")
Back to Journals » Journal of Inflammation Research » Volume 13
Periodontal Disease and Periodontal Disease-Related Bacteria Involved in the Pathogenesis of Alzheimer’s Disease
Authors Matsushita K, Yamada-Furukawa M, Kurosawa M, Shikama Y
Received 2 April 2020
Accepted for publication 29 May 2020
Published 30 June 2020 Volume 2020:13 Pages 275—283
DOI https://doi.org/10.2147/JIR.S255309
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Kenji Matsushita, Masae Yamada-Furukawa, Mie Kurosawa, Yosuke Shikama
Department of Oral Disease Research, National Center for Geriatrics and Gerontology, Obu, Aichi 474-8511, Japan
Correspondence: Kenji Matsushita
Department of Oral Disease Research, National Center for Geriatrics and Gerontology, 7-430 Morioka-Chou, Obu, Aichi 474-8511, Japan
Email [email protected]
Abstract: Alzheimer’s disease (AD) is the most common cause of dementia, and it exhibits pathological properties such as deposition of extracellular amyloid β (Aβ) and abnormally phosphorylated Tau in nerve cells and a decrease of synapses. Conventionally, drugs targeting Aβ and its related molecules have been developed on the basis of the amyloid cascade hypothesis, but sufficient effects on the disease have not been obtained in past clinical trials. On the other hand, it has been pointed out that chronic inflammation and microbial infection in the brain may be involved in the pathogenesis of AD. Recently, attention has been focused on the relationship between the periodontopathic bacterium Porphylomonas gingivalis and AD. P. gingivalis and its toxins have been detected in autopsy brain tissues from patients with AD. In addition, pathological conditions of AD are formed or exacerbated in mice infected with P. gingivalis. Compounds that target the toxins of P. gingivalis ameliorate the pathogenesis of AD triggered by P. gingivalis infection. These findings indicate that the pathological condition of AD may be regulated by controlling the bacteria in the oral cavity and the body. In the current aging society, the importance of oral and periodontal care for preventing the onset of AD will increase.
Keywords: Porphylomonas gingivalis, cognitive decline, amyloid β, blood-brain barrier, vascular inflammation
Introduction
Dementia is the most frequent neurological disease in the world and is recognized as a global public health priority by the World Health Organization. Although various methods for prevention and treatment of dementia have yet been studied, no effective method has been established. If risk factors for dementia and factors that suppress its onset and progression could be identified that information could be used effectively, it might be possible to prevent dementia and extend the healthy life span. Recently, the associations between dementia and systemic diseases have been focused on. The pathogenesis of Alzheimer’s disease (AD), which accounts for the largest number of cases of dementia, and the relationships of the pathogenesis of AD with periodontitis and periodontitis-related bacteria are described in this review.
Alzheimer’s Disease
It is estimated that about 44 million people worldwide are currently suffering from dementia. Treatment costs exceed US $600 billion annually in the United States alone. Due to the aging of the population, treatment costs are expected to more than triple by 2050. Dementia is a generic term for diseases that make social life difficult due to impairment of cognitive function. AD is a typical disease, but there are other types of disease such as cerebrovascular dementia, Lewy body dementia, and frontotemporal dementia. AD, the most common form of dementia, was first reported by Dr. Aloysius Alzheimer in Germany in 1907.1 The brain of a female patient who had severe memory loss was analyzed after death. As a result of contraction of the brain, there was abnormal deposition of protein around brain neurons, and this disease was named AD. It has been reported that about 27 million people worldwide suffer from AD.
Late-onset AD may be caused by the complex interaction of genetic and environmental factors. Currently, genetic factors are considered to account for about 70% of the risk for AD, and the APOE gene, which has three variants (ε2, ε3 and ε4) is the greatest risk for sporadic AD. Compared to non-ε4 carriers, the odds ratios (ORs) for AD are up to 3-times higher in carriers of ε4 heterozygotes and up to 12-times higher in carriers ofε4 homozygotes. Previous genome-wide association studies have revealed over 20 genetic risk factors for AD including inflammation, cholesterol metabolism, and endosomal vesicle recycling pathways.2 In particular, microglial activation by amyloid deposition plays an important role in the pathogenesis of AD. The combination of these factors is thought to significantly increase the risk of AD. The results of epidemiological studies suggest that education and exercise may be effective for preventing AD. Middle-aged hypertension and diabetes also increase the risk of AD.3 Risk factors for vascular disease or vascular disease itself may also directly affect the progression of AD pathology.
