")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Personalizing the Care and Treatment of Alzheimer’s Disease: An Overview
Authors Svob Strac D , Konjevod M, Sagud M , Nikolac Perkovic M, Nedic Erjavec G, Vuic B, Simic G , Vukic V, Mimica N, Pivac N
Received 17 January 2021
Accepted for publication 5 May 2021
Published 28 May 2021 Volume 2021:14 Pages 631—653
DOI https://doi.org/10.2147/PGPM.S284615
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Dubravka Svob Strac,1,* Marcela Konjevod,1,* Marina Sagud,2,3 Matea Nikolac Perkovic,1 Gordana Nedic Erjavec,1 Barbara Vuic,1 Goran Simic,4 Vana Vukic,4 Ninoslav Mimica,5 Nela Pivac1
1Laboratory for Molecular Neuropsychiatry, Division of Molecular Medicine, Rudjer Boskovic Institute, Zagreb, Croatia; 2Department of Psychiatry, Clinical Hospital Centre Zagreb, Zagreb, Croatia; 3University of Zagreb Medical School, Zagreb, Croatia; 4Department of Neuroscience, Croatian Institute for Brain Research, Zagreb, Croatia; 5Clinics for Psychiatry Vrapce, Zagreb, Croatia
*These authors contributed equally to this work
Correspondence: Nela Pivac
Laboratory for Molecular Neuropsychiatry, Division of Molecular Medicine, Rudjer Boskovic Institute, Bijenicka Cesta 54, Zagreb, 10 000, Croatia
Tel +385 1 4571 207
Fax +385 1 456 1010
Email [email protected]
Abstract: Alzheimer’s disease (AD) is a progressive, complex, and multifactorial neurodegenerative disorder, still without effective and stable therapeutic strategies. Currently, available medications for AD are based on symptomatic therapy, which include acetylcholinesterase (AChE) inhibitors and N-methyl-D-aspartate (NMDA) receptor antagonist. Additionally, medications such as antipsychotic drugs, antidepressants, sedative, and hypnotic agents, and mood stabilizers are used for the management of behavioral and psychological symptoms of dementia (BPSD). Clinical research has been extensively investigated treatments focusing on the hallmark pathology of AD, including the amyloid deposition, tau hyperphosphorylation, neuroinflammation, and vascular changes; however, so far without success, as all new potential drugs failed to show significant clinical benefit. The underlying heterogeneous etiology and diverse symptoms of AD suggest that a precision medicine strategy is required, which would take into account the complex genetic, epigenetic, and environmental landscape of each AD patient. The article provides a comprehensive overview of the literature on AD, the current and potential therapy of both cognitive symptoms as well as BPSD, with a special focus on gut microbiota and epigenetic modifications as new emerging drug targets. Their specific patterns could represent the basis for novel individually tailored approaches aimed to optimize precision medicine strategies for AD prevention and treatment. However, the successful application of precision medicine to AD demands a further extensive research of underlying pathological processes, as well as clinical and biological complexity of this multifactorial neurodegenerative disorder.
Keywords: Alzheimer disease, therapy, precision medicine, gut microbiota, epigenetics
Alzheimer Disease
Alzheimer’s disease (AD) is a slow, irreversible, but progressive, complex and multifactorial neurodegenerative disorder and the most common form of dementia, corresponding from 60% to 80% of all dementia cases.1 Cases occurring before the age of 65 are considerably rarer, constituting less than 5% of all AD cases and are termed the early-onset AD (EOAD), while the majority of cases occur after age 65, representing the late-onset AD (LOAD) or sporadic AD. Estimated 1–2% of AD cases have very early age of disease onset, with a more rapid rate of progression and sometimes associated with other neurologic symptoms less frequently present in sporadic AD, and it is inherited in an autosomal dominant fashion.2
AD is not an age-related disorder; however, older age is a major risk factor for AD.3 Specifically, the risk of developing AD significantly increases after 65 years of age, and it reaches up to 31% for individuals beyond the age of 85.3,4 According to the age-dependent hypothesis, cognitive functions are slowly reduced during normal aging. However, certain alterations, such as neuroinflammation, changes in the cell biology, or injuries, might cause a switch from normal aging to AD pathophysiology.5 It is assumed that age affects inflammatory processes in the brain, resulting in an imbalance between anti- and pro-inflammatory factors and leading to chronic low-grade inflammation.6
Furthermore, loss of sex steroid hormones, estrogens and androgens with age represents one of the significant risk factors for AD development.7 In general, women show slower cognitive decline during aging and better results in verbal tasks, while men are better in coordination and visuospatial tasks.8 On the other hand, women with AD show faster decline in cognitive functions and lower score in verbal tasks, compared to men. Moreover, it has been reported that women over 89 years of age had a higher prevalence of AD in comparison to the male subjects.9 Such differences might be a result of different rate of brain atrophy in men and women. Specifically, it has been observed that brain atrophy rate is 1–1.5% faster in women than in men. In addition, higher levels of neurofibrillary tangles in certain brain areas were found in women, with no observed differences in Aβ burden between subjects of different gender.8 The gender differences are not affecting only disease progression, but comorbidity as well.10 Although female patients with AD show larger neuroanatomical and cognitive reduction, men have higher comorbidity burden than women, which might be a reason for higher mortality of male subjects with AD.10 Sex differences in behavioral symptoms of AD have also been observed. Specifically, while women tend to exhibit symptoms like depression, delusions, maniac symptoms and emotional lability, men more often express apathy, abusive and inappropriate behavior.8
In addition, AD variability has been associated with different ethnicity. For example, according to some authors, African Americans had a higher prevalence of AD and greater disease progression, compared to Caucasians.11 On the other hand, the study by Mehta et al12 demonstrated that African Americans and Latino population with AD might have better survival rates than Caucasians and American Indians, whereas the lowest AD-related mortality was reported for Asian subjects.12
Other risk factors of developing AD are genetic13 and acquired factors. While the mutations in genes for amyloid precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2) usually cause early AD, late-form AD is mainly associated with a polymorphism in apolipoprotein E gene (APOE), especially the presence of ε4 allele.14 APOE gene encodes apoE, a key lipid transport protein in the brain that is produced by astrocytes and plays a critical role in the synthesis and delivery of cholesterol in the brain. Humans have three apoE isoforms (apoE2, apoE3 and apoE4) and the carriers of a ε4 allele have an increased risk of developing AD, while ε2 allele acts protectively for its carriers.15 Acquired risk factors include cerebrovascular diseases as the most commonly reported risk factor, type 2 diabetes mellitus, hypertension, obesity, dyslipidemia, marital status, stress, depression, inadequate sleep and smoking.16 Some of the protective factors that can lower the risk for developing AD are physical activity, Mediterranean diet and vitamin D.17
Due to the accelerated aging of human population, the number of AD patients rises each year.3 This high prevalence contributes to the great societal burden, especially for the caregivers, and increases medical health expenses, which is why it is recognized as a major economic problem of the modern society and a fatal global epidemic worldwide.18 The onset of the disease occurs between 15 (for the EOAD) and 20–30 (for the LOAD) years before the appearance of any clinical symptoms. The silent and asymptomatic initial stage of AD, also called preclinical AD, is characterized by the development of pathophysiological hallmarks, which can be analyzed in the cerebrospinal fluid (CSF) or in the brain with amyloid positron emission tomography (PET) imaging.17
AD typically manifests through a progressive loss of episodic memory and cognitive function, leading to language and visuospatial skills deficiencies.19 These deficiencies are often accompanied by behavioral symptoms, such as apathy, aggressiveness and depression.16 Mental status examination plays a crucial role in the determination of clinical manifestations of neurologic and psychiatric diseases.20 Subjective cognitive decline (SCD) is a self-experienced cognitive decline, not detectable on standardized testing, which often occurs in the late stage of preclinical AD. SCD is considered a preclinical manifestation of AD and one of the earliest noticeable symptoms of dementia.21,22 At some critical point, when a level of cognitive decline cannot be compensated, it passes into mild cognitive impairment (MCI), which can be manifested on standardized tests.23 However, although the risk for MCI and dementia is increased in individuals with SCD, it is not always prodromal to AD and subjects with SCD may not show progressive cognitive decline.24 Mini Mental State Examination (MMSE), Clinical Dementia Rating Scale (CDR), Global Deterioration Scale (GDS), Neuropsychiatric Inventory (NPI), Barthel Index of Activities of Daily Living (ADL), and Seoul Neuropsychological Screening Battery (SNSB) are neuropsychological measures used for testing these cognitive symptoms.25 The diagnosis of AD is still made post-mortem and post-mortem studies demonstrated wide variability in terms of accuracy of clinical diagnosis and highlighted the challenges and limitations. Frequently, there are only serial assessments and long-term follow-up demonstrate the progression typical for AD. In addition to the basic assessment of a patient with suspected AD and the early neuropsychological profile of the AD amnestic presentation, there are also cognitive screening instruments for dementia such as Addenbrooke´s Cognitive Examination (ACE-III), Montreal Cognitive Assessment (MoCA), Neuropsychiatry Unit Cognitive Assessment Tool (NUCOG), Rowland Universal Dementia Assessment Scale (RUDAS) and the previously mentioned Mini Mental State Examination.26
Several hypotheses try to explain the development of AD: the hypotheses based on protein deposits, which include the beta-amyloid (Aβ) cascade hypothesis and the tau hypothesis, the hypothesis of reactive processes of neuroinflammation as the first event in AD and the group of hypotheses based on the loss of function, which include calcium disbalance hypothesis, vascular hypothesis and oxidative stress hypothesis.27 AD pathology is very complex with four core features. The first core feature is hyperphosphorylation of tau protein. Tau is an intracellular microtubule-associated protein within neurons, important for structural support and axonal transport. Its hyperphosphorylation leads to microtubule collapse and aggregation into neurofibrillary tangles (NFTs). Further, cleavage of APP by β- and γ-secretase enzymes causes the extracellular accumulation and aggregation of Aβ protein fragments, visible as amyloid plaques in the AD brain.28
Although accumulation of Aβ plaques and formation of NFTs have been associated with the etiology of AD for over a century, neither the exact pathophysiological mechanism of the disease nor definite treatment options have been found so far. The amyloid cascade hypothesis has been challenged with the amyloid β oligomer (AβO) hypothesis proposing that AD is instigated by soluble, ligand-like AβO, that were shown to be neurotoxic.29 Besides Aβ, other products are generated following β- and γ- secretase cleavage of APP in the amyloidogenic processing pathway, including soluble APPβ, C99, and AICD.30 These potentially important APP fragments have not been studied in detail yet. Moreover, γ-secretase has over 90 known substrates that all could interfere with Aβ production, making the idea that APP metabolism and Aβ production in sporadic AD cases depend on interaction and competition among γ-secretase substrates, very likely.31 One of the currently most investigated concepts proposes that the development of AD is triggered by impairment of APP metabolism and then further progresses through tau pathology, rather than through Aβ accumulation.29,32 This concept is supported by the fact that tau pathology is also a major age-related event in persons with Down syndrome, 88% of whom develop dementia before the age of 65, caused by an extra copy of the APP gene.33,34 Therefore, it has been hypothesized that tau pathology within selected projection neurons with susceptible microenvironments may, by itself, initiate sporadic AD.35 This is in line with the findings of late amyloid accumulation in accelerated-senescence non-transgenic OXYS rats,36 used as an animal AD model, and with the finding that functional integrity of synapses in the central nervous system (CNS) of cognitively intact individuals with high AD neuropathology is associated with the absence of synaptic tau oligomers.37 Hence, in comparison to Braak stage-specific readouts, connectivity-based, personalized tau-PET readouts reduced the sample size of planned patient-centered simulated tau-targeting clinical trial interventions by approximately 40%.38
CNS inflammation starts in the very early stages of AD, probably decades before its clinical manifestation, and the characteristics and the intensity of the inflammatory process change with the progression of the disease. We still know very little about the initial process of inflammation compared to the neuroinflammatory processes present in the later stages of AD.39 Cuello suggests that neurons burdened with Aβ could be the initiators of inflammation and activators of disease-aggravating inflammatory process mediated by mobilized microglia.39 This early process could be manageable by the anti-inflammatory agents, unlike the CNS inflammatory process in the late stages of AD. Microglia, the resident macrophages of the CNS, which are activated in AD, produce cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and nitric oxide (NO) that affect neuroinflammation.28 Several recent findings revealed that the probable major links between disordered APP metabolism and tau pathology are microglial and inflammasome activation and transmissible exosomes containing tau oligomers.40 Microglia are the primary innate immune cells of the brain, acting nonspecifically against any foreign antigen, and are considered to be implicated very early in AD pathogenesis. Microglia can acquire two functional states depending on environmental cues: pro-inflammatory (cytotoxic, M1) and anti-inflammatory (immunosuppressive, M2). However, it has been observed that microglia, although abundant in the AD brain, fail to efficiently eliminate AD deposits.41 Despite the onset of AD being dominated by anti-inflammatory microglial phenotype, pro-inflammatory cytokines eventually induce a transition to pro-inflammatory microglial type in the course of the disease progression.41 M1 microglia release pro-inflammatory cytokines such as IL-1α, TNF-α, and complement components that subsequently induce neurotoxic A1 reactive astrocytes. As pro-inflammatory cytokines such as IL-1α, IL-1β, IL-6 and TNF-α, promote inflammation and neurodegeneration, while anti-inflammatory cytokines, such as IL-10, oppose their action,41–43 single nucleotide polymorphisms of the genes, coding for those proteins, influence the susceptibility to the AD pathology.44
However, the evidence suggests that the link between neurodegeneration and microglia could be more complex than was initially thought. Different neurodegenerative diseases and aging have been associated with the loss of microglial homeostatic functions, resulting in chronic neuroinflammation.45 In addition, disease-associated microglia (DAM) were recently identified as a subset of microglia that can be found at sites of neurodegeneration.46 This subset of microglia is characterized by a specific transcriptional and functional profile,47 which includes the expression of a gene coding for triggering receptor expressed on myeloid cells 2 (TREM2), a receptor which is necessary for the activation of DAM.47,48 In addition, TREM2 mutations were previously associated with AD development,49,50 while TREM2 deficiency was shown to potentiate accumulation of Aβ and loss of neurons in the mouse model of AD.51,52 Transcriptome profiling additionally identified APOE and transforming growth factor beta (TGF-β) as important regulators of DAM phenotype.53 Krasemann et al identified the TREM2-APOE pathway as a key regulator of microglia phenotypic change in neurodegenerative diseases.53 The activation of this pathway led to the microglial phenotype switch from a homeostatic to neurodegenerative phenotype and resulted in the inability of microglia to maintain brain homeostasis. Therefore, understanding and recognizing the different roles of microglia in the process of AD initiation and progression is invaluable for future potential therapeutic strategies and application of precision medicine approaches.
