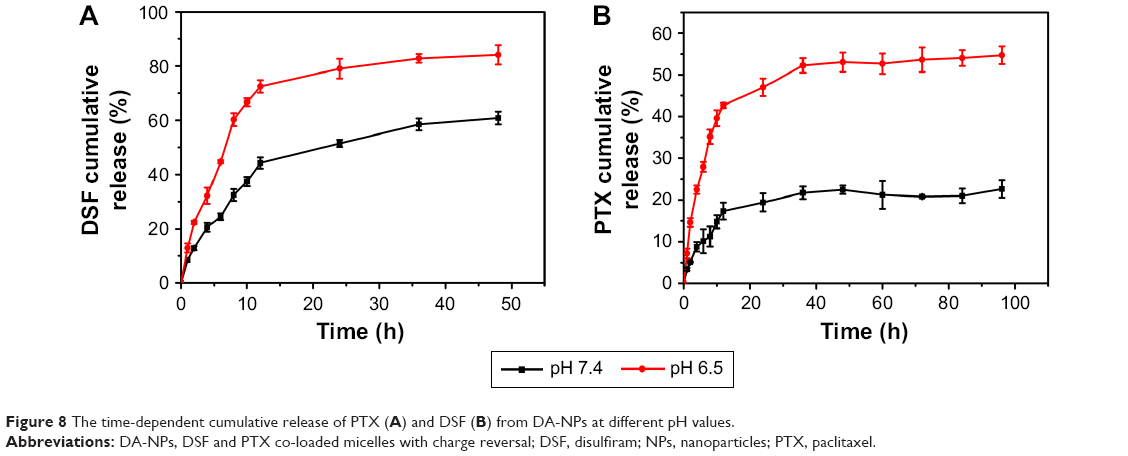 | Figure 8 The time-dependent cumulative release of PTX (A) and DSF (B) from DA-NPs at different pH values. |
Cellular uptake of DA-NPs
To test whether the charge conversion feature of DA-NPs could enhance their cellular internalization, the cellular uptake of DA-NPs and SA-NPs in MCF-7/ADR cells was observed quantitatively by fluorescence microscope and using coumarin-6 as fluorescent marker. As shown in Figure 9, after 2 h of incubation at pH 7.4, little green fluorescence of DA-NPs was observed in the cytoplasm of MCF-7/ADR cells, but abundant green fluorescence could be measured when incubated at pH 6.5. However, whether at pH 7.4 or 6.5, little green fluorescence of SA-NPs was shown in the cytoplasm of MCF-7/ADR cells. These results confirmed that the surface charge-reversal capability of DA-NPs could effectively enhance their internalization into tumor cells.
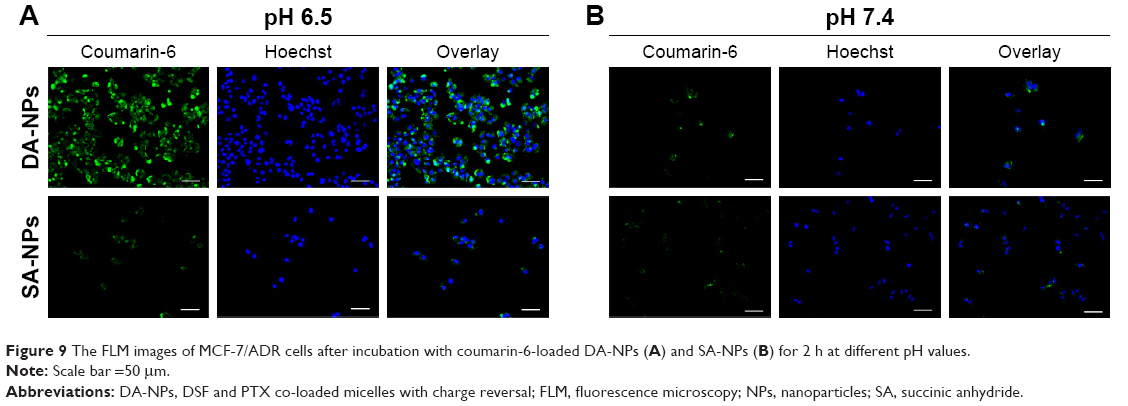 | Figure 9 The FLM images of MCF-7/ADR cells after incubation with coumarin-6-loaded DA-NPs (A) and SA-NPs (B) for 2 h at different pH values. |
The intracellular accumulation of PTX was also evaluated in both PTX-sensitive MCF-7 cells and PTX-resistant MCF-7/ADR cells at pH 7.4 and 6.5 by HPLC method. From the results, the cellular uptake of DA-NPs in MCF-7/ADR cells at pH 6.5 was 1.2-, 1.5- and 1.5-fold higher than that of SA-NPs after incubation for 1, 2 and 4 h, respectively (Figure 10A). In addition, the cellular uptake of DA-NPs in MCF-7 cells at pH 6.5 was 1.6-, 1.8- and 1.5-fold higher than that of SA-NPs after incubation for 1, 2 and 4 h, respectively (Figure 10B). However, under the physiological pH conditions (pH 7.4), the intracellular accumulation of PTX of DA-NPs and SA-NPs in both MCF-7/ADR cells and MCF-7 cells exhibited slight accumulation and no obvious difference. All these results further demonstrated that the acid-triggered surface charge reversal of the micelles could effectively increase cellular uptake.