The main features of Alzheimer’s pathology are the formation of amyloid plaques4 and formation of neurofibrillary tangles (NFTs)5. In these pathological processes, loss of synapses and neurons leading to macroscopic atrophy is observed.6 Amyloid plaques are extracellular deposits composed mainly of misfolded Amyloid β (Aβ) (Aβ40 and Aβ42) with 40 or 42 amino acids that are two byproducts of APP metabolism. Aβ42 is more abundant than Aβ40 in plaques because of its rapid infiltration rate and insolubility. Neurofibrillary tangles are mainly observed as hyperphosphorylated tau paired helical filaments. Although the clinical features and severity of AD correlate well with NFT pathology, β-amyloid deposition reaches a plateau early in the symptomatic phase of AD. The amyloid hypothesis is a general theory of AD pathogenesis.4 Specifically, accumulation of Aβ caused by sequential cleavage of amyloid precursor protein (APP) by β- and γ-secretase enzymes in the brain is caused by an imbalance between Aβ production and its clearance. It is believed that inflammation enhances the formation of NFTs and subsequent neurological dysfunction and neurodegeneration.7 Therapy that can cure AD does not exist. As drug therapy, acetylcholine esterase inhibitors (AChEI) (donepezil, galantamine and rivastigmine)8 and an N-Methyl-D-aspartate (NMDA) receptor antagonist (memantine)9 are used. However, the effects of these drugs are limited, and even if there is a temporary improvement with drug treatment, after several years, patients return to the state they were in when they started taking the drugs and they will progress in the direction of deterioration. Clinical trials of AD drug candidates are currently being conducted worldwide, but their results have not been good. Although many candidate drugs target Aβ, sufficient efficacy and safety have not been confirmed in most cases.10 According to the results of the Dominant Inherited Alzheimer Network (DIAN) Study, a follow-up study of families who developed dominantly inherited AD, there was accumulation of Aβ from 25 years before the onset of AD and hippocampal volume started to decrease 15 years before the onset of AD. Five years before the onset, the accumulation of Aβ peaks and then accumulation of tau in neurons and volume reduction of the hippocampus progress, and “light forgetting” comes to be recognized. When cognitive function decline begins, it progresses rapidly and it will be in need of care five years after the onset. AD is a disease that progresses over a long span of 25 years or more, and at the time of onset, hippocampal atrophy has already progressed. Once AD has developed, it is difficult to treat and it is therefore considered to be very important to prevent and delay the progression.
Alzheimer’s Disease and Inflammation
There is a belief that brain inflammation is involved in the pathogenesis of AD. In autopsy brains from Alzheimer’s patients, accumulation of activated microglia is seen around senile plaques.7 In addition, it has been reported that the risk of AD is reduced to about one-sixth in rheumatoid patients who have been taking NASIDs for a long time.11 Inflammation in the brain enhances Aβ accumulation.12 In addition, deposition of Aβ induces an inflammatory response, which results in synaptic damage and neuronal damage.13 Recently, mutations in the TREM2 gene, which is one of the molecules that control the inflammatory response, have been found in patients with AD, and the importance of the inflammatory response in the pathogenesis of AD has been re-recognized.10 The immune system in the central nervous system is extremely simple, there is no acquired immune system, and the immune response is carried by the innate immune system. Microglia are resident macrophages in the CNS.14 Microglia remain dormant when the microenvironment in the brain is stable and maintain normal CNS function.15 When a change in the microenvironment occurs, microglia change to an activated state and perform pruning of synapses and removal of foreign substances.16–18 In addition, microglia form an immune surveillance system in the brain that regulates key processes associated with AD pathology such as clearance of Aβ and aberrant tau protein and production of neurotrophic and neuroinflammatory factors. The activated microglia show changes in cell morphology and phenotype19,20 and have high productivity for cytokines and inflammatory mediators. Substances that contribute to CNS infection and various neuroinflammations are involved in the activation of microglia. Bacterial lipopolysaccharide (LPS)21 and Aβ22,23 are activators of microglia. The activated microglia have proliferative ability and enhance the ability of phagocytosis and antigen presentation, thereby removing damaged nerve cells in the lesion site. Activated microglia also releases various humoral factors including cytokines, chemokines, and BDNF to repair nerves. On the other hand, excessive release of these factors causes neuropathy.24,25 In fact, it has been observed that active microglia accumulate in lesions of neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease. Thus, microglia may be deeply involved in the pathogenesis of AD (Figure 1). It has been reported that mild systemic inflammation reduces cognitive function and decreases hippocampal capacity and that systemic inflammation increases the risk of AD.26–28 Tumor necrosis factor (TNF)-α levels in blood are elevated in patients with AD and are also correlated with the decline in cognitive function.29,30 LPS increases the saturable transport of insulin across the blood-brain barrier (BBB).31 TNF-α increases the permeability of the BBB through inducing reorganization of actin filaments to stress fibers, leading to increased paracellular clearance of sucrose and inulin.32 It is possible that these inflammatory mediators are transmitted to the brain and activate microglia in the brain.