Moreover, vascular pathology and blood-brain barrier (BBB) disruption have been linked to AD too. Damage to the blood vessels consequently leading to BBB dysfunction and causing impaired brain perfusion is hypothesized to precede neurodegeneration and exist long before other characteristic features of the disease appear.41,54 The first affected area in AD-related neurodegeneration is the entorhinal cortex, spreading to the subiculum, CA1 hippocampal subregion and basal forebrain networks. As the disease progresses, neurodegeneration expands throughout the temporal lobes, affecting the majority of cortical layers,28 where mass neuronal and synaptic loss correlates with cognitive decline in AD.
Despite accumulating data about AD etiology and pathophysiology, there is still an incomplete characterization of the molecular mechanisms underlying AD due to its significant clinical, pathological and biological complexity. Specifically, the risk factors, biomolecular profiles, as well as specific underlying pathophysiological processes in AD patients demonstrate high variation. In addition to age, ethnicity and sex, the disease heterogeneity is also influenced by the extensive genetic variation underlying AD. So far, a variety of enrolled genes has been identified, with individual genetic variants having a small effect on disease risk.55 However, there are still many unknown genetic factors contributing to AD and further sequencing studies are necessary in order to identify genetic risk variants and to determine the specific molecular pathways involved. In addition, AD has a considerable non-genetic component, with different environmental factors, including cerebrovascular disease or traumatic brain injury and lifestyle factors, such as intellectual activity, affecting the risk for AD development and significantly interacting with individual genetic background. Specific latent pathophysiologic processes in AD patients are now more accessible by the application of brain imaging technologies and by quantification of blood and CSF biomarkers. Therefore, successful application of precision medicine to AD is needed, in order to specifically target underlying molecular and clinical heterogeneity of AD and to administer a preventive or therapeutic intervention that is personalized to the identified molecular pattern of risk and disease processes.55
Pharmacological Treatment of AD
The effective and stable therapeutic strategies for AD still do not exist and the failure rate in clinical trials is higher than for any other disease (99.5%). Starting of treatments late during the course of AD progress, inappropriate drug doses, invalid target selection, and predominantly an insufficient knowledge of the diverse pathophysiology of AD are the reasons for failures of disease-modifying treatments for AD.56 It has been proposed that some promising drugs that failed to show clinical improvement in subjects with MCI or moderate stage of AD were administered too late in the disease course.57 In addition, so far most of the clinical trials have neglected the underlying clinical and molecular heterogeneity of AD and considered AD as a homogeneous disease, which could be one of the main reasons for their failure to identify an AD effective therapy.
Currently, treatment of AD is targeted toward symptomatic therapy, although clinical research is developing towards a more definitive treatment of the hallmark pathology in AD, with the expectation that these therapeutic options will attenuate the progressive decline associated with AD. Various trials are underway that aim to reduce the production and overall burden of pathology within the brain.58 Finally, precision medicine is now beginning to be incorporated into AD clinical trials, which are focusing on subgroups of subjects with known genetic risk for AD and specific biofluid or neuroimaging biomarkers.55
However, the treatment of AD has not been significantly changed or improved in the last decade. It includes four approved medications:59 acetylcholinesterase (AChE) inhibitors donepezil, galantamine and rivastigmine, and N-methyl-D-aspartate (NMDA) receptor antagonist memantine (Figure 1), which were approved by US Food and Drug Administration (FDA). Out of three AChE inhibitors, galantamine and donepezil are AChE inhibitors, and rivastigmine inhibits both AChE and butyrylcholinesterase (BChE). These medications are based on the assumption that, according to the cholinergic theory, loss of acetylcholine in the brain is responsible for the development of AD symptoms since the cholinergic system plays an important role in cognitive processes.60 Therefore, inhibition of the enzymes that degrade acetylcholine, AChE and BChE (Figure 2), is the first line of AD treatment.61
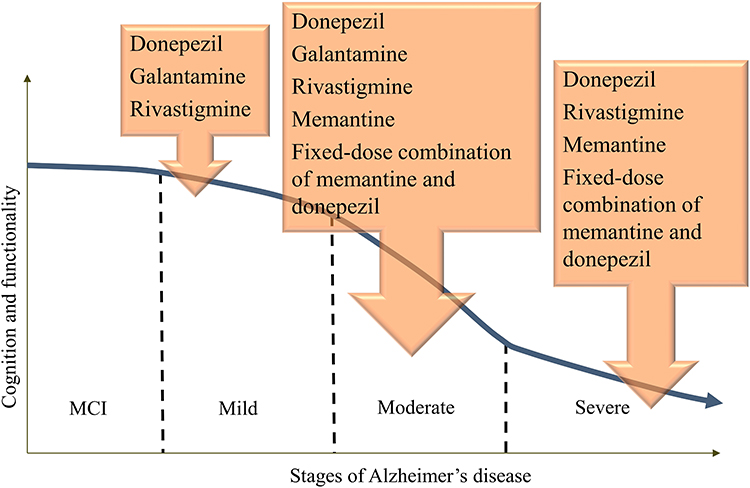 |
Figure 1 FDA-approved treatments across phases of AD. |
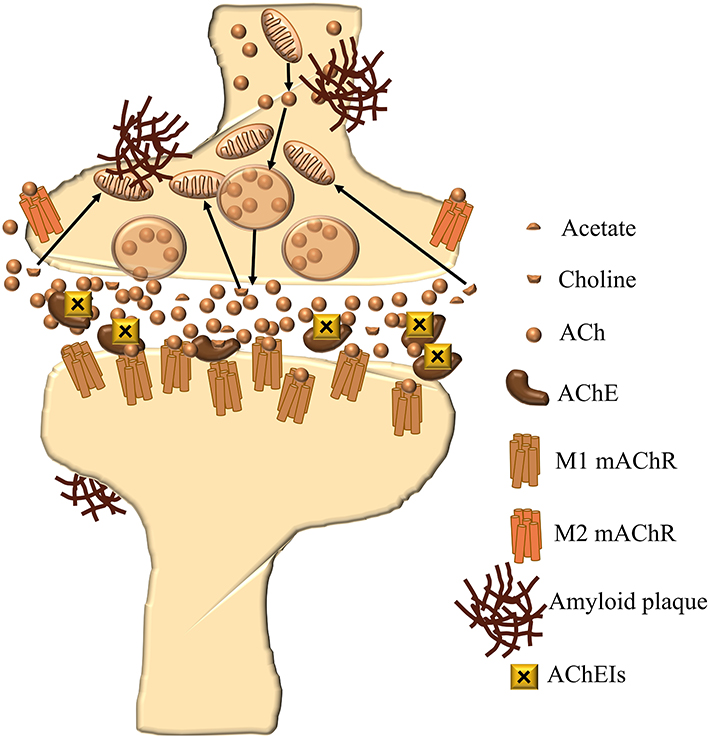 |
Figure 2 Cholinergic synapse in AD during treatment with AChE inhibitors. |
These medications have been shown to improve cognitive functions and some of them may also have positive effects on behavioral symptoms.62 However, none of these drugs affect the course and the outcome of the disease.63 The pharmacological treatment of AD differs according to the stage of disease (Figure 1), and AChE inhibitors are used for mild to moderate AD, while the antagonist of NMDA receptors, memantine, is used to treat moderate to severe AD.64 These drugs provide symptom relief; however, sometimes they induce side effects. Unfortunately, they are not disease-modifying and they do not represent a cure.65 They cannot slow down the disease progression, but only temporarily improve brain function, and slow down the cognitive deterioration via reduction of AChE activity, resulting in higher acetylcholine levels.66 Other investigated AChE inhibitors were characterized with significant side effects: physostigmine (diarrhea, stomach cramps, increased salivation, excessive sweating), tacrine (nausea, vomiting, loss of appetite, diarrhea, clumsiness, hepatotoxicity, impractical dosing schedule (4 times daily) due to the short half-life) and metrifonate (neuromuscular and respiratory side effects after the long-term use), and therefore were not approved for the treatment of AD.61
The results of a Cochrane study for donepezil, which included 8257 subjects with mild, moderate, and severe AD, showed statistically significant improvement in cognitive functions, activities of daily living, and clinician-rated global impression of change scale, with no significant effects on behavioral symptoms. Both doses (5 mg and 10 mg) of donepezil were effective, with more adverse effects (Table 1), mainly gastrointestinal effects, reported for the higher dose.67
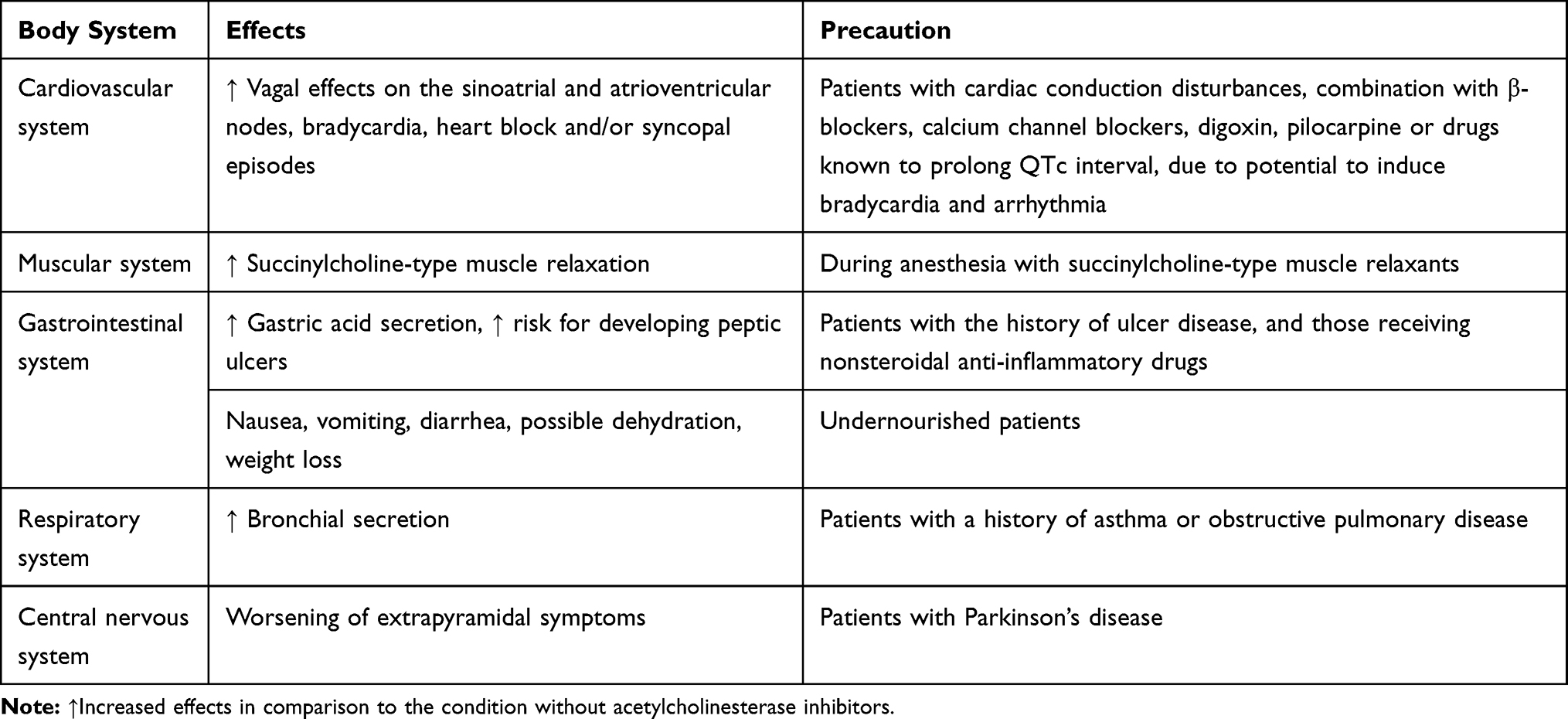 |
Table 1 Adverse Reactions of Acetylcholinesterase Inhibitors |
The other AChE inhibitor, rivastigmine (1.5–6 mg twice daily), was approved by the FDA for all stages of AD. It was associated with cognitive improvement/stabilization, especially if the treatment started in the early phases of AD.68 Rivastigmine has led to statistically significant improvement in cognitive functions in patients with mild to moderate AD when administered in high doses (6–12 mg). It had no significant effect on behavioral symptoms. Adverse effects (Table 1) were quite common with high doses, mostly gastrointestinal symptoms, although headaches and syncope were also reported. The rivastigmine transdermal patch was also tested. A smaller patch with a lower dosage was as effective as both the bigger patch (higher dosage) and the perioral form of the medicine, which was given in the equivalent daily dose but had less adverse effects.68
The effects of galantamine (4–12 mg twice daily) were also analyzed in a meta-analysis, and the results showed a statistically significant improvement in cognitive functions in mild to moderate AD. The side effects (Table 1) were similar to those of other AChE inhibitors.69 This review suggested that, according to the stage of AD, clinical characteristics and tolerability, titration of the dose and switching between AChE inhibitors might improve the response rate. Switching between AChE inhibitors may prevent issues related to the lack of efficacy or safety/tolerability in patients with AD.69
In AD, the amyloid plaques affect glutamatergic neurotransmission, elevate synaptic and extrasynaptic glutamate levels and elicit excitotoxicity. This leads to hypoactivity, neuronal loss, progression of AD, and cognitive deficits. The activation of synaptic NMDA glutamate receptor is critical for the survival of neurons,64 but excessive glutamatergic stimulation leads to excitotoxicity, which causes loss of synaptic function and neuronal death, processes believed to be a part of the pathogenesis leading to neurodegenerative changes in AD.70 Memantine is a non-competitive NMDA receptor antagonist, which binds to the receptors when high concentrations of glutamate are present in the synaptic cleft (Figure 3), as is the case of AD.71
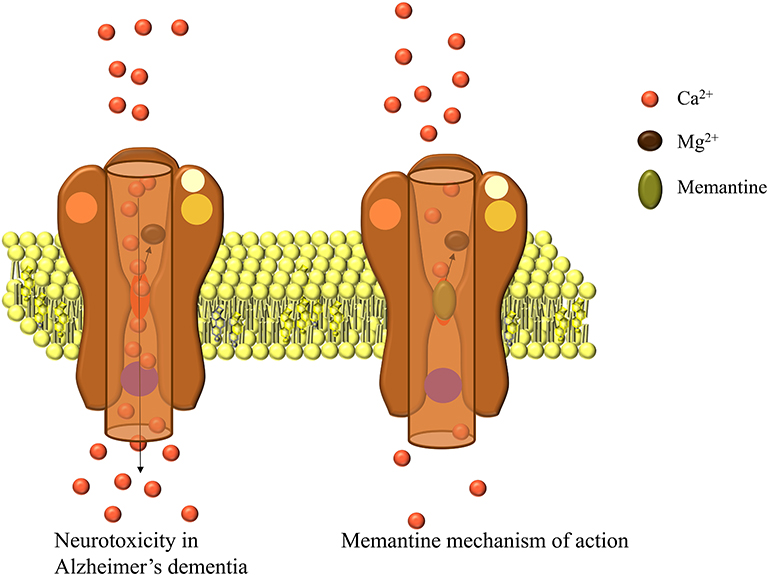 |
Figure 3 Overstimulation of NMDA receptors in AD and memantine mechanism of action. |
A 2017 meta-analytic study compared the efficacy and safety between monotherapy of donepezil and combined therapy with donepezil and memantine in subjects with moderate and severe AD. The study analyzed cognition, behavioral and psychological symptoms, and global functions. The results showed the combination therapy to be superior in all domains, without significant adverse effects.72 A 2018 meta-analysis compared the safety and effectiveness of AChE and memantine, and according to this study,70 the most effective approach to improve cognitive functions in mild to moderate AD was (individually) donepezil 10 mg and galantamine 24 mg or 32 mg daily. For moderate to severe AD, the most effective therapy was a combination of memantine 20 mg and donepezil 10 mg. Memantine had the best acceptability profile.70 No effects on behavioral symptoms were registered.73
Although many new drugs with novel mechanism of action were effective in animal models, only a few of them showed efficacy in improving cognitive decline. In addition, until now, most of the studied drugs (nicotinic receptor agonists, glutamate receptor modulators, γ-secretase inhibitors, growth factors, statins, monoclonal antibodies, tau inhibitors, serotonin receptor modulators, etc.) are either under development or suspended in phases 1–3 of clinical trials. As a result, many new potential drugs failed to show clinical benefit in double-blind placebo-controlled trials and were discontinued due to the serious side effects, resulting in no new AD drugs in the last ten years.