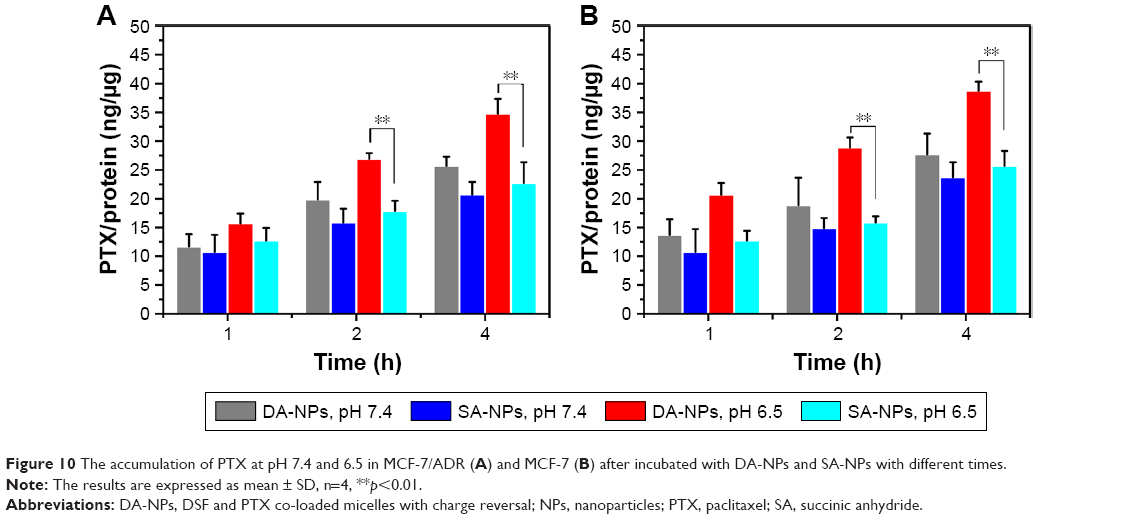 | Figure 10 The accumulation of PTX at pH 7.4 and 6.5 in MCF-7/ADR (A) and MCF-7 (B) after incubated with DA-NPs and SA-NPs with different times. |
Inhibition of P-gp analysis
DSF is a member of the dithiocarbamate family which was traditionally used in the treatment of alcoholism by irreversibly inhibiting the acetaldehyde dehydrogenase.43,44 Recently, it is reported that DSF could permanently inhibit P-gp activity by covalently modifying the cysteine residues in the nucleotide-binding domains of P-gp and/or block its maturation.45,46 The influence of DSF on the intracellular accumulation of PTX and the P-gp-mediated drug efflux was investigated in both MCF-7 cells and MCF-7/ADR cells.
As shown in Figure 11A, free PTX the accumulation of PTX in MCF-7/ADR were very low (11.07±1.71 ng/μg protein), but the amount significantly increased to 18.83±1.72 ng/μg protein when co-incubation with DSF after 4 h. Furthermore, DA-NPs and SA-NPs were found to increase intracellular PTX concentrations compared with free drug and this may be caused by internalized of these micelles into the cells via endocytic pathways. Moreover, the free PTX accumulation in MCF-7/ADR cells was lower than that in MCF-7 cells, which may be one of the reasons that MCF-7/ADR was resistant to PTX. However, there was no significant difference of the concentration of PTX among each group in MCF-7 cells (Figure 11B). These results suggested that DSF may increase the intracellular accumulation of PTX.