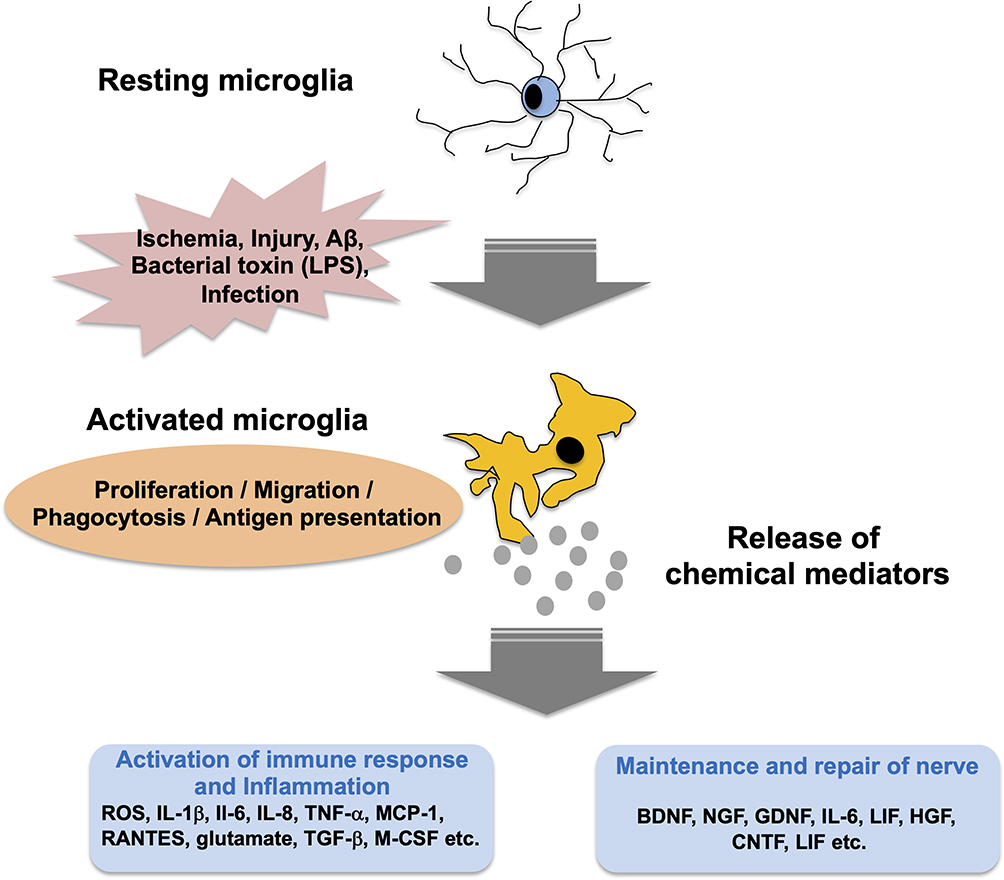 |
Figure 1 Activation of microglia and its role. |
Periodontal Disease, Periodontal Disease-Related Bacteria and AD
Correlations of the number of possessed teeth and the presence or absence of periodontal disease with cognitive function and AD have been reported. People with edentulous jaws and those with few teeth have a higher prevalence and incidence of dementia.33 In addition, the results of a meta-analysis showed that there were significant correlations between dementia and various clinical parameters of periodontal disease such as periodontal probing depth (PPD), bleeding on probing (BOP), gingival bleeding index (GBI), clinical attachment level (CAL), and plaque index (PI).33 After 10 years of follow-up of periodontal patients and healthy volunteers over 50 years of age, it was shown that periodontal disease patients have a 1.7-times higher risk of developing AD than do healthy people.34 In addition, it has been reported that periodontal disease sufferers have a faster rate of decline in cognitive function than do healthy people.35 Chronic inflammation in peripheral organs can exacerbate the molecular pathology of AD. Periodontal disease is also a chronic inflammatory disease and is involved in the development and progression of various diseases such as arteriosclerosis and diabetes as well as obesity, preterm birth and low birth weight childbirth.36 Inflammatory cytokines including interleukin (IL)-1β, IL-6, and TNF-α are elevated in peripheral blood in periodontitis patients, and these inflammatory mediators may exacerbate cerebral inflammation in AD.
Differences in gut microbiota have been shown to be associated with lifestyle-related diseases such as obesity, cardiovascular disease, and diabetes.37–40 Recently, an association between the gut microbiome and dementia has been confirmed,41,42 and the gut microbiome may regulate host brain function through the microbiome–gut–brain axis.43 Specifically, brain inflammation is caused by changes in gut microbiota,44 which can cause deposition of amyloid β in the brain.45,46 Therefore, enterobacteria may be involved in the pathogenesis of Alzheimer’s disease. Saji et al reported that different compositions of gut microbiota were found in patients with dementia and those without dementia. Briefly, the proportion of enterotype I bacteria in the gut flora from patients with dementia was lower than that in the gut flora from patients without dementia.47 Intestinal Bacteroides species are increased in gut microbiota in patients with mild cognitive impairment (MCI). In addition, white matter hyperintensity and parahippocampal atrophy were seen in patients with a large number of Bacteroides species.48 These findings suggested that an increase of specific gut bacteria is involved in the pathogenesis of dementia.