In addition to AChE and NMDA receptor inhibitors, other neurotransmitters-based therapies have been also investigated for AD in preclinical studies and clinical trials, including GABAergic modulators, serotonin and adenosine receptor modulators, as well as histaminergic modulators.74,75
One of the most extensively investigated therapeutic strategies for AD is the inhibition of Aβ cascade, either by inhibiting β-secretase (verubecestat, lanabecestat, elenbecestat, atabecestat, CNP520) or γ-secretase (tarenflurblil, semagacestat, avagacestat), or more recently by increasing α-secretase activity.76–78 Passive immunotherapy using the anti-Aβ antibodies destabilizing or clearing Aβ oligomers (bapineuzumab, gantenerumab, aducanumab) has been another developing strategy targeting Aβ pathology. In addition, the metal protein attenuating compounds (clioquino l- PBT1, PBT2) have emerged as potential anti-Aβ aggregates preventing Aβ oligomerization. The potential of nilvadipine, Ca2+ channel antagonist, for reducing the Aβ levels in the brain is also investigated.76–78 Limitations and inadequacies of targeting most popular amyloid cascade and AβO hypotheses may originate from disregarding the physiological function of Aβ. Indeed, ancestral APP gene arose hundreds of millions of years ago, and would not be so highly conserved throughout evolution unless being important.41 Firstly, it has been observed that Aβ resembles molecules from the group of so-called “antimicrobial peptides” and subsequently demonstrated that it could inhibit growth of eight human pathogens, thus providing evidence that Aβ might function as a part of the innate immune system.41 Secondly, previously mentioned BBB breakdown manifests in the form of cerebral microbleeds.54 Amyloid plaques form at sites of these microbleeds and seal vascular ruptures thus preventing major bleeding and hemorrhagic strokes.41 Additionally, Aβ, likewise APP containing Kunitz protease inhibitor domain called protease-nexin 2 (PN-2), is able to inhibit serine proteases, like trypsin and coagulation factors, acting as an anticoagulant.41 The Aβ is demonstrated to exhibit apolipoprotein-like function thereby enhancing cholesterol efflux from cells and reducing cellular cholesterol levels through interaction with ABCA1 transmembrane protein. It enhances memory in physiological, picomolar concentrations, while it impairs cognition only when present in excess amounts.79 Finally, Aβ is associated with a reduced risk of cancer. Experimental evidence suggests that Aβ may inhibit tumor cell growth and slow tumor growth by retarding neovascularization and even by slowing down the proliferation of tumor cells by limiting the availability of essential micronutrients due to its high binding affinity for iron, copper, and zinc. It is also assumed that Aβ may act by intercepting oncogenic viruses.80 Hence, still underappreciated physiological roles of Aβ range from protection against infections, prevention of strokes, promotion of recovery after brain injury, cholesterol transport, to synaptic function, regulation and possible contribution to tumor suppression.
Therefore, Aβ immunization or any other procedure aimed at removal, neutralization or suppression of Aβ production should be preceded by an assessment of potential activators of Aβ production and deposition, such as infections, brain injuries, type 2 diabetes mellitus, tumors, etc. This could be a part of the routine clinical evaluation when checking for reversible causes of dementia, such as pseudodementia due to depression and B12 hypovitaminosis (treated with antidepressants and vitamin B12, respectively). Subsequently, on the basis of such an assessment of a patient with suspected or overt dementia, and before considering any amyloid removal interventions, we suggest that the next step should be aimed to treat: 1) possible infection, 2) other established, but treatable risk factors that accelerate amyloid production and deposition, especially type 2 diabetes mellitus, and 3) imminent neuroinflammation due to possible microglial activation. So far, a single clinical trial has introduced antibiotic minocycline to AD subjects but failed due to the inclusion of the small number of patients only with advanced dementia.41 In addition, AD proof-of-principle clinical trials with antiviral drugs, such as acyclovir, and antifungal drugs, such as ketoconazole, together with likely beneficial immune-selective anti-inflammatory derivatives, are required in the near future.
The inhibition of tau hyper-phosphorylation, aggregation, or tau expression level and activity, by using tau expression inhibitors (antisense oligonucleotides), phosphatase modifiers (PPZ, OKA, memantine), tau kinase inhibitors (tideglusib, lithium, amino-thiazole, oxindolequinazoline, sirenade, R-roscovitine, aloisine), tau acetylation inhibitors (salsalate), tau deglycosylation inhibitors (phosphodiesterase type 4 inhibitor), tau aggregation inhibitors (methylene blue, LMTX), or microtubule stabilizers (epothilone D, NAP, TPI 287), could also be potentially effective therapeutic approaches preventing AD progression.76–78,81
In addition, the inhibition of inflammatory response, targeting the innate immunity by inhibiting cascades such as JAK2/STAT3, NF-κB/NLRP3, p38 MAPK, calcium/calcineurin/NFAT, TREM2, TLR, or RAGE/CSF1R/P2Y1R pathways (stattic, SB203580, SB202190, SP600125, NJK14047, VIVIT peptide, tacrolimus, anti-apoE, anti-TREM2, GW2580, MRS2179), as well as targeting adaptive immune system, by treatment with Aβ-specific Th2 lymphocytes and anti-Aβ IgG, could represent novel strategies to reduce symptoms in AD associated with neuroinflammation. Nonsteroidal anti-inflammatory drugs, such as aspirin and ibuprofen, are currently used for the treatment of AD patients.76–78,81 In addition, vitamin E (an antioxidant) supplementation is currently administered to AD patients since neuroprotective effects of antioxidants represent a promising preventive and therapeutic strategy.77
Additional potential therapeutic strategy for AD is neuroregeneration by direct supplementation of neurotrophins (hNGFp, AAV2-NGF, BDNF) or indirectly via increase of neurotrophic effects by peptide mimetics that target neurotrophin receptors (7,8-DHF, doxygedunin, LM22A-4, bicyclic BDNF loop mimetic). Another strategy to address AD is to promote neuroregeneration. This could be achieved by supplementing the functional neuronal cells either by the glia-neuron conversion (using neurogenin 2, Dlx2, the combination of various molecules) or by the transplantation of stem cells that are genetically modified to encode neurotrophic factor genes or pre-treated with neurotrophic or growth factors.76–78,81
Complementing traditional drug discovery with drug repositioning and repurposing is another approach aiming to maximize the efforts for AD drug development.78,82 For instance, metformin, type 2 diabetes drug, was repurposed for AD treatment, due to its anti-inflammatory and neuroprotective properties in AD.78 Five classes of compounds for repurposing as treatments for AD are suggested; tetracycline antibiotics, calcium channel blockers, angiotensin receptor blockers (ARBs), glucagon-like peptide 1 (GLP1) analogues and retinoid therapy. However, the three highest priority candidates for repurposing in AD, which were most recently selected are ROCK2 inhibitor fasudil, the cholinesterase inhibitor phenserine, with novel anti- apoptotic properties, and the antiviral drugs: aciclovir, valaciclovir and famciclovir.82
Multi target directed ligand (MTDLs) is a novel approach in AD therapeutic strategies targeting several molecular targets synergistically. For example, new compound, named RPEL, is a piperazine derivative that contains the pharmacophore fragment of tacrine. It is a dual-action compound found effective in preventing cognitive impairments, which showed the inhibitory potency against cholinesterase, reduced the phosphorylation of tau protein and inhibited the release of the Aβ peptide.78,83
Treatment of Behavioral and Psychological Symptoms of Dementia
Besides memory and learning impairments, as well as other cognitive disturbances,84 AD patients also show “non-cognitive” symptoms, known as behavioral and psychological symptoms of dementia (BPSD). BPSD are heterogeneous group of behaviors, psychiatric symptoms and mood disorders,85 diagnosed in approximately 90% of AD patients. BPSD encompass symptoms such as sleep disturbances, anxiety, aggression, depression, wandering, delusion, hallucinations, and disinhibition,84–87 which are clustered as 1) apathy; 2) affective (anxiety, depression); 3) psychosis (paranoia, delusions, hallucinations); and 4) disinhibition (motor hyperactivity, impulsivity, aggression).84,86 More than 50% of AD patients show more than four neuropsychiatric symptoms at the same time. Therefore, the presence of such symptoms affects normal functioning, as well as further cognitive decline,88 and represents one of the main reasons for the institutionalization of AD patients.84 Gender differences have been observed between female and male AD patients with BPSD. It has been noted that females are more prone to develop AD compared to males, whereas female AD patients usually develop depressive symptoms, while male AD patients more often express aggressive behavior.87 Likewise, differences between patients with EOAD and LOAD with diagnosed BPSD were noticed. LOAD patients had higher BPSD severity compared with EOAD patients.89
The development of BPSD in AD patients is a result of neuronal loss in various brain areas, including the hippocampus, brain stem nuclei, and parahippocampal gyrus, as well as glutamate reduction; changes in cholinergic and dopaminergic neurotransmission in the frontal and temporal cortex assumed to be associated with motor hyperactivity and aggression, and altered GABA and serotonin concentrations that might lead to the development of apathy and depression.90 Management of BPSD is highly individualized and requires detailed evaluation of presented symptoms in order to determine appropriate, effective treatment strategy.91 However, due to complex etiopathogenesis of BPSD and AD, the choice of adequate treatment is often challenging.90
Due to the complex etiology and symptoms diversity, the treatment of BPSD involves different therapy approaches.92 The part of BPSD treatment includes pain management, as well as treatment of comorbid somatic disorders. Although pharmacotherapy for BPSD is provided, it might cause certain side effects and it should be prescribed only when it is necessary. Hence, non-pharmacological approaches for BPSD are suggested before any pharmacological treatment.90,91 Behavioral, individual, caregiver and environmental interventions92 aim to educate and provide support to the family members and other caregivers. It includes different trainings, development of communication skills, dementia care mapping, psychosocial interventions, physical activity, sensory stimulation, reality orientation, cognitive stimulation, validation therapy, psychotherapeutic interventions, and reminiscence therapy.90,91
Psychoeducational trainings for family members have shown benefit in more than 86% of studies, while support groups were beneficial for both, caregivers and patients. Physical activity has many beneficial effects on the human organism, improving cognition, fitness and behavior.90,91 Therapy that involves sensory stimulation might have beneficial effect in patients with BPSD, especially in the reduction of agitation and behavioral disturbances. Several studies showed that music sessions help with the reduction of agitation, delirium, irritability, depression and anxiety, as well as improve the memory and orientation in AD patients.93,94 Bright Light Therapy (BLT) has a positive outcome in patients with BPSD, by reducing agitation, aggression and improving cognitive performance and motor behavior.93 Furthermore, problem solving and reminiscence therapy for treating depression and anxiety have been shown beneficial in patients with dementia.91 Psychological interventions are mostly effective in later stages of dementia, especially when both, the caregivers and the patients, are involved in the therapy. Such approach helps with dealing with aggression, depression, anxiety and agitation.90 However, due to heterogeneity of BPSD in AD, these approaches sometimes do not show adequate efficacy, leading to strategies based on drug treatment.92
Pharmacological treatment is usually administered if none of the non-pharmacological interventions lead to improvement, in large patients’ distress, or in subjects who represent a danger to themselves or others.90,91 For BPSD, antidepressants are usually prescribed since they show low side effects.90 Some of the prescribed antidepressants include citalopram, sertraline, escitalopram, trazodone and mirtazapine.90,92 However, their efficacy is questionable. For instance, mirtazapine, which is usually used for the treatment of patients with depression, has not demonstrated therapeutic effects in AD patients.91 Antidementia drugs, AChE inhibitors and memantine, might also be useful in BPSD treatment. AChE inhibitors, such as donepezil, rivastigmine and galantamine are usually prescribed for BPSD treatment in patients with dementia. Starting and recommended doses for rivastigmine are 4.6 mg/24h patch, 9.5 mg/24h patch and 13.3 mg/24h patch.88 However, treatment with donepezil was not effective in BPSD reduction in AD patients, compared with placebo.88,90
Another group of medication, which has been prescribed for BPSD, are antipsychotics. They are often administered together with anti-dementia drugs or antidepressants.95 Atypical antipsychotics showed some therapeutic effect in patients with BPSD, while most effective are risperidone, olanzapine, quetiapine and aripiprazole. However, antipsychotic drugs often cause extrapyramidal side effects, including parkinsonism.16 For certain BPSD symptoms, especially aggression, antipsychotics might be useful in the alleviation of symptoms; however, their use must be limited and controlled.90 Specifically, evidence suggests that some other serious side effects, such as cerebrovascular adverse effects, cognitive decline and even death, also occur when antipsychotics are used among individuals with BPSD.88 Therefore, antipsychotic treatment should be prescribed only in the case of severe or refractory symptoms, where non-pharmacological or other pharmacological approaches were not effective or possible. After a risk-benefit analysis, the lowest effective doses of antipsychotics should be used for the shortest possible time, with adequate oversight in order to maximize the gain and minimize the harm from antipsychotic treatment.96 Special caution should be taken when administrating antipsychotic drugs to female patients since some serious side effects of these medications are more often seen among women than among men.97
Benzodiazepines are commonly used in patients with BPSD, although their efficacy is usually blurred with caused side effects. However, occasional use of certain benzodiazepines might help with aggression, agitation and sleep disturbances,91 while mood stabilizers are usually not recommended.90 Combination of different pharmacotherapeutics might be more beneficial in treatment of AD subjects with BPSD. For example, the combination of memantine and citalopram led to symptom reduction in AD subjects. Sleep disturbances, aggression, agitation, irritability, caregiver distress, apathy and anxiety were lower after the combined treatment.98 Furthermore, AD patients who were receiving donepezil in combination with memantine showed improvement in BPSD, global functions, as well as in cognitive functions compared to patients that were only receiving donepezil.72 Patients who were treated with donepezil and choline alphoscerate (n=57) showed significant improvement in certain BPSD symptoms, including depression, apathy, agitation and anxiety, compared with patients treated with donepezil and placebo (n=58).99 Therefore, in AD patients with BPSD, the combination of several medications might have a beneficial effect in comparison with monotherapeutic approach.