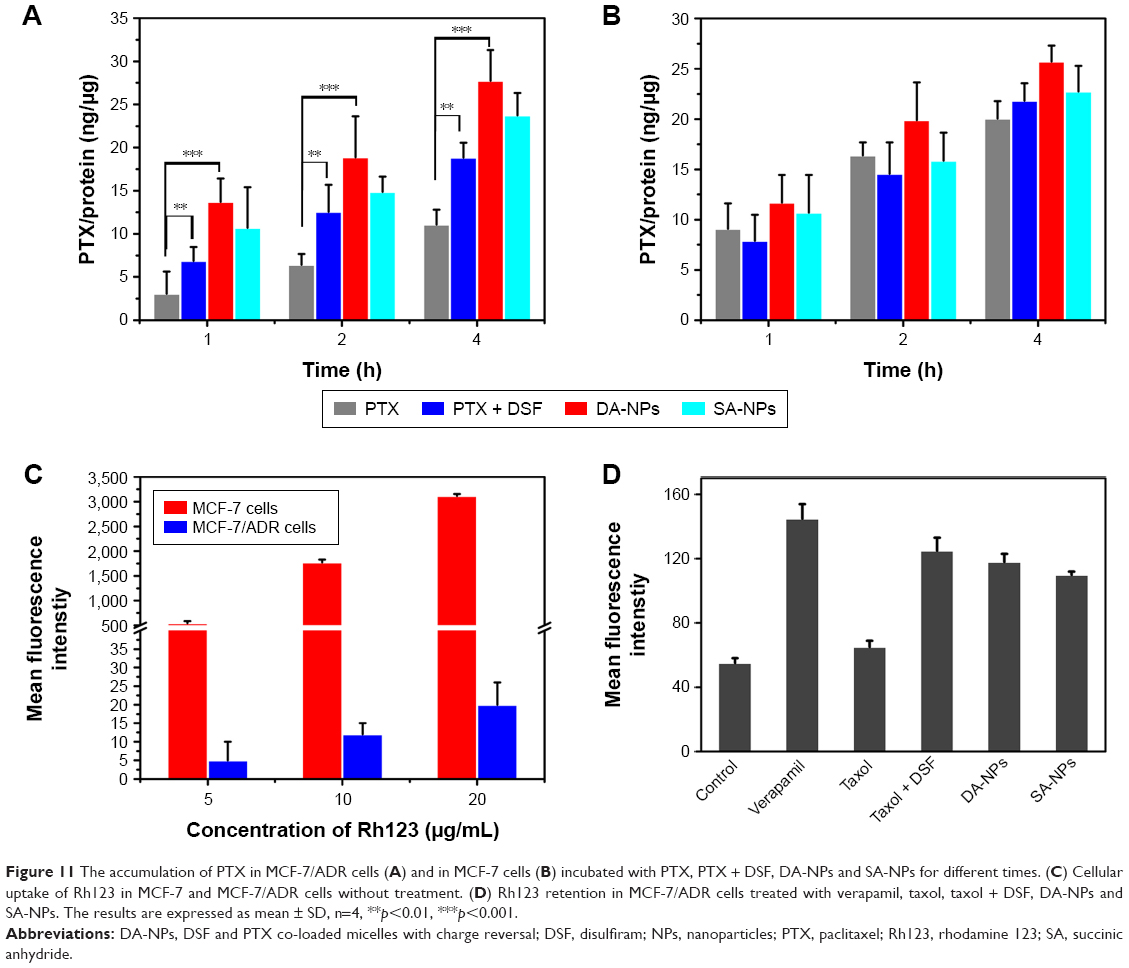 | Figure 11 The accumulation of PTX in MCF-7/ADR cells (A) and in MCF-7 cells (B) incubated with PTX, PTX + DSF, DA-NPs and SA-NPs for different times. (C) Cellular uptake of Rh123 in MCF-7 and MCF-7/ADR cells without treatment. (D) Rh123 retention in MCF-7/ADR cells treated with verapamil, taxol, taxol + DSF, DA-NPs and SA-NPs. The results are expressed as mean ± SD, n=4, **p<0.01, ***p<0.001. |
Overexpressed P-gp on the membrane of MCF-7/ADR cells was one of the major causes of the MDR phenotype.12,47 To overcome this drug efflux mechanism, it is necessary to either inhibit its expression, inhibit its function, or bypass transporter recognition.48 The potential effect of DSF on the P-gp activity in MCF-7/ADR cells was investigated using Rh123 as a P-gp substrate. As shown in Figure 11C, when the concentration of Rh123 was 5, 10 and 20 μg/mL, the mean fluorescence intensity of the MCF-7 cells was 108.5-, 147.8- and 155.9-folds higher than that of MCF-7/ADR cells, respectively, which indicated that MCF-7/ADR cells retained strong MDR properties, while the MCF-7 cells were relatively sensitive.35 Furthermore, the drug efflux inhibition was evaluated on different NPs using MCF-7/ADR cells. As shown in Figure 11D, the mean fluorescence intensity of Rh123 in the cells treated with taxol did not alter significantly. However, the intracellular accumulation of Rh123 in MCF-7/ADR cells treated with taxol + DSF, DA-NPs, SA-NPs or verapamil (positive control group) was significantly higher than that in untreated cells. This phenomenon suggested the P-gp transport function was greatly inhibited by DSF.
In vitro cytotoxicity
The vitro cytotoxicity of various PTX-loaded NPs was further investigated by MTT method.6 It reported that MCF-7/ADR cells, a typical resistant cancer cell line of DOX, exhibit a cross drug resistant to PTX.49,50 This cell line has been used to study the reversal of MDR for PTX in this study. Figure 12A revealed that the cell viability of taxol in MCF-7/ADR cells were above that in MCF-7 cells at each dose, suggesting a drug-resistant property against PTX.12 Compared with taxol, the combination of PTX and DSF could effectively improve the killing effect of drug-resistant cells at each dose, which may be due to the P-gp inhibitor effect of DSF. This phenomenon was