It has been reported that some microorganisms were detected in autopsy brains of AD patients. HSV1, Chlamydia pneumonie, spirochetes, and fungi have been reported, and these microorganisms cause brain inflammation that results in synaptic dysfunction and neuronal cell death.49–51 In order to enclose the invading microbes in the brain, amyloid β (Aβ) is secreted from neurons and has been shown to fold and kill pathogens and protect the brain.52,53 Thus, Aβ may function as an innate immune molecule responsible for infection in the brain. On the other hand, it is thought that Aβ-containing microorganisms are deposited in brain tissue, resulting in the formation of senile plaques that cause damage to cranial nerves and exacerbate the condition of AD.54 In addition to these bacteria, it is thought that several periodontal pathogens such as Porphyromonas gingivalis, Prevotella intermedia, Aggregatibacter actinomycetemcomitans, Fusobacterium nucleatum, Tannerella forsythensis, and Eikenella corrodens are involved in the development of several inflammatory diseases at remote organ sites like AD.55 Especially, the association between Porphyromonas gingivalis and AD has attracted attention.56,57 P. gingivalis, a type of periodontitis-related bacteria, was frequently detected in autopsy brain tissues of patients who died of AD. On the other hand, the bacteria were not detected in normal human brain tissue.58 Recently, it has also been reported that gingipain, a toxin produced by the same bacteria, was frequently detected in the brains of patients with AD and it has been shown by using a mouse model that the toxin may be involved in the pathogenesis of AD.59 We administered P. gingivalis directly into the oral cavities of AD model mice (APP-Tg mice) to cause experimental periodontitis, and then we conducted a novel object recognition test to evaluate the cognitive functions in the P. gingivalis administration group and non-administration group.60 We found that cognitive function in the P. gingivalis administration group was significantly reduced compared to that in the non-administration group. In addition, increased Aβ deposition, increased TNF-α and IL-1β production, and an increase in LPS concentration in the brain were found in the P. gingivalis administration group, compared to those in the non-administration group. It was also found that P. gingivalis LPS induced the production of Aβ in neurons and that the coexistence of LPS with Aβ enhanced the production of TNF-α and IL-1βin microglia in cultures. These results suggest that infection with P. gingivalis and the resulting inflammation aggravate the pathology of AD (Figure 2).
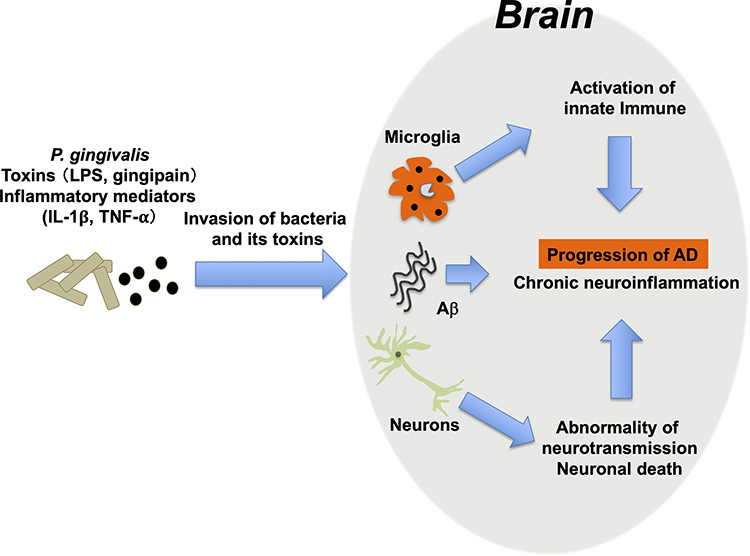 |
Figure 2 Possible mechanisms by which periodontal disease and P. gingivalis aggravate Alzheimer’s disease. |
Induction of Cerebral Small Vessel Disease by P. Gingivalis and AD
Cerebrovascular disease is known to be an important risk factor for AD.61 From histopathological analysis based on necropsy, 80% of patients diagnosed with AD showed cortical infarcts, lacunar infarction, cerebral microbleeds and multiple microinfarcts that are indicative of small vessel disease (SVD), intracranial atherosclerosis, and cerebral amyloid angiopathy (CAA).62 It has been suggested that these disorders result in decreased cerebral blood flow and increased BBB permeability, exacerbating cognitive dysfunction. One of the mechanisms by which P. gingivalis exacerbates AD may be exacerbation of those cerebrovascular disorders. Periodontal disease has been reported to be an independent risk factor for ischemic stroke.63 It has been reported that there is a correlation between an increase in the antibody titer to P. gingivalis and the onset of stroke.64 P. gingivalis tends to adhere to inflamed blood vessels. P. gingivalis is frequently detected in peripheral arteries of patients with Burger’s disease.65 Recent studies have also shown that P. gingivalis is detected with 100% probability from the coronary or femoral artery in patients with atherosclerotic cardiovascular disease. P. gingivalis adheres to E-selectin via its outer membrane proteins (OMPs). It also invades inflamed epithelial cells and vascular