Personalizing the Care and Treatment of AD
Personalizing the care and treatment of AD intends to change the current treatment and social care of AD patients, by implementing precision medicine approach. Precision medicine is an approach that takes into account individual differences in biology, lifestyle and environment, aiming to optimize maximally the effectiveness of disease prevention and treatment. It has a multidisciplinary character and integrates a series of mechanisms to investigate, prevent, treat and cure complex diseases, such as AD. This approach needs accurate diagnosis and aims to minimize side effects,55 and is based on the knowledge of the specific biological and genetic background, but also on the biomarker, phenotypic and psychosocial characteristics of the individual patient. Improved understanding of different factors responsible for development of AD onset and progression allows for better disease prevention and personalized treatment strategies.55 For application of precision medicine in AD, it is also necessary to identify and validate disease-specific, mechanism-based, or (epi)genetics-dependent biomarkers of AD, which can improve and increase the accuracy of AD clinical diagnosis and patients’ susceptibility, monitor disease progression and therapeutic efficacy, as well as to analyze the benefits and adverse effects of particular drugs.
Personalizing the care and treatment should allow independence, choice and control over the services for the patients with AD. The underlying heterogeneous pathways for AD development and diverse AD symptoms require precision medicine strategy.100 This strategy includes the collection of demographic information for each patient (ie, APOE genotype, age, gender, education, environmental exposure, lifestyle, medical history), as a basis for patient stratification and incorporation of in-depth information to facilitate the design of precision medicine strategies, such as physical activity, diet, brain stimulation, social communication and medication.100 Assessment of the patients with AD, but also their family members, caregivers and care partners, should be person-centered. According to the recent study,101 systematic, comprehensive and temporary assessments should be done using a collaborative team approach, utilizing files, documentation and communication systems to ease the delivery of person-centered information, with aim to gather information, and build relationship, education, and support. These person-centered assessments should result in better planning to optimize physical, psychosocial and fiscal wellbeing, with a main goal to enlarge the knowledge of the possible care options, palliative care and hospice.101 Precision medicine approach is necessary since neither the exact pathophysiological mechanism of AD, nor the definite treatment options that are suitable for all AD patients, have been found so far.
Nevertheless, progress in life sciences and emergence of multi-omics studies (such as genomics, transcriptomics, epigenomics, proteomics, metabolomics, microbiomics, etc.) have shifted focus to precision medicine, promising improvements in both diagnostics and treatment of AD, as in other still untreatable or undertreated diseases.41 However, for the development of customized diagnostics and therapeutic interventions in precision medicine, big omics data should be integrated with clinical information, increasing the understanding of heterogeneous pathophysiology of AD. Taking into account the complex genetic, epigenetic, and environmental landscape of AD,102 in order to create the most effective implementation of precision medicine for patients with AD, patient stratification needs to be established based on comprehensive collection of both demographic (APOE genotype, age, gender, education, environmental exposure, life style, medical history) and in-depth information (genomic variations, brain imaging, central and peripheral biomarkers, functional assays on patient-derived neurons).100 Regarding environmental impact, a study was designed to determine a correlation between living near major roads and incidence of neurological diseases103 in two population-based adult cohorts and the results showed that living near major roadways was significantly associated with higher incidence of dementia. Suspected factors responsible for observed association were traffic-related air pollutants (ultrafine particles, nitrogen oxides, etc.) probably leading to neuroinflammation and oxidative stress, and noise, possibly through effects of sleep fragmentation and impaired cognition.103
Simultaneously, emerging technologies, such as induced pluripotent stem cells (iPSC), are highly valuable in the search for genetic determinants and environmental modulators of the disease, as well as in the screening of potential drugs that will, hopefully, revolutionize the treatment of AD patients.104 Some interventions have been already recognized as significant in prevention and treatment in early stages of AD: physical activity, brain stimulation through music and art, social communication, diet, supplements for energy supply, and drugs targeting neural transmission. For moderate and severe stages of AD, stage-specific intervention strategies in recent years move towards drugs targeting neural inflammation and neural regeneration therapy.100
However, in the next chapters, we focus on gut microbiota and epigenetic modifications as new emerging targets for AD management. Their immense variation and specific patterns could represent the basis for novel individually tailored approaches aimed to optimize precision medicine strategies for AD prevention and treatment.
Gut Microbiota as a Target for AD Treatment
The human gut is a habitat for around 1000 species, with the bacterial density estimated to cca 1011 per mL.105 Gut microbiota are different between individuals, which is a phenomenon affecting the differences in biochemical and genetic individuality, as well as the resistance to diseases in humans.106,107 In adult individuals, the composition of gut microbiota is influenced by a variety of individual factors such as host genetics, age, the status of the host immune system, emotional stress, diet, and exercise,108 resulting in immense compositional variation of gut microbiota between and within individuals.109 Considering the great number of metabolic enzymes synthesized by the gut microbiota, it is no wonder that gut microbiota is regarded as an additional (metabolic) organ in the human organism.110
It is bidirectional, constant communication between the gastrointestinal tract and the CNS called the brain-gut axis or more recently, due to the major role of the gut microbiota, the brain-gut-microbiota axis.107 This communication is conducted through neural, immune, endocrine and metabolic signaling.111 A chronic inflammation in the elderly, caused by the hyperstimulation of the immune system,112 may also be associated with a persistent inflammatory state of the gut mucosa leading to the gut barrier breakdown, further increase of proinflammatory cytokines in the circulation, the BBB impairment and neuroinflammation.113 Besides, there is a direct synthesis of various neurotransmitters and neuromodulators, including serotonin and dopamine, by the gut microbiota.114
Therefore, a contribution of the brain-gut-microbiota axis disturbances to the pathogenesis of neurodegenerative disorders, including AD, is more than expected,114 with a growing body of experimental and clinical data confirming it. Since bacterial cells produce amyloids to bind to each other, the gut microbiota is a source of a significant amount of those proteins.115 Bacterial and human CNS amyloids differ in their primary structure, but share similarities in their tertiary structure.116 It is assumed that the exposure to gut bacterial amyloids could cause priming of the host’s immune system, thus enhancing inflammatory responses to endogenous neuronal amyloids.115 Besides amyloids, gut microbiota produces lipopolysaccharides which activate toll-like receptor (TLR) 4 promoting inflammatory response through the interactions with CD14.116 The same pathway is included in the inflammatory response to Aβ.117 Furthermore, a marker of intestinal inflammation, calprotectin, is a heterodimer of S100A8 and A9, the two proteins that can form amyloid oligomers and fibrils similar to Aβ and α-synuclein and even induce Aβ fibrilization.118 In the CSF and the brain of AD patients, calprotectin levels are significantly higher, promoting amyloid aggregation and co-aggregation with Aβ.118 Additionally, calprotectin levels are elevated also in feces of AD patients, with the assumption that it can enter the circulation and contribute to neuroinflammation.119
Obviously, gut-derived inflammatory response in aging and poor diet in the elderly may promote the pathogenesis of dementia,120 suggesting that the therapy directed to the modification of the gut microbiota composition represents a new therapeutic option in AD. Manipulation of gut microbiota by antibiotics in animal models showed an association of consequential dysbiosis with changes in behavior and brain chemistry.121 In humans, it was shown that the antibiotic treatment of Helicobacter pylori is associated with neurological disorders including anxiety, panic attacks, major depression, psychosis and delirium.122 On the other hand, the use of probiotics and prebiotics seems to reduce the risk of AD123 and improve cognition in AD patients.124 It was already shown that the diet might influence the development of AD. For example, antioxidant polyphenols from coffee beans can reduce oxidative stress induced by brain injuries and lower the risk of AD,125 but the bioavailability and the activity of polyphenols depend strongly on healthy gut microbiota.126 Additionally, there are evidence that high-fat diet induces changes in gut microbiota leading to an increase of intestinal permeability and lipopolysaccharide absorption, which causes endotoxemia and systemic inflammation.127 Caloric restriction, on the other hand, can optimize the gut microbiota composition by increasing the proportion of Lactobacillus.128
There are some clinical studies dealing with the new potential anti-AD therapeutics directed to gut microbiota. For example, a Phase 3 clinical trial in China reported sodium oligomannate suppressing gut dysbiosis and the associated phenylalanine/isoleucine accumulation, suppressing neuroinflammation and reversing the cognitive impairment.129 A recent study, conducted on AD-like pathology with amyloid and neurofibrillary tangles (ADLPAPT) transgenic mouse model of AD, showed that the ADLPAPT mouse differed from the wild-type (WT) mouse in the composition of the gut microbiota and was characterized by the loss of epithelial barrier integrity and chronic intestinal and systemic inflammation.130 In this study, a frequent transfer or transplantation of the fecal microbiota from WT mice into ADLPAPT mice reduced the formation of amyloid β plaques and neurofibrillary tangles and improved glial reactivity and cognitive impairment.130 There is growing evidence suggesting that AD is closely related to the imbalance of gut microbiota or may even begin in the gut. Considering an immense compositional variation of gut microbiota between and within individuals, therapeutic interventions targeting microbiota have a great potential to become a new tool of precision medicine approach in AD.
Potential Epigenetic Therapy for AD
Various findings suggest that epigenetic editing might offer future therapeutic approaches in AD,131 targeting DNA methylation, chromatin remodeling, and non-coding RNAs.132 Among the common epigenetic mechanisms, histone modifications are involved in the regulation of gene expression and play an important role in AD pathogenesis.133,134 Histone acetylation is the most-studied histone posttranslational modification, and the results of the studies using cellular and animal models of AD demonstrate a consistent reduction in histone acetylation.135,136 Therefore, a variety of histone deacetylase inhibitors (HDACi) have been tested for their potential in reversing pathological hallmarks of AD both in vitro and in vivo.133 HDACi have been shown to affect AD pathology, including Aβ accumulation and tau phosphorylation, as well as to improve synaptic plasticity, learning and memory.137,138 The pan-HDACi such as vorinostat (SAHA), trichostatin A (TSA), valproic acid (VPA), sodium butyrate (NaB), sodium 4-phenylbutyrate (4-PBA) and entinostat (MS-275), which affect class I, II and IV HDACs, as well as nicotinamide, a specific inhibitor for sirtuin class III HDACs have been shown to reverse the hypoacetylation and have beneficial effects in AD animal studies.137,138 The efficacy of HDAC-inhibitor, vorinostat in attenuating the expression of transcription factor PU.1 in human microglia has been shown, suggesting that it may be useful to reduce microglial-mediated immune responses, such as the excess inflammation observed in AD.139 In addition, treatment with some recently developed HDACi, such as hydroxamide-based class I and II HDACi (I2) and mercaptoacetamide-based class II HDACi (W2) with longer half-life and better ability to cross the blood brain barrier, restored learning and memory in AD mice.140 These compounds have shown some promise in the studies of AD since they appear to reduce AD pathology in vitro and memory impairments in vivo.141
Some of HDACi, such as VPA, nicotinamide, and 4-PBA, have been investigated in clinical trials for AD treatment.132,142 However, HDACi and especially pan-HDACi usually exhibit toxic effects, limiting their clinical application,143 and different strategies can be employed to overcome these drawbacks. The HDACi are usually non-selective and affect not only histones but also other cytoplasmic proteins.144 It has been observed that increased HDAC2 and HDAC3 activity exerts a negative impact on cognition, while reduced HDAC1 activity may be neurotoxic; therefore, HDAC-based therapy inhibiting HDAC2 or HDAC3, but not HDAC1 could represent potential therapeutic target for AD.141 For instance, RGFP-966, a selective HDAC3 inhibitor has been shown to decrease pathological tau phosphorylation and Aβ protein expression, improve learning and memory and normalize a number of AD-related genes in cellular and animal AD model.145 Other examples of specific HDACi are tubacin, a selective HDAC6 inhibitor, and suramin, a selective S1RT1 and S1RT2 inhibitor.137 Improved spatial memory and cognitive performance, decreased expression of APOEε4, β-amyloid, β-secretase and phosphorylated tau, as well as elevated levels of BDNF, ADAM10, SIRT1, REST, BIN1, MINT2 have been observed in mice after treatment with HDACi M344 with substrate selectivity for HDAC6.140,146
An additional strategy, which suits AD very well due to its multifactorial origin, is the development of multitarget drugs (MTDs).143 The examples of HDACi-based MTDs include the concomitant modulation of HDACs with phosphodiesterase 5 (PDE5),147,148 antioxidant properties,149 transglutaminase 2 (TG2)150 and glycogen kinase synthase 3β (GSK3β).151 In the AD mouse model, chronic treatment with CM-414 that acts as a dual inhibitor of PDE5 and HDACs, rescued the impaired long-term potentiation in hippocampal slices, diminished brain Aβ and tau phosphorylation levels, increased the inactive form of GSK3β, reverted the decrease in dendritic spine density on hippocampal neurons and reversed cognitive deficits.152
Another potential therapeutic approach might be the increase in histone acetyltransferases (HATs).153 Activation of specific HATs may reinstate general acetylation balance and activate gene expression programs involved in neuroprotection. Several HATs, including CBP (cAMP-response element binding protein), p300 and PACAF (p300/CBP-associated factor) showed more specific performance than non-selective HDACi. However, the poor solubility and membrane permeability of HAT activators make them rather unsuitable for AD treatment.132 Nevertheless, a recent study demonstrated that CSP-TTK21, a small-molecule activator of CBP/p300 HAT efficiently reverses epigenetic, transcriptional, synaptic plasticity, and behavioral deficits in a mouse model of AD.154 Alternative strategies also consider natural products, such as curcumin and derivatives, which inhibit the formation of Aβ oligomers and tau aggregation, and have anti-inflammatory and antioxidative properties.155 Curcumin has been considered as a selective inhibitor of the p300/CREB binding protein HAT activity.156 Several clinical trials investigated the combinations of curcumin with other natural compounds as potential treatment for AD and mild cognitive impairment.132,142 Other phytochemicals such as resveratrol and epigallocatechin gallate (EGCG) have been shown to regulate histone acetylation. Resveratrol was identified as activator of the conserved HDAC class III family of the sirtuins. Resveratrol has antioxidant, anti-inflammatory, and neuroprotective properties and can decrease the toxicity and aggregation of Aβ peptides in the hippocampus of AD patients, promote neurogenesis, and prevent hippocampal damage.157 The key neuroprotective mechanism of resveratrol in AD seems to be linked with SIRT1 activation.158 Although, clinical trials are evaluating the potential of resveratrol in the prevention of cognitive impairment and cerebrovascular dysfunction in AD,132,142 evidence-based clinical studies are still insufficient. Citicoline or cytidine-5ʹ-diphosphate-choline is a naturally occurring cholinergic compound in human cells, with particularly high abundance in the brain tissue where it serves as a precursor to the neurotransmitter acetylcholine. It was proven to have beneficial effects on cognitive functions in AD patients when combined with memantine treatment,159 treatment with AChE inhibitors,160 or as triple therapy with memantine and AChE inhibitors.161 Citicoline is also an important intermediate in the biosynthesis of phospholipids, essential components in neuronal membranes, and a potential neuroprotective agent due to its positive effect on sirtuin 1 (SIRT1) expression.162 Hypermethylation of the SIRT1 gene and decreased expression of SIRT1 is a common finding in AD, related to pathogenic mechanisms such as abnormal APP processing, neuroinflammation, neurodegeneration, and mitochondrial dysfunction.163 Neuroprotective effects of citicoline could also be associated with its ability to modulate the activity and expression of certain MAP-kinase family members, which are involved in neuronal death.164
Histone methyltransferase inhibitors, such as S-adenosyl methionine (SAM), which is one of the main methyl donors in the body, as well as DNA and histone methylation activator, might also have potential therapeutic effects in AD.132 However, large loss of methyltransferase function has been associated with learning deficiencies in both AD patients and mouse models of AD. Therefore, only partial histone methyltransferase inhibition could help restore balanced enzyme function.141 In summary, despite substantial progress, the role of histone epigenetic modifications in AD and their potential for AD treatment require further investigation. In the future, obtained pharmacoepigenetic data could help to optimize the precision medicine approach to AD.