also observed in DA-NPs and SA-NPs. However, in MCF-7 cells, the cell viability of combination of PTX and DSF was obviously lower than that of other drug forms, which could be due to the effect of DSF on cell apoptosis (Figure 12B). Moreover, the results showed that the cytotoxic effect of the DA-NPs was pH dependent on both normal and drug-resistant cells (Figure 12C and D). At pH 6.5, the cytotoxicity of DA-NPs was significantly higher than that of SA-NPs, this may be due to the acid-triggered surface charge reversal.24,29 In addition, all the results indicated that DA-NPs with charge reversal could co-deliver PTX and DSF, and showed strong cytotoxicity to drug-resistant MCF-7/ADR cells.
 | Figure 12 The cell viability of MCF-7/ADR cells (A) and MCF-7 cells (B) incubated with PTX, DSF, PTX + DSF, DA-NPs and SA-NPs for 48 h; and the cell viability of MCF-7/ADR (C) and MCF-7 cells (D) treated with DA-NPs and SA-NPs at pH 6.5 or 7.4 for 4 h and further incubated of 48 h. |
Conclusion
In this study, we designed and constructed a surface charge-switchable polymer-based multidrug delivery system, which provided promising approaches to reverse MDR and achieved more effective treatment. The nanostructure exhibited a variety of attractive functions, such as tumorous acid-triggered surface charge reversal and sequential release of two drugs in tumor cells, which could enhance intracellular drug delivery and produce good synergistic effects of P-gp inhibitors and cytotoxic agents, respectively. As a result, this system showed stronger antitumor effect to multidrug-resistant cells and has a great potential to achieve better therapeutic effects in cancer treatment.
Acknowledgments
This study was financially supported by the Nature Science Foundation of Jiangsu Province, People’s Republic of China (grant no BK20160704), the National Natural Science Foundation of China (grant no 81341134), the Cooperation Foundation of Southeast University and Nanjing Medical University, People’s Republic of China (grant no 2242017K3DN40), the Key Project of Natural Science Research for Colleges and Universities of the Anhui Province of China (grant no KJ2015A274), the Scientific Research Projects of the Bengbu Medical College of Anhui Province (Byycx1638) and Jiangsu Pharmaceutical Association-Shire Biopharmaceutical Foundation (S201601).