endothelial cells.66 P. gingivalis produces trypsin-like cysteine proteases such as gingipains (lysine gingipain (Kgp), arginine gingin dipain A (RgpA), and arginine gingin dipain B (RgpB)) and is involved in the formation of vascular lesions together with the pathogenesis of periodontal disease.67,68 Gingipains are present in the outer membranes of bacteria, but some are released as outer membrane vesicles (OMV).69,70 Kgp and RgpA/B are deeply involved in the survival and virulence of P. gingivalis.71 Gingipains produced by the bacterium activate the blood coagulation system72 and degrade the anticoagulant factor thrombomodulin that is expressed on vascular endothelial cells,73 so that thrombi are easily formed in blood vessels. In addition, gingipains directly damage endothelial cells and epithelial cells.67,68,74 Such an action of P. gingivalis may be involved in the pathogenesis of cerebral amyloid angiopathy (CAA), which often accompanies AD.75 From the above, the following exacerbation mechanism of AD is inferred (Figure 2). P. gingivalis and its toxins in the oral cavity are transferred to the brain through the bloodstream or intestine. Normally, there is a blood-brain barrier (BBB), and so it is thought that they cannot be transferred into the brain. However, the increase of inflammatory mediators in the blood, senescence of the cerebrovasculature, or direct action of bacterial toxins on blood vessels causes vascular inflammation and thrombosis, resulting in decreased cerebral blood flow. The permeability of the BBB is also enhanced, and then the bacteria and their toxins may enter the brain. P. gingivalis and its toxins that have migrated to the brain parenchyma enhance the production of Aβ and activate microglia in conjunction with Aβ and Tau. Then, an innate immune response in the brain is induced and damages neurons. Such chronic brain inflammation and neuron degeneration may exacerbate the pathological condition of AD.
Conclusions
Based on previous reports and our findings, we have described the relationships between periodontal disease, periodontal disease-related bacteria and AD (Figure 3). The conditions of periodontal disease (bacterial infection and chronic inflammation) weaken the blood-brain barrier and pose a risk for cerebrovascular disease. The conditions also cause inflammation in the brain. Periodontal disease may indirectly make AD pathology worse through exacerbation of diabetes.76,77 Furthermore, tooth loss leads to deterioration of cognitive function.78,79 Therefore, it is possible that periodontal disease directly and indirectly exacerbates the condition of dementia (Figure 4). Therefore, periodontal care is considered to be essential for dementia control. On the other hand, in periodontal disease patients, bacteremia often occurs due to regular brushing and chewing.80,81 Although it is important to treat periodontitis for the prevention of AD, it is also necessary to take measures against bacteremia by invasive treatment such as scaling and root planing in the future. In addition, it may be useful for the prevention of dementia by evaluation of gut microbiota and oral microbiota as potential risk factors for dementia may be useful for prevention of dementia. Changes in bacterial flora are associated with quantitative and qualitative changes in diet.60,82–85 By clarifying the causal relationships of nutrition with bacterial flora and dementia, the possibility of a new method for prevention of dementia by improving eating habits may be expanded.
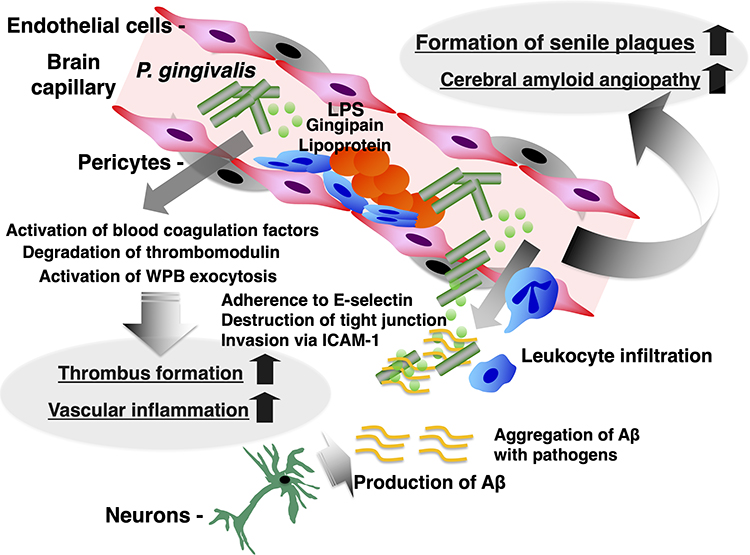 |
Figure 3 Induction of blood–brain barrier breakdown and amyloid deposition by P. gingivalis in cerebral blood capillaries. |
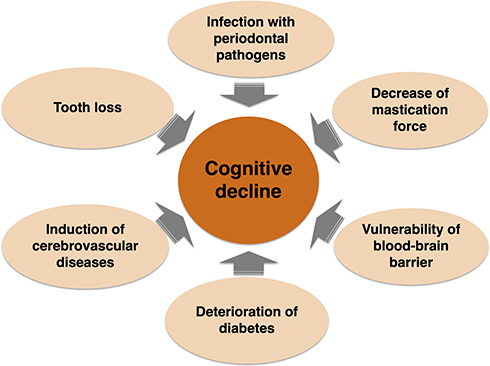 |
Figure 4 Deterioration of cognitive function caused by periodontal disease. |
The incidence of AD onset increases in the late 70s. On the other hand, accumulation of Aβ in the brain has started after the age of 50 years. Care for periodontal disease and control of oral bacteria in that period is important to regulate the onset and exacerbation of AD. Prevention of periodontal disease and maintenance of oral health will become increasingly important in the aging society.