Numerous studies have reported dysregulation of specific miRNAs associated with AD pathology and suggested that this dysregulated miRNA pattern could be used in order to improve AD diagnostics and serve as the basis for a novel and more effective therapeutic approach.165–167 Parsi et al168 suggested miR-16 as a good candidate for future development of AD therapy. Using brain delivery of miR-16 mimics in mice, they achieved the downregulation of APP, BACE1, and tau in a region-dependent manner. They also identified additional targets of miR-16, including nicastrin, γ-secretase, α-synuclein and transferrin receptor 1 (TfR1). Similar effect on tau protein expression was observed in Neuro2a cells for miR-132 mimics.169 Treatment with miR-132 mimics had a beneficial effect on memory function in mice and supported the role of miR-132/212 in the regulation of tau pathology.169 The importance of miR-132 in AD pathology and its therapeutic potential were confirmed by intracerebroventricular injections of miR-132 mimics, which resulted in upregulation of inositol 1,4,5-trisphosphate 3-kinase B (ITPKB), an enzyme involved in Aβ deposition and tau phosphorylation.170 Transfection of PC12 and SH-SY5Y cells with miR-193a-3p mimics reduced Aβ-induced neurotoxicity by targeting the expression of phosphatase and tensin homolog (PTEN) gene.171 The miR-107 was shown to have neuroprotective effects in PC12 cells and in mice after the treatment with 6-hydroxydopamine, suggesting that miR-107 plays an important role against neurotoxicity by inhibiting the expression of programmed cell death 10 (PDCD10) protein.172
Administration of miR-326 lentiviral vectors into AD mice inhibited c-Jun N-terminal kinase (JNK) signaling pathway, downregulated the expression of proto-oncogene VAV1, and inhibited tau phosphorylation, leading to improvements in cognitive function.173 Based on these results, miR-326 might also be a promising target for AD treatment. The use of miR‐34c antagomir (AM34c) ameliorated the cognitive function in SAMP8 mice by negatively regulating the expression of synaptotagmin 1.174 A similar effect was achieved with miR-188-5p oligonucleotide transfection in 5XFAD mouse model of AD.175 Zolochevska et al176 used in vivo mouse model and intracerebroventricular injections to demonstrate that miR-149, miR-485 and miR-4723 could prevent Aβ oligomer binding to the synapses. The miR-200b and miR-200c mimics were also able to prevent Aβ-derived toxicity in mice.177 Injection of lentiviral particles encoding miR-31 in 3xTg-AD mice resulted in APP and BACE1 downregulation improved cognitive functions and decreased anxiety levels.178
Recently, the potential of blocking miR-592 in order to lower oxidative stress injury in astrocytes was reported and it was suggested to be mediated by dyslexia-associated protein KIAA0319 and (Kelch-like ECH-associating protein 1) nuclear factor erythroid 2 related factor 2-antioxidant response element (Keap1/Nrf2/ARE) signaling pathway.179 Wang et al180 detected upregulation of miR-33 expression in SH-SY5Y cells after treatment with Aβ25–35. Downregulation of miR-33 suppressed inflammation, oxidative stress, and cell apoptosis, while also improving synaptic plasticity.180 The protective effect of miR-33 downregulation was achieved by suppressing Akt/mammalian target of rapamycin (mTOR) signaling pathway activation.180 Intra-hippocampal injection of miR-342-3p antagomir in 3xTg-AD mice confirmed the association between miR-342-3p and AD, suggesting that miR-342-3p inhibition can improve cognitive deficits.181 Downregulation of miR-342-3p also attenuated the hippocampal Aβ-plaque burden revealing the important effect of miR-342-3p on Aβ metabolism pathways in AD and suggesting its therapeutical potential.181 The miR-299-5p treatment was shown to affect the levels of autophagy related 5 (ATG5) protein and attenuate autophagy in different cell lines, while the injection of miR-299-5p into cerebral ventricles of AD mice model inhibited autophagy and apoptosis, resulting in improved cognitive performance and pointing to miR-299-5p as a potential neuroprotective factor in AD.182
Pereira et al183 made a big step towards the usability of miRNA-based therapeutics by successfully encapsulating pre-miR-29b into polyplexes and by verifying that chitosan/pre-miR-29b and polyethylenimine/pre-miR-29b systems efficiently deliver pre-miR-29b to cell cytoplasm and reduce BACE1 expression and Aβ42 levels. Gabr and Brogi184 used high-throughput screening in order to identify multitargeted therapeutics for AD. They focused on ligands capable of inhibiting AChE and miR-15b biogenesis. The dual screening strategy yielded a multitarget-directed ligand, MG-6267, which acts through dual inhibition of AChE and miR-15b biogenesis.184 Cellular assays confirmed effectiveness of MG-6267 in protecting neuroblastoma SH-SY5Y cells from Aβ-induced cytotoxicity.184
Berberine, which is most commonly used to help treat diabetes, obesity, and inflammation, was shown to increase circRNA histone deacetylase 9 (circHDAC9) expression and decrease miR-142-5p level in human neuronal cells treated with Aβ42.185 Zhang et al185 suggested that berberine has neuroprotective effect in AD since it protected human neuronal cells from neuronal damage induced by Aβ42 by regulating the circHDAC9/miR-142-5p axis. Berberine treatment, along with lncRNA β-amyloid cleaving enzyme 1 antisense RNA (BACE1-AS) interference, protected neuronal cells treated with Aβ25–35 and recovered their viability.186 Berberine was suggested to act, at least partly, through BACE1-AS/miR-132-3p axis, suggesting the potential of combined berberine treatment and BACE1-AS depletion in AD therapy.186 Osthole, a naturally active coumarin, was shown to upregulate the expression of miR-101a-3p.187 In APP/PS1 mice, osthole increased learning and memory abilities187 and enhanced cell viability, prevented cell death, and reversed the downregulation of synapsin-1, synaptophysin, and postsynaptic density-95 (PSD-95) protein in a cellular model of AD.188 This effect of osthole was associated with the upregulation of miR-9 and consequent decrease in calcium/calmodulin dependent protein kinase kinase 2 (CAMKK2) and phospho-AMP-activated kinase α (p-AMPKα) levels. Jiao et al189 additionally demonstrated an effect of osthole treatment on miR-107 expression. Administration of osthole to APP/PS1 mice increased miR-107 expression, resulting in BACE1 inhibition, significant decrease of Aβ in hippocampal and cortex regions, and in the improvement of memory and learning abilities.189 These results suggested protective effect of osthole and its potential to delay the development of AD.
Memantine which is traditionally used to treat patients with AD, was demonstrated to ameliorate BBB permeability.190 This NMDA receptor antagonist downregulated the expression of long non‐coding RNA LINC00094 and decreased the permeability of BBB by upregulating miR‐224‐4p/miR‐497‐5p and thus inhibiting the expression of endophilin‐1, a multifunctional protein which, among other things, regulates BBB permeability.190 Therefore, a combination of LINC00094 silencing and memantine treatment could be a novel target in AD therapy research. Donepezil, AChE inhibitor used for AD treatment, probably achieves its effect partially through its interaction with miR-206. Upregulation of miR-206-3p was detected in the hippocampus and cortex of APP/PS1 mice and it was normalized after administration of donepezil, suggesting that miR-206-3p could be a novel pharmacological target in AD and a mediator of anti-dementia effects of donepezil.191
There are studies showing that resveratrol, a natural polyphenol found in red wine, can reduce neuroinflammation and Aβ deposition in patients with mild or moderate AD.192 The 52-week phase-II clinical trial gave evidence that resveratrol reduced Aβ40 in CSF and plasma samples of individuals with mild to moderate AD.193 It is interesting that resveratrol might exert its effect by modulating the expression of specific miRNAs, including the downregulation of pro-inflammatory miR-155 and the upregulation of anti-inflammatory miR-663.194 All the above mentioned evidence led Kou and Chen195 to suggest resveratrol as a potential target in the prevention and/or treatment of AD. Simvastatin, used to lower cholesterol levels, was demonstrated to have a positive effect on memory deficits in patients with AD and in animal model of AD.196 Simvastatin reduced the expression of inflammatory cytokines and mediators, suppressed the apoptosis and contributed to the survival of neurons in the study conducted by Huang et al,196 suggesting that simvastatin could be used as an anti-AD treatment, achieving its effect through modulation of miR-106b expression. Therefore, miRNA-based therapy has a great therapeutical potential in complex diseases such as AD, in which the cause is related to a number of genes and biological processes. Future studies will try to implement anti-miRNAs or miRNA mimics as precision medicine approaches for the treatment of AD patients; however, there is still a long way to go before certain miRNAs will be available for AD therapy. Nevertheless, epigenetics represents a new powerful tool for precision medicine, by applying the knowledge of epigenetic mechanisms and epigenetic profiles of AD patients to personalized diagnostics and treatments in AD.
Conclusion
Currently available medications for AD are based only on symptomatic therapy, and include AChE inhibitors and NMDA receptor antagonist. Clinical research has been extensively investigating treatments focusing of the hallmark pathology of AD, including amyloid deposition, tau hyperphosphorylation, neuroinflammation and vascular changes; however, so far without success since all new potential drugs failed to show significant clinical benefit. Due to the large heterogeneity of AD etiology, cognitive, behavioral and other symptoms, somatic comorbidities, and patient lifestyle, there is no preventive or therapeutic intervention suitable for all AD patients. Therefore, precision medicine strategy is urgently required to guide clinical practice. In addition to factors such as dementia stage, medical and psychiatric comorbidity, as well as major cognitive symptoms and BPSD, the complex interplay between genetic variability and environmental factors moderated by epigenetic changes, should be taken into account for each individual AD patient. Both gut microbiota and epigenetic modifications represent new emerging drug targets, whose specific patterns could form the basis for novel individually tailored, patient-centered, multidisciplinary approaches aimed to optimize personalized prevention and treatment of AD. However, the successful application of precision medicine to AD demands a further extensive research of underlying pathological processes, as well as clinical and biological complexity of this multifactorial neurodegenerative disorder.
Funding
This study was funded by the Croatian Science Foundation project no. IP-2019-04-6100.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Crous-Bou M, Minguillόn C, Gramunt N, Molinuevo JL. Alzheimer´s disease prevention: from risk factors to early intervention. Alzheimers Res Ther. 2017;9(1):71. doi:10.1186/s13195-017-0297-z
2. Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179(2):312–339.