Disclosure
The authors report no conflicts of interest in this work.
References
Yin Q, Shen J, Zhang Z, Yu H, Li Y. Reversal of multidrug resistance by stimuli-responsive drug delivery systems for therapy of tumor. Adv Drug Deliv Rev. 2013;65(13–14):1699–1715. | ||
Gupta B, Ramasamy T, Poudel BK, et al. Development of bioactive PEGylated nanostructured platforms for sequential delivery of doxorubicin and imatinib to overcome drug resistance in metastatic tumors. ACS Appl Mater Interfaces. 2017;9(11):9280–9290. | ||
Li Y, Xu X, Zhang X, Li Y, Zhang Z, Gu Z. Tumor-specific multiple stimuli-activated dendrimeric nanoassemblies with metabolic blockade surmount chemotherapy resistance. ACS Nano. 2017;11(1):416–429. | ||
Wang X, Liow SS, Wu Q, et al. Codelivery for paclitaxel and Bcl-2 conversion gene by PHB-PDMAEMA amphiphilic cationic copolymer for effective drug resistant cancer therapy. Macromol Biosci. In 2017. | ||
Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer: mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 2000;11(4):265–283. | ||
Qiu L, Qiao M, Chen Q, et al. Enhanced effect of pH-sensitive mixed copolymer micelles for overcoming multidrug resistance of doxorubicin. Biomaterials. 2014;35(37):9877–9887. | ||
Chen Z, Shi T, Zhang L, et al. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: a review of the past decade. Cancer Lett. 2016;370(1):153–164. | ||
Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2(1):48–58. | ||
Muddineti OS, Kumari P, Ghosh B, Torchilin VP, Biswas S. d-alpha-tocopheryl succinate/phosphatidyl ethanolamine conjugated amphiphilic polymer-based nanomicellar system for the efficient delivery of curcumin and to overcome multiple drug resistance in cancer. ACS Appl Mater Interfaces. 2017;9(20):16778–16792. | ||
Zhang H, Fan X, Li F, et al. Thermo and pH dual-controlled charge reversal amphiphilic graft copolymer micelles for overcoming drug resistance in cancer cells. J Mater Chem B. 2015;3(22):4585–4596. | ||
Chen W, Wang F, Zhang X, et al. Overcoming ABCG2-mediated multidrug resistance by a mineralized hyaluronan–drug nanocomplex. J Mater Chem B. 2016;4(41):6652–6661. | ||
Chen F, Wu J, Zheng C, et al. TPGS modified reduced bovine serum albumin nanoparticles as a lipophilic anticancer drug carrier for overcoming multidrug resistance. J Mater Chem B. 2016;4(22):3959–3968. | ||
Li N, Huang C, Luan Y, Song A, Song Y, Garg S. Active targeting co-delivery system based on pH-sensitive methoxy-poly (ethylene glycol) 2K-poly (ε-caprolactone) 4K-poly (glutamic acid) 1K for enhanced cancer therapy. J Colloid Interface Sci. 2016;472:90–98. | ||