Acknowledgments
This work was supported by The Research Funding for Longevity Sciences (29-25) from National Center for Geriatrics and Gerontology (NCGG), Japan and by the Ministry of Education, Science, Sports and Culture, Grant-in Aid for Scientific Research (B), 2020–2022 (20H03904, Kenji Matsushita).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Alzheimer A. Über eine eigenartige Erkrankung der hirnrinde. Allgemeine Zeitschrift fur Psychiatrie und Psychisch-gerichtliche Medizin. 1907;64:146–148.
2. Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77(1):43–51. doi:10.1016/j.biopsych.2014.05.006
3. Xu W, Tan L, Wang HF, et al. Meta-analysis of modifiable risk factors for Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2015;86(12):1299–1306. doi:10.1136/jnnp-2015-310548
4. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi:10.1126/science.1072994
5. Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16(3):
6. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–292. doi:10.1016/j.jalz.2011.03.003
7. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405. doi:10.1016/S1474-4422(15)70016-5
8. Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev. 2006;(1):CD005593.
9. McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev. 2006;(2):CD003154.
10. Cao J, Hou J, Ping J, Cai D. Advances in developing novel therapeutic strategies for Alzheimer’s disease. Mol Neurodegener. 2018;13(1):64.
11. McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology. 1996;47(2):425–432. doi:10.1212/WNL.47.2.425
12. Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421. doi:10.1016/S0197-4580(00)00124-X
13. Cai Z, Hussain MD, Yan LJ. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int J Neurosci. 2014;124(5):307–321. doi:10.3109/00207454.2013.833510
14. Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6(4):193–201. doi:10.1038/nrneurol.2010.17
15. Salter MW, Beggs S. Sublime microglia: expanding roles for the guardians of the CNS. Cell. 2014;158(1):15–24. doi:10.1016/j.cell.2014.06.008
16. Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi:10.1016/j.neuron.2012.03.026
17. Norden DM, Muccigrosso MM, Godbout JP. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology. 2015;96(Pt A):29–41. doi:10.1016/j.neuropharm.2014.10.028
18. Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339(6116):156–161. doi:10.1126/science.1227901
19. Walker DG, Lue LF. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther. 2015;7(1):56. doi:10.1186/s13195-015-0139-9
20. Michell-Robinson MA, Touil H, Healy LM, et al. Roles of microglia in brain development, tissue maintenance and repair. Brain. 2015;138(5):1138–1159. doi:10.1093/brain/awv066
21. Pei Z, Pang H, Qian L, et al. MAC1 mediates LPS-induced production of superoxide by microglia: the role of pattern recognition receptors in dopaminergic neurotoxicity. Glia. 2007;55(13):1362–1373. doi:10.1002/glia.20545
22. Rojanathammanee L, Floden AM, Manocha GD, Combs CK. Attenuation of microglial activation in a mouse model of Alzheimer’s disease via NFAT inhibition. J Neuroinflammation. 2015;12(1):42. doi:10.1186/s12974-015-0255-2
23. Wu Z, Sun L, Hashioka S, et al. Differential pathways for interleukin-1β production activated by chromogranin A and amyloid β in microglia. Neurobiol Aging. 2013;34(12):2715–2725. doi:10.1016/j.neurobiolaging.2013.05.018
24. Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40(2):140–155. doi:10.1002/glia.10161
25. Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol. 2006;84(1):49–59. doi:10.1139/Y05-143
26. Engelhart MJ, Geerlings MI, Meijer J, et al. Inflammatory proteins in plasma and the risk of dementia: the Rotterdam study. Arch Neurol. 2004;61(5):668–672. doi:10.1001/archneur.61.5.668
27. Marsland AL, Gianaros PJ, Abramowitch SM, Manuck SB, Hariri AR. Interleukin-6 covaries inversely with hippocampal grey matter volume in middle-aged adults. Biol Psychiatry. 2008;64(6):484–490. doi:10.1016/j.biopsych.2008.04.016
28. Tobinick EL. Re: inflammatory markers and the risk of Alzheimer disease: the Framingham Study. Neurology. 2008;70(14):
29. Alvarez A, Cacabelos R, Sanpedro C, Garcia-Fantini M, Aleixandre M. Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol Aging. 2007;28(4):533–536. doi:10.1016/j.neurobiolaging.2006.02.012