3. Hou Y, Dan X, Babbar M, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15(10):565–581. doi:10.1038/s41582-019-0244-7
4. Guerreiro R, Bras J. The age factor in alzheimer’s disease. Genome Med. 2015;7:106. doi:10.1186/s13073-015-0232-5
5. Herrup K. Reimagining alzheimer’s disease-an age-based hypothesis. J Neurosci. 2010;30(50):16755–16762. doi:10.1523/JNEUROSCI.4521-10.2010
6. Hoozemans JJM, Rozemuller AJM, van Haastert ES, Eikelenboom P, van Gool WA. Neuroinflammation in alzheimer’s disease wanes with age. J Neuroinflammation. 2011;8:171. doi:10.1186/1742-2094-8-171
7. Vest RS, Pike CJ. Gender, sex steroid hormones, and alzheimer’s disease. Horm Behav. 2013;63(2):301–307. doi:10.1016/j.yhbeh.2012.04.006
8. Ferretti MT, Iulita MF, Cavedo E, et al. Sex differences in alzheimer disease - the gateway to precision medicine. Nat Rev Neurol. 2018;14(8):457–469. doi:10.1038/s41582-018-0032-9
9. Fratiglioni L, Grut M, Forsell Y, et al. Prevalence of alzheimer’s disease and other dementias in an elderly urban population: relationship with age, sex, and education. Neurology. 1991;51:1886–1892. doi:10.1212/WNL.41.12.1886
10. Sinforiani E, Citterio A, Zucchella C, et al. Impact of gender differences on the outcome of alzheimer’s disease. Dement Geriatr Cogn Disord. 2010;30(2):147–154. doi:10.1159/000318842
11. Weiner MF. Perspective on race and ethnicity in alzheimer’s disease research. Alzheimers Dement. 2008;4(4):233–238. doi:10.1016/j.jalz.2007.10.016
12. Mehta KM, Yaffe K, Perez-Stable EJ, et al. Race/ethnic differences in AD survival in US alzheimer’s disease centers. Neurology. 2008;70(14):1163–1170. doi:10.1212/01.wnl.0000285287.99923.3c
13. Nikolac Perkovic M, Pivac N. Genetic markers of alzheimer’s disease. In: Kim YK, editor. Advances in Experimental Medicine and Biology. Frontiers in Psychiatry: Artificial Intelligence, Precision Medicine, and Other Paradigm Shifts. Singapore: Springer Publ.; 2019:27–52.
14. Cacace R, Sleegers K, Van Broeckhoven C. Molecular genetics of early-onset Alzheimer's disease revisited. Alzheimers Dement. 2016;12(6):733–748. doi:10.1016/j.jalz.2016.01.012
15. Suidan GL, Ramaswamy G. Targeting apolipoprotein E for alzheimer´s disease: an industry perspective. Int J Mol Sci. 2019;20(9):2161. doi:10.3390/ijms20092161
16. Silva MVF, Loures CMG, Alves LCV, de Souza LC, Borges KBG, Carvalho MDG. Alzheimer´s disease: risk factors and potentially protective measures. J Biomed Sci. 2019;26(1):33. doi:10.1186/s12929-019-0524-y
17. Dubois B, Hampel H, Feldman HH, et al. Preclinical Alzheimer´s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12(3):292–323.
18. Hampel H, O’Bryant S, Durrleman S, et al. A precision medicine initiative for Alzheimer’s disease: the road ahead to biomarker-guided integrative disease modeling. Climacteric. 2017;20(2):107–118. doi:10.1080/13697137.2017.1287866
19. Nikolac Perkovic M, Svob Strac D, Tudor L, Konjevod M, Nedic Erjavec G, Pivac N. Catechol-O-methyltransferase, cognition and alzheimer’s disease. Curr Alzheimer Res. 2018;15:408–419. doi:10.2174/1567205015666171212094229
20. Finney GR, Minagar A, Heilman KM. Assessment of mental status. Neurol Clin. 2016;34(1):1–16. doi:10.1016/j.ncl.2015.08.001
21. Bredesen DE, Amos EC, Canick J, et al. Reversal of cognitive decline in Alzheimer´s disease. Aging. 2016;8(6):1250–1258. doi:10.18632/aging.100981
22. Lin Y, Shan PY, Jiang WJ, Sheng C, Ma L. Subjective cognitive decline: preclinical manifestation of alzheimer’s disease. Neurol Sci. 2019;40(1):41–49. doi:10.1007/s10072-018-3620-y
23. Rabin LA, Smart CM, Amariglio RE. Subjective cognitive decline in preclinical alzheimer´s disease. Annu Rev Clin Psychol. 2017;13:369–396. doi:10.1146/annurev-clinpsy-032816-045136
24. Jessen F, Amariglio RE, Buckley RF, et al. The characterisation of subjective cognitive decline. Lancet Neurol. 2020;19(3):271–278. doi:10.1016/S1474-4422(19)30368-0
25. Park SJ, Lee JE, Lee KS, Kim JS. Comparison of odor identification among amnestic and non-amnestic mild cognitive impairment, subjective cognitive decline, and early alzheimer´s dementia. Neurol Sci. 2018;39(3):557–564. doi:10.1007/s10072-018-3261-1
26. Eratne D, Loi SM, Farrand S, Kelso W, Velakoulis D, Looi JCL. Alzheimer´s disease paper 1: clinical update on epidemiology, pathophysiology and diagnosis. Australas Psychiatry. 2018;26(4):347–357. doi:10.1177/1039856218762308
27. Lloret A, Esteve D, Monllor P, Cervera-Ferri A, Lloret A. The effectiveness of vitamin E treatment in alzheimer´s disease. Int J Mol Sci. 2019;20(4):879. doi:10.3390/ijms20040879
28. Duncan T, Valenzuela M. Alzheimer´s disease, dementia, and stem cell therapy. Stem Cell Res Ther. 2017;8:111. doi:10.1186/s13287-017-0567-5
29. Kametani F, Hasegawa M. Reconsideration of amyloid hypothesis and tau hypothesis in alzheimer’s disease. Front Neurosci. 2018;12:25. doi:10.3389/fnins.2018.00025
30. Zhang T, Chen D, Lee TH. Phosphorylation signaling in APP processing in alzheimer’s disease. Int J Mol Sci. 2020;21(1):209. doi:10.3390/ijms21010209
31. Simic G, Spanic E, Langer Horvat L, Hof PR. Blood-brain barrier and innate immunity in the pathogenesis of alzheimer’s disease. Prog Mol Biol Transl Sci. 2019;168:99–145.
32. Simic G, Babić Leko M, Wray S, et al. Tau protein hyperphosphorylation and aggregation in alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules. 2016;6(1):6. doi:10.3390/biom6010006
33. McGlinchey E, McCallion P, McCarron M. Down syndrome and dementia: advances in the field. Curr Opin Psychiatry. 2020;33(3):278–283. doi:10.1097/YCO.0000000000000589
34. Lemoine L, Ledreux A, Mufson EJ, et al. Regional binding of tau and amyloid PET tracers in down syndrome autopsy brain tissue. Mol Neurodegener. 2020;15:68. doi:10.1186/s13024-020-00414-3
35. Arnsten AFT, Datta D, Del Tredici K, Braak H. Hypothesis: tau pathology is an initiating factor in sporadic alzheimer’s disease. Alzheimers Dement. 2020;17(1):115–124. doi:10.1002/alz.12192
36. Stefanova NA, Muraleva NA, Korbolina EE, Kiseleva E, Maksimova KY, Kolosova NG. Amyloid accumulation is a late event in sporadic alzheimer’s disease-like pathology in nontransgenic rats. Oncotarget. 2015;6(3):1396–1413. doi:10.18632/oncotarget.2751
37. Singh A, Allen D, Tumurbaatar B, et al. Functional integrity of synapses in the central nervous system of cognitively intact individuals with high alzheimer’s disease neuropathology is associated with absence of synaptic tau oligomers. J Alzheimers Dis. 2020;78(4):1661–1678. doi:10.3233/JAD-200716
38. Franzmeier N, Dewenter A, Frontzkowski L, et al. Patient-centered connectivity-based prediction of tau pathology spread in alzheimer’s disease. Sci Adv. 2020;6(48):eabd1327. doi:10.1126/sciadv.abd1327
39. Cuello AC. Early and late CNS inflammation in alzheimer’s disease: two extremes of a continuum? Trends Pharmacol Sci. 2017;38(11):956–966. doi:10.1016/j.tips.2017.07.005
40. Spanic E, Langer Horvat L, Hof PR, Šimić G. Role of microglial cells in alzheimer’s disease tau propagation. Front Aging Neurosci. 2019;11:271. doi:10.3389/fnagi.2019.00271
41. Simic G. Rare diseases and omics-driven personalized medicine. Croat Med J. 2019;60:485–487. doi:10.3325/cmj.2019.60.485
42. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. doi:10.1038/nature21029
43. Culjak M, Nikolac Perkovic M, Uzun S, et al. The association between TNFα, IL-1α and IL-10 with alzheimer’s disease. Curr Alzheimer Res. 2020;17(11):972–984. doi:10.2174/1567205017666201130092427
44. Babic Leko M, Nikolac Perković M, Klepac N, et al. IL-1α, IL-6, IL-10, and TNFα single nucleotide polymorphisms in human influence the susceptibility to alzheimer’s disease pathology. J Alzheimers Dis. 2020;75(3):1029–1047. doi:10.3233/JAD-200056
45. Hemonnot AL, Hua J, Ulmann L, Hirbec H. Microglia in alzheimer disease: well-known targets and new opportunities. Front Aging Neurosci. 2019;11:233. doi:10.3389/fnagi.2019.00233
46. Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell. 2018;173(5):1073–1081. doi:10.1016/j.cell.2018.05.003
47. Keren-Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of alzheimer’s disease. Cell. 2017;169(7):1276–1290.e17. doi:10.1016/j.cell.2017.05.018
48. Yeh FL, Hansen DV, Sheng M. TREM2, microglia, and neurodegenerative diseases. Trends Mol Med. 2017;23(6):512–533. doi:10.1016/j.molmed.2017.03.008
49. Colonna M, Wang Y. TREM2 variants: new keys to decipher alzheimer disease pathogenesis. Nat Rev Neurosci. 2016;17(4):201–207. doi:10.1038/nrn.2016.7
50. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. doi:10.1056/NEJMoa1211851
51. Wang Y, Ulland TK, Ulrich JD, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016;213(5):667–675. doi:10.1084/jem.20151948
52. Yuan P, Condello C, Keene CD, et al. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. 2016;90(4):724–739. doi:10.1016/j.neuron.2016.05.003
53. Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47(3):566–581.e9. doi:10.1016/j.immuni.2017.08.008
54. Sweeney MD, Sagare AP, Zlokovic BV. Blood–brain barrier breakdown in alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133–150.
55. Reitz C. Toward precision medicine in alzheimer’s disease. Ann Transl Med. 2016;4(6):107. doi:10.21037/atm.2016.03.05
56. Barman NC, Khan NM, Islam M, et al. CRISPR-Cas9: a promising genome editing therapeutic tool for alzheimer´s disease-a narrative review. Neurol Ther. 2020;9(2):419–434. doi:10.1007/s40120-020-00218-z
57. Amanatkar H, Papagiannopoulos B, Grossberg G. Analysis of recent failures of disease modifying therapies in alzheimer’s disease suggesting a new methodology for future studies. Expert Rev Neurother. 2016;17(1):5–25.
58. Weller J, Budson A. Current understanding of alzheimer´s disease diagnosis and treatment. F1000Res. 2018;7:F1000. doi:10.12688/f1000research.14506.1
59. Briggs R, Kennelly SP, O’Neill D. Drug treatment in alzheimer’s disease. Clin Med. 2016;16(3):247–253. doi:10.7861/clinmedicine.16-3-247
60. Ferreira-Vieira TH, Silva FR, Ribeiro FM. Targeting the cholinergic system. Curr Neuropharmacol. 2016;14(1):101–115. doi:10.2174/1570159X13666150716165726
61. Sharma K. Cholinesterase inhibitors as alzheimer’s therapeutics. Mol Med Rep. 2019;20(2):1479–1487. doi:10.3892/mmr.2019.10374
62. Krizaj Grden A, Mimica N. Treatment approach to alzheimer’s disease. Socijalna psihijatrija. 2019;47(3):325–334. doi:10.24869/spsih.2019.325
63. Sultan S, Al-Hamady M, Hesham Hafiz A, Mashhoor Al-Barakati A, Al-Huthali RK, Al-Jahdali NH. An update on treatment of alzheimer disease - a literature review. Eur J Pharm Med Res. 2018;5(7):9–18.
64. Wang R, Reddy PH. Role of glutamate and NMDA receptors in alzheimer’s disease. J Alzheimers Dis. 2017;57(4):1041–1048. doi:10.3233/JAD-160763
65. Chertkow H, Hogan DB, Black S, et al. An action plan to face the challenge of dementia: international statement on dementia from IAP for health. J Prev Alzheimers Dis. 2018;5(3):207–212. doi:10.14283/jpad.2018.27
66. Hung SY, Fu WM. Drug candidates in clinical trials for alzheimer’s disease. J Biomed Sci. 2017;24(1):47. doi:10.1186/s12929-017-0355-7
67. Birks J, Harvey RJ. Donepezil for dementia due to alzheimer’s disease. Cochrane Database Syst Rev. 2018;6(6):CD001190. doi:10.1002/14651858.CD001190.pub3
68. Nguyen K, Hoffman H, Chakkamparambil B, Grossberg GT. Evaluation of rivastigmine in alzheimer’s disease. Neurodegener Dis Manag. 2021;11(1):35–48. doi:10.2217/nmt-2020-0052
69. Blesa R, Toriyama K, Ueda K, Knox S, Grossberg G. Strategies for continued successful treatment in patients with alzheimer’s disease: an overview of switching between pharmacological agents. Curr Alzheimer Res. 2018;15(10):964–974. doi:10.2174/1567205015666180613112040
70. Kishi T, Matsunaga S, Oya K, Nomura I, Ikuta T, Iwata N. Memantine for alzheimer’s disease: an updated systematic review and meta-analysis. J Alzheimers Dis. 2017;60(2):401–425. doi:10.3233/JAD-170424
71. Folch J, Busquets O, Ettcheto M, Sanchez-Lopez E, Castro-Torres RD, Verdaguer E. Memantine for the treatment of dementia: a review on its current and future applications. J Alzheimers Dis. 2018;62(3):1223–1240. doi:10.3233/JAD-170672
72. Chen R, Chan PT, Chu H, et al. Treatment effect between monotherapy of donepezil versus combination with memantine for alzheimer disease: a meta-analysis. PLoS One. 2017;12(8):e0183568.
73. Dou KX, Tan MS, Tan CC, et al. Comparative safety and effectiveness of cholinesterase inhibitors and memantine for alzheimer’s disease: a network meta-analysis of 41 randomized controlled trials. Alzheimers Res Ther. 2018;10(1):126. doi:10.1186/s13195-018-0457-9
74. Kandimalla R, Reddy PH. Therapeutics of neurotransmitters in alzheimer’s disease. J Alzheimers Dis. 2017;57(4):1049–1069. doi:10.3233/JAD-161118
75. Di Giovanni G, Svob Strac D, Sole M, et al. Monoaminergic and histaminergic strategies and treatments in brain diseases. Front Neurosci. 2016;10:541.