Wang AT, Liang DS, Liu YJ, Qi XR. Roles of ligand and TPGS of micelles in regulating internalization, penetration and accumulation against sensitive or resistant tumor and therapy for multidrug resistant tumors. Biomaterials. 2015;53:160–172. | ||
Yao Q, Gutierrez DC, Hoang NH, et al. Efficient codelivery of paclitaxel and curcumin by novel bottlebrush copolymer-based micelles. Mol Pharm. 2017;14(7):2378–2389. | ||
Wang N, He T, Shen Y, et al. Paclitaxel and tacrolimus coencapsulated polymeric micelles that enhance the therapeutic effect of drug-resistant ovarian cancer. ACS Appl Mater Interfaces. 2016;8(7):4368–4377. | ||
Duan X, Xiao J, Yin Q, et al. Smart pH-sensitive and temporal-controlled polymeric micelles for effective combination therapy of doxorubicin and disulfiram. ACS Nano. 2013;7(7):5858–5869. | ||
Wu L, Wu M, Lin X, Zhang X, Liu X, Liu J. Magnetite nanocluster and paclitaxel-loaded charge-switchable nanohybrids for MR imaging and chemotherapy. J Mater Chem B. 2017;5:849–857. | ||
Han SS, Li ZY, Zhu JY, et al. Dual-pH sensitive charge-reversal polypeptide micelles for tumor-triggered targeting uptake and nuclear drug delivery. Small. 2015;11(21):2543–2554. | ||
Zhao X, Liu P, Song Q, Gong N, Yang L, Wu WD. Surface charge-reversible polyelectrolyte complex nanoparticles for hepatoma-targeting delivery of doxorubicin. J Mater Chem B. 2015;3(30):6185–6193. | ||
Li L, Sun W, Zhong J, et al. Multistage nanovehicle delivery system based on stepwise size reduction and charge reversal for programmed nuclear targeting of systemically administered anticancer drugs. Adv Funct Mater. 2015;25(26):4101–4113. | ||
Qiu N, Liu X, Zhong Y, et al. Esterase-activated charge-reversal polymer for fibroblast-exempt cancer gene therapy. Adv Mater Deerfield. 2016;28(48):10613–10622. | ||
Li Q, Yao W, Yu X, Zhang B, Dong J, Jin Y. Drug-loaded pH-responsive polymeric micelles: simulations and experiments of micelle formation, drug loading and drug release. Colloids Surf B Biointerfaces. 2017;158:709–716. | ||
Yuan YY, Mao CQ, Du XJ, Du JZ, Wang F, Wang J. Surface charge switchable nanoparticles based on zwitterionic polymer for enhanced drug delivery to tumor. Adv Mater Deerfield. 2012;24(40):5476–5480. | ||
Zhu J, Niu Y, Li Y, et al. Stimuli-responsive delivery vehicles based on mesoporous silica nanoparticles: recent advances and challenges. J Mater Chem B. 2017;5:1339–1352. | ||