30. Holmes C, Cunningham C, Zotova E, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73(10):768–774. doi:10.1212/WNL.0b013e3181b6bb95
31. Xaio H, Banks WA, Niehoff ML, Morley JE. Effect of LPS on the permeability of the blood-brain barrier to insulin. Brain Res. 2001;896(1–2):36–42. doi:10.1016/S0006-8993(00)03247-9
32. Deli MA, Descamps L, Dehouck MP, et al. Exposure of tumor necrosis factor-α to luminal membrane of bovine brain capillary endothelial cells cocultured with astrocytes induces a delayed increase of permeability and cytoplasmic stress fiber formation of actin. J Neurosci Res. 1995;41(6):717–726. doi:10.1002/jnr.490410602
33. Fang WL, Jiang MJ, Gu BB, et al. Tooth loss as a risk factor for dementia: systematic review and meta-analysis of 21 observational studies. BMC Psychiatry. 2018;18(1):345. doi:10.1186/s12888-018-1927-0
34. Chen CK, Wu YT, Chang YC. Association between chronic periodontitis and the risk of Alzheimer’s disease: a retrospective, population-based, matched-cohort study. Alzheimers Res Ther. 2017;9(1):56. doi:10.1186/s13195-017-0282-6
35. Ide M, Harris M, Stevens A, et al. Periodontitis and cognitive decline in Alzheimer’s disease. PLoS One. 2016;11(3):e0151081. doi:10.1371/journal.pone.0151081
36. Cullinan MP, Seymour GJ. Periodontal disease and systemic illness: will the evidence ever be enough? Periodontol 2000. 2013;62(1):271–286. doi:10.1111/prd.12007
37. Jiang C, Li G, Huang P, Liu Z, Zhao B. The gut microbiota and Alzheimer’s disease. J Alzheimers Dis. 2017;58(1):1–15. doi:10.3233/JAD-161141
38. Emoto T, Yamashita T, Sasaki N, et al. Analysis of gut microbiota in coronary artery disease patients: a possible link between gut microbiota and coronary artery disease. J Atheroscler Thromb. 2016;23(8):908–921. doi:10.5551/jat.32672
39. Yamashita T, Kasahara K, Emoto T, et al. Intestinal immunity and gut microbiota as therapeutic targets for preventing atherosclerotic cardiovascular diseases. Circ J. 2015;79(9):1882–1890. doi:10.1253/circj.CJ-15-0526
40. Sharma S, Tripathi P. Gut microbiome and type 2 diabetes: where we are and where to go? J Nutr Biochem. 2019;63:101–108. doi:10.1016/j.jnutbio.2018.10.003
41. Alkasir R, Li J, Li X, Jin M, Zhu B. Human gut microbiota: the links with dementia development. Protein Cell. 2017;8(2):90–102. doi:10.1007/s13238-016-0338-6
42. Vogt NM, Kerby RL, Dill-McFarland KA, et al. Gut microbiome alterations in Alzheimer’s disease. Sci Rep. 2017;7(1):13537. doi:10.1038/s41598-017-13601-y
43. Solas M, Milagro FI, Ramirez MJ, Martinez JA. Inflammation and gut-brain axis link obesity to cognitive dysfunction: plausible pharmacological interventions. Curr Opin Pharmacol. 2017;37:87–92. doi:10.1016/j.coph.2017.10.005
44. Wu SC, Cao ZS, Chang KM, Juang JL. Intestinal microbial dysbiosis aggravates the progression of Alzheimer’s disease in Drosophila. Nat Commun. 2017;8(1):24. doi:10.1038/s41467-017-00040-6
45. Pistollato F, Sumalla Cano S, Elio I, Masias Vergara M, Giampieri F, Battino M. Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr Rev. 2016;74(10):624–634. doi:10.1093/nutrit/nuw023
46. Cattaneo A, Cattane N, Galluzzi S, et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging. 2017;49:60–68. doi:10.1016/j.neurobiolaging.2016.08.019
47. Saji N, Niida S, Murotani K, et al. Analysis of the relationship between the gut microbiome and dementia: a cross-sectional study conducted in Japan. Sci Rep. 2019;9(1):1008. doi:10.1038/s41598-018-38218-7
48. Saji N, Murotani K, Hisada T, et al. The relationship between the gut microbiome and mild cognitive impairment in patients without dementia: a cross-sectional study conducted in Japan. Sci Rep. 2019;9(1):19227. doi:10.1038/s41598-019-55851-y
49. Itzhaki RF, Lathe R, Balin BJ, et al. Microbes and Alzheimer’s disease. J Alzheimers Dis. 2016;51(4):979–984. doi:10.3233/JAD-160152
50. Alonso R, Pisa D, Marina AI, Morato E, Rabano A, Carrasco L. Fungal infection in patients with Alzheimer’s disease. J Alzheimers Dis. 2014;41(1):301–311.
51. Pisa D, Alonso R, Rabano A, Rodal I, Carrasco L. Different brain regions are infected with fungi in Alzheimer’s disease. Sci Rep. 2015;5:15015.