76. Kang YJ, Diep YN, Tran M, Cho H. Therapeutic targeting strategies for early- to late-staged alzheimer’s disease. Int J Mol Sci. 2020;21(24):9591. doi:10.3390/ijms21249591
77. Abeysinghe AA, Deshapriya RD, Udawatte C. Alzheimer’s disease; a review of the pathophysiological basis and therapeutic interventions. Life Sci. 2020;256:117996. doi:10.1016/j.lfs.2020.117996
78. Husna Ibrahim N, Yahaya MF, Mohamed W, Teoh SL, Hui CK, Kumar J. Pharmacotherapy of alzheimer’s disease: seeking clarity in a time of uncertainty. Front Pharmacol. 2020;11:261. doi:10.3389/fphar.2020.00261
79. Morley JE, Farr SA. The role of amyloid-beta in the regulation of memory. Biochem Pharmacol. 2014;88(4):479–485. doi:10.1016/j.bcp.2013.12.018
80. Brothers HM, Gosztyla ML, Robinson SR. The physiological roles of amyloid-β peptide hint at new ways to treat alzheimer’s disease. Front Aging Neurosci. 2018;10(118):1–16. doi:10.3389/fnagi.2018.00118
81. Galimberti D, Scarpini E. Disease-modifying treatments for alzheimer’s disease. Ther Adv Neurol Disord. 2011;4(4):203–216. doi:10.1177/1756285611404470
82. Ballard C, Aarsland D, Cummings J, et al. Drug repositioning and repurposing for alzheimer disease. Nat Rev Neurol. 2020;16(12):661–673. doi:10.1038/s41582-020-0397-4
83. Jadhav S, Avila J, Schöll M, et al. A walk through tau therapeutic strategies. Acta Neuropathol Commun. 2019;7(1):22.
84. Scassellati C, Ciani M, Maj C, et al. Behavioral and Psychological Symptoms of Dementia (BPSD): clinical characterization and genetic correlates in an Italian Alzheimer’s disease cohort. J Pers Med. 2020;10(3):90. doi:10.3390/jpm10030090
85. Hashimoto M, Yatabe Y, Ishikawa T, et al. Relationship between dementia severity and behavioral and psychological symptoms of dementia in dementia with lewy bodies and alzheimer’s disease patients. Dement Geriatr Cogn Dis Extra. 2015;5(2):244–252. doi:10.1159/000381800
86. Chakraborty S, Lennon JC, Malkaram SA, et al. Serotonergic system, cognition, and BPSD in alzheimer’s disease. Neurosci Lett. 2019;704:36–44. doi:10.1016/j.neulet.2019.03.050
87. Lee J, Lee KJ, Kim H. Gender differences in behavioral and psychological symptoms of patients with alzheimer’s disease. Asian J Psychiatr. 2017;26:124–128. doi:10.1016/j.ajp.2017.01.027
88. Deardorff WJ, Grossberg GT. Behavioral and psychological symptoms in alzheimer’s dementia and vascular dementia. Handb Clin Neurol. 2019;165:5–32.
89. Mushtaq R, Pinto C, Tarfarosh SFA, et al. A comparison of the Behavioral and Psychological Symptoms of Dementia (BPSD) in early-onset and late-onset alzheimer’s disease - a study from South East Asia (Kashmir, India). Cureus. 2016;8(5):e625.
90. Tible OP, Riese F, Savaskan E, von Gunten A. Best practice in the management of behavioural and psychological symptoms of dementia. Ther Adv Neurol Disord. 2017;10(8):297–309. doi:10.1177/1756285617712979
91. Bessey LJ, Walaszek A. Management of behavioral and psychological symptoms of dementia. Curr Psychiatry Rep. 2019;21(8):66. doi:10.1007/s11920-019-1049-5
92. Preuss UW, Wong JWM, Koller G. Treatment of behavioral and psychological symptoms of dementia: a systematic review. Psychiatr Pol. 2016;50(4):679–715. doi:10.12740/PP/64477
93. de Oliveira AM, Radanovic M, Cotting Homem de Mello P, et al. Nonpharmacological interventions to reduce behavioral and psychological symptoms of dementia: a systematic review. Biomed Res Int. 2015;2015:218980. doi:10.1155/2015/218980
94. Gomez Gallego M, Gomez Garcia J. Music therapy and alzheimer’s disease: cognitive, psychological, and behavioural effects. Neurologia. 2017;32(5):300–308. doi:10.1016/j.nrl.2015.12.003
95. Ohno Y, Kunisawa N, Shimizu S. Antipsychotic treatment of Behavioral and Psychological Symptoms of Dementia (BPSD): management of extrapyramidal side effects. Front Pharmacol. 2019;10:1045. doi:10.3389/fphar.2019.01045
96. Tampi RR, Tampi DJ, Rogers K, Alagarsamy S. Antipsychotics in the management of behavioral and psychological symptoms of dementia: maximizing gain and minimizing harm. Neurodegener Dis Manag. 2020;10(1):5–8. doi:10.2217/nmt-2019-0036
97. Seeman MV. Men and women respond differently to antipsychotic drugs. Neuropharmacology. 2020;163:107631. doi:10.1016/j.neuropharm.2019.05.008
98. Zhou T, Wang J, Xin C, Kong L, Wang C. Effect of memantine combined with citalopram on cognition of BPSD and moderate alzheimer’s disease: a clinical trial. Exp Ther Med. 2019;17(3):1625–1630. doi:10.3892/etm.2018.7124
99. Carotenuto A, Rea R, Traini E, et al. The effect of the association between donepezil and choline alphoscerate on behavioral disturbances in alzheimer’s disease: interim results of the ASCOMALVA trial. J Alzheimers Dis. 2017;56(2):805–815. doi:10.3233/JAD-160675
100. Peng X, Xing P, Li X, et al. Towards personalized intervention for alzheimer’s disease. Genom Proteom Bioinf. 2016;14(5):289–297. doi:10.1016/j.gpb.2016.01.006
101. Molony SL, Kolanowski A, Van Haitsma K, Rooney KE. Person-centered assessment and care planning. Gerontologist. 2018;58(S1):S32–S47. doi:10.1093/geront/gnx173
102. Simic G, Babic Leko M, Wray S, et al. Monoaminergic neuropathology in alzheimer’s disease. Prog Neurobiol. 2017;151:101–138.
103. Chen H, Kwong JC, Copes R, et al. Living near major roads and the incidence of dementia, parkinson’s disease, and multiple sclerosis: a population-based cohort study. Lancet. 2017;389:718–726. doi:10.1016/S0140-6736(16)32399-6
104. Alic I, Goh PA, Murray A, et al. Patient-specific alzheimer-like pathology in trisomy 21 cerebral organoids reveals BACE2 as a gene dose-sensitive AD suppressor in human brain. Mol Psychiatry. 2020. doi:10.1038/s41380-020-0806-5
105. Hu X, Wang T, Jin F. Alzheimer’s disease and gut microbiota. Sci China Life Sci. 2016;59(10):1006–1023. doi:10.1007/s11427-016-5083-9
106. Aziz Q, Dore J, Emmanuel A, Guarner F, Quigley EM. Gut microbiota and gastrointestinal health: current concepts and future directions. Neurogastroenterol Motil. 2013;25(1):4–15. doi:10.1111/nmo.12046
107. Dinan TG, Cryan JF. Gut instincts: microbiota as a key regulator of brain development, ageing and neurodegeneration. J Physiol. 2017;595(2):489–503. doi:10.1113/JP273106
108. Quigley EMM. Gut microbiome as a clinical tool in gastrointestinal disease management: are we there yet? Nat Rev Gastroenterol Hepatol. 2017;14(5):315–320. doi:10.1038/nrgastro.2017.29
109. The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(207–214). doi:10.1038/nature11234
110. Scheline RR. Metabolism of foreign compounds by gastrointestinal microorganisms. Pharmacol Rev. 1973;25:451–523.
111. Quigley EMM. Microbiota-brain-gut axis and neurodegenerative diseases. Curr Neurol Neurosci Rep. 2017;17(12):94. doi:10.1007/s11910-017-0802-6
112. Frasca D, Blomberg BB. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology. 2016;17(1):7–19. doi:10.1007/s10522-015-9578-8
113. Köhler CA, Maes M, Slyepchenko A, et al. The gut-brain axis, including the microbiome, leaky gut and bacterial translocation: mechanisms and pathophysiological role in alzheimer’s disease. Curr Pharm Des. 2016;22(40):6152–6166. doi:10.2174/1381612822666160907093807
114. Bhattacharjee S, Lukiw WJ. Alzheimer’s disease and the microbiome. Front Cell Neurosci. 2013;7(10):153. doi:10.3389/fncel.2013.00153
115. Friedland RP, Chapman MR. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017;13(12):e1006654. doi:10.1371/journal.ppat.1006654
116. Zhao Y, Dua P, Lukiw WJ. Microbial sources of amyloid and relevance to amyloidogenesis and Alzheimer’s Disease (AD). J Alzheimers Dis Parkinsonism. 2015;5(1):177. doi:10.4172/2161-0460.1000177
117. Fassbender K, Walter S, Kühl S, et al. The LPS receptor (CD14) links innate immunity with alzheimer’s disease. FASEB J. 2004;18(1):203–205. doi:10.1096/fj.03-0364fje
118. Wang C, Klechikov AG, Gharibyan AL, et al. The role of pro-inflammatory S100A9 in alzheimer’s disease amyloid-neuroinflammatory cascade. Acta Neuropathol. 2014;127(4):507–522. doi:10.1007/s00401-013-1208-4
119. Leblhuber F, Geisler S, Steiner K, Fuchs D, Schütz B. Elevated fecal calprotectin in patients with alzheimer’s dementia indicates leaky gut. J Neural Transm. 2015;122(9):1319–1322. doi:10.1007/s00702-015-1381-9
120. Caracciolo B, Xu W, Collins S, Fratiglioni L. Cognitive decline, dietary factors and gut–brain interactions. Mech Ageing Dev. 2014;136(137):59–69. doi:10.1016/j.mad.2013.11.011
121. Fröhlich EE, Farzi A, Mayerhofer R, et al. Cognitive impairment by antibiotic-induced gut dysbiosis: analysis of gut microbiota-brain communication. Brain Behav Immun. 2016;56:140–155. doi:10.1016/j.bbi.2016.02.020
122. Neufeld NH, Mohamed NS, Grujich N, Shulman K. Acute neuropsychiatric symptoms associated with antibiotic treatment of Helicobacter pylori infections. J Psychiatr Pract. 2017;23(1):25–35. doi:10.1097/PRA.0000000000000205
123. Pistollato F, Iglesias RC, Ruiz R, et al. Nutritional patterns associated with the maintenance of neurocognitive functions and the risk of dementia and alzheimer’s disease: a focus on human studies. Pharmacol Res. 2018;131:32–43. doi:10.1016/j.phrs.2018.03.012
124. Akbari E, Asemi Z, Kakhaki RD, et al. Effect of probiotic supplementation on cognitive function and metabolic status in alzheimer’s disease: a randomized, double-blind and controlled trial. Front Aging Neurosci. 2016;8:256. doi:10.3389/fnagi.2016.00256
125. Eskelinen MH, Ngandu T, Tuomilehto J, Soininen H, Kivipelto M. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009;16(1):85–91. doi:10.3233/JAD-2009-0920
126. Moco S, Martin FPJ, Rezzi S. Metabolomics view on gut microbiome modulation by polyphenol-rich foods. J Proteome Res. 2012;11(10):4781–4790. doi:10.1021/pr300581s
127. Cani PD, Bibiloni R, Knauf C, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–1481. doi:10.2337/db07-1403
128. Wang T, Hu X, Liang S, et al. Lactobacillus fermentum NS9 restores the antibiotic induced physiological and psychological abnormalities in rats. Benef Microbes. 2015;6(5):707–717. doi:10.3920/BM2014.0177
129. Wang X, Sun G, Feng T, et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit alzheimer’s disease progression. Cell Res. 2019;29(10):787–803. doi:10.1038/s41422-019-0216-x
130. Kim MS, Kim Y, Choi H, et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an alzheimer’s disease animal model. Gut. 2020;69(2):283–294. doi:10.1136/gutjnl-2018-317431
131. Berson A, Nativio R, Berger SL, et al. Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 2018;41(9):587–598. doi:10.1016/j.tins.2018.05.005
132. Teijido O, Cacabelos R. Pharmacoepigenomic interventions as novel potential treatments for alzheimer’s and parkinson’s diseases. Int J Mol Sci. 2018;19(10):3199. doi:10.3390/ijms19103199
133. Cacabelos R, Torrellas C. Epigenetics of aging and alzheimer’s disease: implications for pharmacogenomics and drug response. Int J Mol Sci. 2015;16(12):30483–30543. doi:10.3390/ijms161226236
134. Stoccoro A, Coppede F. Role of epigenetics in alzheimer’s disease pathogenesis. Neurodegener Dis Manag. 2018;8(3):181–193. doi:10.2217/nmt-2018-0004
135. Narayan P, Dragunow M. Alzheimer’s disease and histone code alterations. Adv Exp Med Biol. 2017;978:321–336.
136. Sharma S, Sarathlal KC, Taliyan R. Epigenetics in neurodegenerative diseases: the role of histone deacetylases. CNS Neurol Disord Drug Targets. 2019;18(1):11–18. doi:10.2174/1871527317666181004155136
137. Lardenoije R, Latrou A, Kenis G, et al. The epigenetics of aging and neurodegeneration. Prog Neurobiol. 2015;131:21–64.