Wei X, Wang Y, Xiong X, et al. Codelivery of a π-π stacked dual anticancer drug combination with nanocarriers for overcoming multidrug resistance and tumor metastasis. Adv Funct Mater. 2016;26(45):8266–8280. | ||
Du JZ, Mao CQ, Yuan YY, Yang XZ, Wang J. Tumor extracellular acidity-activated nanoparticles as drug delivery systems for enhanced cancer therapy. Biotechnol Adv. 2014;32(4):789–803. | ||
Noh I, Kim HO, Choi J, et al. Co-delivery of paclitaxel and gemcitabine via CD44-targeting nanocarriers as a prodrug with synergistic antitumor activity against human biliary cancer. Biomaterials. 2015;53:763–774. | ||
Wang Y, Lv S, Deng M, Tang Z, Chen X. A charge-conversional intracellular-activated polymeric prodrug for tumor therapy. Polym Chem. 2016;7(12):2253–2263. | ||
Wu L, Zou Y, Deng C, Cheng R, Meng F, Zhong Z. Intracellular release of doxorubicin from core-crosslinked polypeptide micelles triggered by both pH and reduction conditions. Biomaterials. 2013;34(21):5262–5272. | ||
Ma P, Zhang X, Ni L, et al. Targeted delivery of polyamidoamine-paclitaxel conjugate functionalized with anti-human epidermal growth factor receptor 2 trastuzumab. Int J Nanomedicine. 2015;10:2173–2190. | ||
Zhong Y, Goltsche K, Cheng L, et al. Hyaluronic acid-shelled acid-activatable paclitaxel prodrug micelles effectively target and treat CD44-overexpressing human breast tumor xenografts in vivo. Biomaterials. 2016;84:250–261. | ||
Deng H, Zhao X, Liu J, et al. Reactive oxygen species (ROS) responsive PEG–PCL nanoparticles with pH-controlled negative-to-positive charge reversal for intracellular delivery of doxorubicin. J Mater Chem B. 2015;3(48):9397–9408. | ||
Jiang Y, Wang X, Liu X, et al. Enhanced antiglioma efficacy of ultrahigh loading capacity paclitaxel prodrug conjugate self-assembled targeted nanoparticles. ACS Appl Mater Interfaces. 2017;9(1):211–217. | ||
Yin M, Bao Y, Gao X, et al. Redox/pH dual-sensitive hybrid micelles for targeting delivery and overcoming multidrug resistance of cancer. J Mater Chem B. 2017;5(16):2964–2978. | ||
Li J, Cheng D, Yin T, et al. Copolymer of poly(ethylene glycol) and poly(L-lysine) grafting polyethylenimine through a reducible disulfide linkage for siRNA delivery. Nanoscale. 2014;6(3):1732–1740. | ||
Sousa-Herves A, Wurfel P, Wegner N, et al. Dendritic polyglycerol sulfate as a novel platform for paclitaxel delivery: pitfalls of ester linkage. Nanoscale. 2015;7(9):3923–3932. | ||
Caron J, Maksimenko A, Wack S, et al. Improving the antitumor activity of squalenoyl-paclitaxel conjugate nanoassemblies by manipulating the linker between paclitaxel and squalene. Adv Healthc Mater. 2013;2(1):172–185. | ||