138. Liu X, Jiao B, Shen L. The epigenetics of alzheimer’s disease: factors and therapeutic implications. Front Genet. 2018;9:579. doi:10.3389/fgene.2018.00579
139. Rustenhoven J, Smith AM, Smyth LC, et al. PU.1 regulates alzheimer’s disease-associated genes in primary human microglia. Mol Neurodegener. 2018;13(1):44. doi:10.1186/s13024-018-0277-1
140. Esposito M, Sherr GL. Epigenetic modifications in alzheimer’s neuropathology and therapeutics. Front Neurosci. 2019;13:476. doi:10.3389/fnins.2019.00476
141. Coneys R, Wood IC. Alzheimer’s disease: the potential of epigenetic treatments and current clinical candidates. Neurodegener Dis Manag. 2020;10(3):543–558. doi:10.2217/nmt-2019-0034
142. ClinicalTrials.gov. US National Institutes of Health. Available from: http://clinicaltrials.gov/.
143. Milelli A, De Simone A. Histone deacetylase inhibitors as multitarget ligands: new players in alzheimer’s disease drug discovery? ChemMedChem. 2019;14(11):1067–1073. doi:10.1002/cmdc.201900174
144. Lu X, Wang L, Yu C, et al. Histone acetylation modifiers in the pathogenesis of alzheimer’s disease. Front Cell Neurosci. 2015;9:226. doi:10.3389/fncel.2015.00226
145. Janczura KJ, Volmar CH, Sartor GC, et al. Inhibition of HDAC3 reverses alzheimer’s disease-related pathologies in vitro and in the 3xTg-AD mouse model. Proc Natl Acad Sci U S A. 2018;115(47):E11148–E11157. doi:10.1073/pnas.1805436115
146. Volmar C-H, Salah-Uddin H, Janczura KJ, et al. M344 promotes nonamyloidogenic amyloid precursor protein processing while normalizing alzheimer’s disease genes and improving memory. Proc Natl Acad Sci U S A. 2017;114(43):E9135–E9144. doi:10.1073/pnas.1707544114
147. Rabal O, Sánchez-Arias JA, Cuadrado-Tejedor M, et al. Design, synthesis, and biological evaluation of first-in-class dual acting Histone Deacetylases (HDACs) and Phosphodiesterase 5 (PDE5) inhibitors for the treatment of alzheimer’s disease. J Med Chem. 2016;59(19):8967–9004. doi:10.1021/acs.jmedchem.6b00908
148. Cuadrado-Tejedor M, Garcia-Barroso C, Sanzhez-Arias J, et al. Concomitant histone deacetylase and phosphodiesterase 5 inhibition synergistically prevents the disruption in synaptic plasticity and it reverses cognitive impairment in a mouse model of alzheimer’s disease. Clin Epigenetics. 2015;7(1):108. doi:10.1186/s13148-015-0142-9
149. Hu J, An B, Pan T, et al. Design, synthesis, and biological evaluation of histone deacetylase inhibitors possessing glutathione peroxidase-like and antioxidant activities against alzheimer’s disease. Bioorg Med Chem. 2018;26(21):5718–5729. doi:10.1016/j.bmc.2018.10.022
150. Basso M, Chen HH, Tripathy D, et al. Designing dual transglutaminase 2/histone deacetylase inhibitors effective at halting neuronal death. ChemMedChem. 2018;13(3):227–230. doi:10.1002/cmdc.201700601
151. De Simone A, La Pietra V, Betari N, et al. Discovery of the first-in-class GSK-3β/HDAC dual inhibitor as disease-modifying agent to combat alzheimer’s disease. ACS Med Chem Lett. 2019;10(4):469–474. doi:10.1021/acsmedchemlett.8b00507
152. Cuadrado-Tejedor M, Garcia-Barroso C, Sánchez-Arias JA, et al. A first-in-class small-molecule that acts as a dual inhibitor of HDAC and PDE5 and that rescues hippocampal synaptic impairment in alzheimer’s disease mice. Neuropsychopharmacology. 2017;42(2):524–539. doi:10.1038/npp.2016.163
153. Ganai SA, Banday S, Farooq Z, et al. Modulating epigenetic HAT activity for reinstating acetylation homeostasis: a promising therapeutic strategy for neurological disorders. Pharmacol Ther. 2016;166:106–122.
154. Chatterjee S, Cassel R, Schneider-Anthony A, et al. Reinstating plasticity and memory in a tauopathy mouse model with an acetyltransferase activator. EMBO Mol Med. 2018;10(11):e8587. doi:10.15252/emmm.201708587
155. Chainoglou E, Hadjipavlou-Litina D. Curcumin in health and diseases: alzheimer’s disease and curcumin analogues, derivatives, and hybrids. Int J Mol Sci. 2020;21(6):1975. doi:10.3390/ijms21061975
156. Hassan FU, Rehman MS, Khan MS, et al. Curcumin as an alternative epigenetic modulator: mechanism of action and potential effects. Front Genet. 2019;10:514. doi:10.3389/fgene.2019.00514
157. Braidy N, Jugder BE, Poljak A, et al. Resveratrol as a potential therapeutic candidate for the treatment and management of alzheimer’s disease. Curr Top Med Chem. 2016;16(17):1951–1960. doi:10.2174/1568026616666160204121431
158. Gomes BAQ, Silva JPB, Romeiro CFR, et al. Neuroprotective mechanisms of resveratrol in alzheimer’s disease: role of SIRT1. Oxid Med Cell Longev. 2018;2018:8152373. doi:10.1155/2018/8152373
159. Gareri P, Cotroneo AM, Orsitto G, Putignano S. The CITIMEM study: a pilot study. Optimizing pharmacological treatment in dementia. Arch Gerontol Geriatr. 2020;89:104073. doi:10.1016/j.archger.2020.104073
160. Gareri P, Castagna A, Cotroneo AM, et al. The Citicholinage Study: citicoline plus cholinesterase inhibitors in aged patients affected with Alzheimer’s Disease Study. J Alzheimers Dis. 2017;56(2):557–565. doi:10.3233/JAD-160808
161. Castagna A, Fabbo A, Manzo C, et al. Study on the benefits of combined citicoline, memantine, and acetylcholinesterase inhibitor treatments in older patients affected with alzheimer’s disease. J Alzheimers Dis. 2021;79(4):1509–1515. doi:10.3233/JAD-201211
162. Hurtado O, Hernández-Jiménez M, Zarruk JG, et al. Citicoline (CDP-choline) increases sirtuin1 expression concomitant to neuroprotection in experimental stroke. J Neurochem. 2013;126(6):819–826. doi:10.1111/jnc.12269
163. Cacabelos R, Carril JC, Cacabelos N, et al. Sirtuins in alzheimer’s disease: SIRT2-related GenoPhenotypes and implications for PharmacoEpiGenetics. Int J Mol Sci. 2019;20(5):1249. doi:10.3390/ijms20051249
164. Krupinski J, Slevin M, Badimon L. Citicoline inhibits MAP kinase signalling pathways after focal cerebral ischaemia. Neurochem Res. 2005;30(8):1067–1073. doi:10.1007/s11064-005-7201-0
165. Angelucci F, Cechova K, Valis M, Kuca K, Zhang B, Hort J. MicroRNAs in alzheimer’s disease: diagnostic markers or therapeutic agents? Front Pharmacol. 2019;10:665. doi:10.3389/fphar.2019.00665
166. Gupta P, Bhattacharjee S, Sharma AR, Sharma G, Lee SS, Chakraborty C. miRNAs in alzheimer disease - a therapeutic perspective. Curr Alzheimer Res. 2017;14(11):1198–1206. doi:10.2174/1567205014666170829101016
167. Silvestro S, Bramanti P, Mazzon E. Role of miRNAs in alzheimer’s disease and possible fields of application. Int J Mol Sci. 2019;20(16):3979. doi:10.3390/ijms20163979
168. Parsi S, Smith PY, Goupil C, Dorval V, Hébert SS. Preclinical evaluation of miR-15/107 family members as multifactorial drug targets for alzheimer’s disease. Mol Ther Nucleic Acids. 2015;4:e256. doi:10.1038/mtna.2015.33
169. Smith PY, Hernandez-Rapp J, Jolivette F, et al. miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum Mol Genet. 2015;24:6721–6735. doi:10.1093/hmg/ddv377
170. Salta E, Sierksma A, Vanden Eynden E, De Strooper B. miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in alzheimer’s brain. EMBO Mol Med. 2016;8:1005–1018. doi:10.15252/emmm.201606520
171. Cao F, Liu Z, Sun G. Diagnostic value of miR-193a-3p in alzheimer’s disease and miR-193a-3p attenuates amyloid-β induced neurotoxicity by targeting PTEN. Exp Gerontol. 2020;130:110814. doi:10.1016/j.exger.2019.110814
172. Sun L, Zhang T, Xiu W, et al. MiR-107 overexpression attenuates neurotoxicity induced by 6-hydroxydopamine both in vitro and in vivo. Chem Biol Interact. 2020;315:108908. doi:10.1016/j.cbi.2019.108908
173. He B, Chen W, Zeng J, Tong W, Zheng P. MicroRNA-326 decreases tau phosphorylation and neuron apoptosis through inhibition of the JNK signaling pathway by targeting VAV1 in alzheimer’s disease. J Cell Physiol. 2020;235:480–493. doi:10.1002/jcp.28988
174. Shi Z, Zhang K, Zhou H, et al. Increased miR-34c mediates synaptic deficits by targeting synaptotagmin 1 through ROS-JNK-p53 pathway in alzheimer’s disease. Aging Cell. 2020;19:e13125. doi:10.1111/acel.13125
175. Lee K, Kim H, An K, et al. Replenishment of microRNA-188-5p restores the synaptic and cognitive deficits in 5XFAD mouse model of alzheimer’s disease. Sci Rep. 2016;6:34433. doi:10.1038/srep34433
176. Zolochevska O, Taglialatela G. Selected microRNAs increase synaptic resilience to the damaging binding of the alzheimer’s disease amyloid beta oligomers. Mol Neurobiol. 2020;57:2232–2243. doi:10.1007/s12035-020-01868-8
177. Higaki S, Muramatsu M, Matsuda A, et al. Defensive effect of microRNA-200b/c against amyloid-beta peptide-induced toxicity in alzheimer’s disease models. PLoS One. 2018;13:e0196929. doi:10.1371/journal.pone.0196929
178. Barros-Viegas AT, Carmona V, Ferreiro E, et al. miRNA-31 improves cognition and abolishes amyloid-β pathology by targeting APP and BACE1 in an animal model of alzheimer’s disease. Mol Ther Nucleic Acids. 2020;19:1219–1236. doi:10.1016/j.omtn.2020.01.010
179. Wu GD, Li ZH, Li X, Zheng T, Zhang DK. microRNA-592 blockade inhibits oxidative stress injury in alzheimer’s disease astrocytes via the KIAA0319-mediated Keap1/Nrf2/ARE signaling pathway. Exp Neurol. 2020;324:113128. doi:10.1016/j.expneurol.2019.113128
180. Wang X, Li X, Huang B, et al. Downregulation of miR-33 has protective effect against Aβ25–35-induced injury in SH-SH-SY5Y cells. Med Sci Monit. 2020;26:e921026.
181. Fu Y, Hu X, Zheng C, et al. Intrahippocampal miR-342-3p inhibition reduces β-amyloid plaques and ameliorates learning and memory in alzheimer’s disease. Metab Brain Dis. 2019;34(5):1355–1363. doi:10.1007/s11011-019-00438-9
182. Zhang Y, Liu C, Wang J, et al. MiR-299-5p regulates apoptosis through autophagy in neurons and ameliorates cognitive capacity in APPswe/PS1dE9 mice. Sci Rep. 2016;6:24566. doi:10.1038/srep24566
183. Pereira PA, Tomás JF, Queiroz JA, Figueiras AR, Sousa F. Recombinant pre- miR-29b for alzheimer´s disease therapeutics. Sci Rep. 2016;6:19946. doi:10.1038/srep19946
184. Gabr MT, Brogi S. MicroRNA-based multitarget approach for alzheimer’s disease: discovery of the first-in-class dual inhibitor of acetylcholinesterase and microRNA-15b biogenesis. J Med Chem. 2020;63(17):9695–9704. doi:10.1021/acs.jmedchem.0c00756
185. Zhang N, Gao Y, Yu S, Sun X, Shen K. Berberine attenuates Aβ42-induced neuronal damage through regulating circHDAC9/miR-142-5p axis in human neuronal cells. Life Sci. 2020;252:117637. doi:10.1016/j.lfs.2020.117637
186. Ge Y, Song X, Liu J, Liu C, Xu C. The combined therapy of berberine treatment with lncRNA BACE1-AS depletion attenuates Aβ25-35 induced neuronal injury through regulating the expression of miR-132-3p in neuronal cells. Neurochem Res. 2020;45(4):741–751. doi:10.1007/s11064-019-02947-6
187. Lin Y, Liang X, Yao Y, Xiao H, Shi Y, Yang J. Osthole attenuates APP-induced alzheimer’s disease through up-regulating miRNA-101a-3p. Life Sci. 2019;225:117–131. doi:10.1016/j.lfs.2019.04.004
188. Li S, Yan Y, Jiao Y, et al. Neuroprotective effect of osthole on neuron synapses in an alzheimer’s disease cell model via upregulation of microRNA-9. J Mol Neurosci. 2016;60(1):71–81. doi:10.1007/s12031-016-0793-9
189. Jiao Y, Kong L, Yao Y, et al. Osthole decreases beta amyloid levels through up-regulation of miR-107 in alzheimer’s disease. Neuropharmacology. 2016;108:332–344. doi:10.1016/j.neuropharm.2016.04.046
190. Zhu L, Lin M, Ma J, et al. The role of LINC00094/miR-224-5p (miR-497-5p)/Endophilin-1 axis in memantine mediated protective effects on blood-brain barrier in AD microenvironment. J Cell Mol Med. 2019;23(5):3280–3292. doi:10.1111/jcmm.14214
191. Wang CN, Wang YJ, Wang H, et al. The anti-dementia effects of donepezil involve miR-206-3p in the hippocampus and cortex. Biol Pharm Bull. 2017;40(4):465–472. doi:10.1248/bpb.b16-00898
192. Martin I. Resveratrol for alzheimer’s disease? Sci Transl Med. 2017;9(375):eaam6055. doi:10.1126/scitranslmed.aam6055
193. Turner RS, Thomas RG, Craft S, et al. A randomized, double-blind, placebo-controlled trial of resveratrol for alzheimer disease. Neurology. 2015;85(16):1383–1391. doi:10.1212/WNL.0000000000002035
194. Tili E, Michaille JJ, Adair B, et al. Resveratrol decreases the levels of miR-155 by upregulating miR-663, a microRNA targeting JunB and JunD. Carcinogenesis. 2010;31(9):1561–1566. doi:10.1093/carcin/bgq143
195. Kou X, Chen N. Resveratrol as a natural autophagy regulator for prevention and treatment of alzheimer’s disease. Nutrients. 2017;9(9):927.
196. Huang W, Li Z, Zhao L, Zhao W. Simvastatin ameliorate memory deficits and inflammation in clinical and mouse model of alzheimer’s disease via modulating the expression of miR-106b. Biomed Pharmacother. 2017;92:46–57. doi:10.1016/j.biopha.2017.05.060
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.