Yu Y, Chen C-K, Law W-C, et al. Well-defined degradable brush polymer–drug conjugates for sustained delivery of paclitaxel. Mol Pharm. 2012;10(3):867–874. | ||
Du J-Z, Du X-J, Mao C-Q, Wang J. Tailor-made dual pH-sensitive polymer–doxorubicin nanoparticles for efficient anticancer drug delivery. J Am Chem Soc. 2011;133(44):17560–17563. | ||
Zhou Z, Shen Y, Tang J, et al. Charge-reversal drug conjugate for targeted cancer cell nuclear drug delivery. Adv Funct Mater. 2009;19(22):3580–3589. | ||
Chen S, Rong L, Lei Q, et al. A surface charge-switchable and folate modified system for co-delivery of proapoptosis peptide and p53 plasmid in cancer therapy. Biomaterials. 2016;77:149–163. | ||
Song W, Tang Z, Lei T, et al. Stable loading and delivery of disulfiram with mPEG-PLGA/PCL mixed nanoparticles for tumor therapy. Nanomedicine. 2016;12(2):377–386. | ||
Kast RE, Boockvar JA, Brüning A, et al. A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the international initiative for accelerated improvement of glioblastoma care. Oncotarget. 2013;4(4):502–530. | ||
Loo TW, Bartlett MC, Clarke DM. Disulfiram metabolites permanently inactivate the human multidrug resistance P-glycoprotein. Mol Pharm. 2004;1(6):426–433. | ||
Loo TW, Clarke DM. Blockage of drug resistance in vitro by disulfiram, a drug used to treat alcoholism. J Natl Cancer Inst. 2000;92(11):898–902. | ||
Murray S, Briasoulis E, Linardou H, Bafaloukos D, Papadimitriou C. Taxane resistance in breast cancer: mechanisms, predictive biomarkers and circumvention strategies. Cancer Treat Rev. 2012;38(7):890–903. | ||
Patel NR, Pattni BS, Abouzeid AH, Torchilin VP. Nanopreparations to overcome multidrug resistance in cancer. Adv Drug Deliv Rev. 2013;65(13):1748–1762. | ||
Baek JS, Cho CW. Controlled release and reversal of multidrug resistance by co-encapsulation of paclitaxel and verapamil in solid lipid nanoparticles. Int J Pharm. 2015;478(2):617–624. | ||
Jia L, Li Z, Shen J, et al. Multifunctional mesoporous silica nanoparticles mediated co-delivery of paclitaxel and tetrandrine for overcoming multidrug resistance. Int J Pharm. 2015;489(1):318–330. |
Supplementary materials
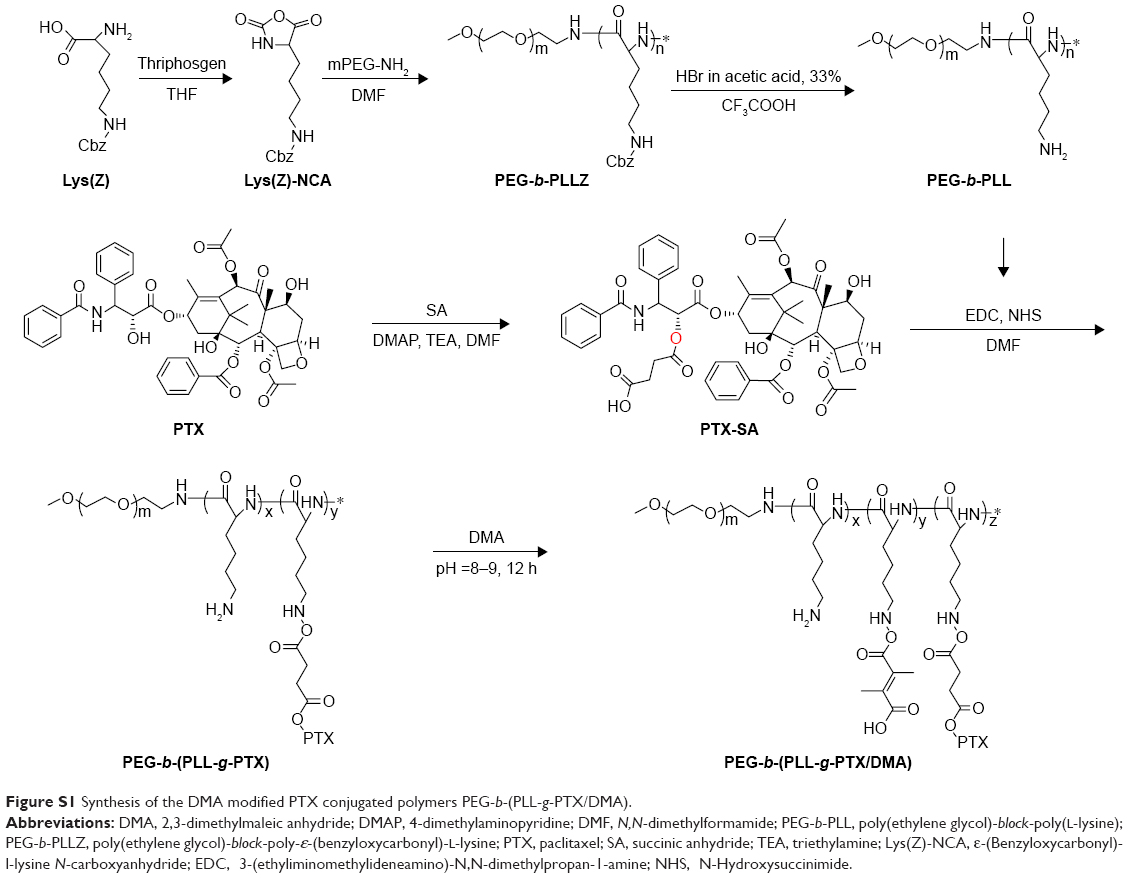 | Figure S1 Synthesis of the DMA modified PTX conjugated polymers PEG-b-(PLL-g-PTX/DMA). |
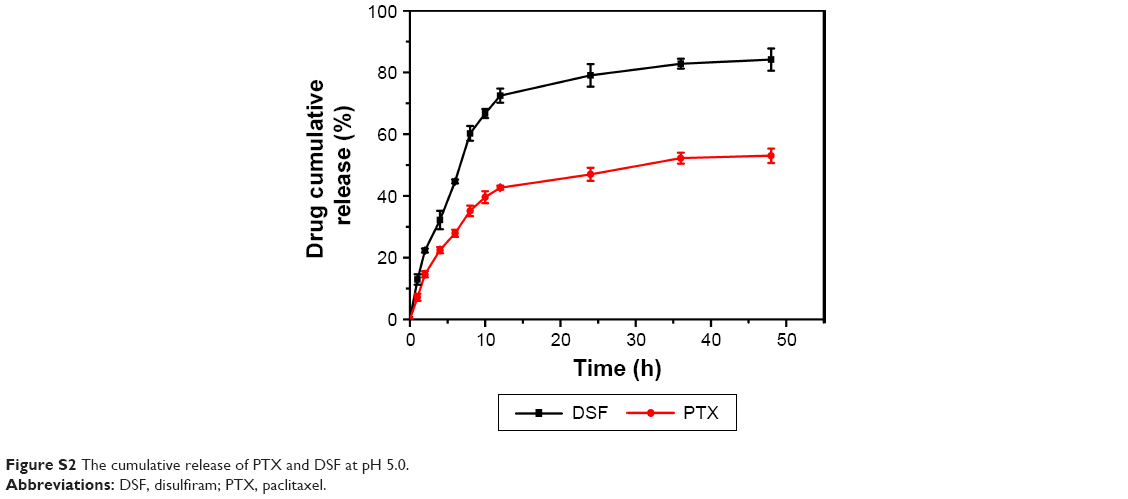 | Figure S2 The cumulative release of PTX and DSF at pH 5.0. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.