52. Kagan BL, Jang H, Capone R, et al. Antimicrobial properties of amyloid peptides. Mol Pharm. 2012;9(4):708–717. doi:10.1021/mp200419b
53. Soscia SJ, Kirby JE, Washicosky KJ, et al. The Alzheimer’s disease-associated amyloid β-protein is an antimicrobial peptide. PLoS One. 2010;5(3):e9505. doi:10.1371/journal.pone.0009505
54. Fulop T, Itzhaki RF, Balin BJ, Miklossy J, Barron AE. Role of microbes in the development of Alzheimer’s disease: state of the art – an international symposium presented at the 2017 IAGG congress in San Francisco. Front Genet. 2018;9:362. doi:10.3389/fgene.2018.00362
55. Kamer AR, Craig RG, Dasanayake AP, Brys M, Glodzik-Sobanska L, de Leon MJ. Inflammation and Alzheimer’s disease: possible role of periodontal diseases. Alzheimers Dement. 2008;4(4):242–250. doi:10.1016/j.jalz.2007.08.004
56. Singhrao SK, Harding A, Poole S, Kesavalu L, Crean S. Porphyromonas gingivalis periodontal infection and its putative links with Alzheimer’s disease. Mediators Inflamm. 2015;2015:137357. doi:10.1155/2015/137357
57. Olsen I, Taubman MA, Singhrao SK. Porphyromonas gingivalis suppresses adaptive immunity in periodontitis, atherosclerosis, and Alzheimer’s disease. J Oral Microbiol. 2016;8(1):33029. doi:10.3402/jom.v8.33029
58. Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis. 2013;36(4):665–677. doi:10.3233/JAD-121918
59. Dominy SS, Lynch C, Ermini F, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5(1):eaau3333. doi:10.1126/sciadv.aau3333
60. Ishida N, Ishihara Y, Ishida K, et al. Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech Dis. 2017;3(1):15. doi:10.1038/s41514-017-0015-x
61. Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol. 2016;15(9):934–943. doi:10.1016/S1474-4422(16)30029-1
62. Toledo JB, Arnold SE, Raible K, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain. 2013;136(9):2697–2706. doi:10.1093/brain/awt188
63. Grau AJ, Becher H, Ziegler CM, et al. Periodontal disease as a risk factor for ischemic stroke. Stroke. 2004;35(2):496–501. doi:10.1161/01.STR.0000110789.20526.9D
64. Pussinen PJ, Alfthan G, Jousilahti P, Paju S, Tuomilehto J. Systemic exposure to Porphyromonas gingivalis predicts incident stroke. Atherosclerosis. 2007;193(1):222–228. doi:10.1016/j.atherosclerosis.2006.06.027
65. Iwai T, Inoue Y, Umeda M, et al. Oral bacteria in the occluded arteries of patients with Buerger disease. J Vasc Surg. 2005;42(1):107–115. doi:10.1016/j.jvs.2005.03.016
66. Komatsu T, Nagano K, Sugiura S, et al. E-selectin mediates Porphyromonas gingivalis adherence to human endothelial cells. Infect Immun. 2012;80(7):2570–2576. doi:10.1128/IAI.06098-11
67. Sheets SM, Potempa J, Travis J, Casiano CA, Fletcher HM. Gingipains from Porphyromonas gingivalis W83 induce cell adhesion molecule cleavage and apoptosis in endothelial cells. Infect Immun. 2005;73(3):1543–1552. doi:10.1128/IAI.73.3.1543-1552.2005
68. Kinane JA, Benakanakere MR, Zhao J, Hosur KB, Kinane DF. Porphyromonas gingivalis influences actin degradation within epithelial cells during invasion and apoptosis. Cell Microbiol. 2012;14(7):1085–1096. doi:10.1111/j.1462-5822.2012.01780.x
69. Guo Y, Nguyen KA, Potempa J. Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontol 2000. 2010;54(1):15–44. doi:10.1111/j.1600-0757.2010.00377.x
70. Gui MJ, Dashper SG, Slakeski N, Chen YY, Reynolds EC. Spheres of influence: porphyromonas gingivalis outer membrane vesicles. Mol Oral Microbiol. 2016;31(5):365–378. doi:10.1111/omi.12134
71. Grenier D, Roy S, Chandad F, et al. Effect of inactivation of the Arg- and/or Lys-gingipain gene on selected virulence and physiological properties of Porphyromonas gingivalis. Infect Immun. 2003;71(8):4742–4748. doi:10.1128/IAI.71.8.4742-4748.2003
72. Imamura T. The role of gingipains in the pathogenesis of periodontal disease. J Periodontol. 2003;74(1):111–118. doi:10.1902/jop.2003.74.1.111
73. Inomata M, Ishihara Y, Matsuyama T, et al. Degradation of vascular endothelial thrombomodulin by arginine- and lysine-specific cysteine proteases from Porphyromonas gingivalis. J Periodontol. 2009;80(9):1511–1517. doi:10.1902/jop.2009.090114
74. Kato Y, Hagiwara M, Ishihara Y, et al. TNF-α augmented Porphyromonas gingivalis invasion in human gingival epithelial cells through Rab5 and ICAM-1. BMC Microbiol. 2014;14(1):229. doi:10.1186/s12866-014-0229-z
75. Freeze WM, Jacobs HIL, Schreuder F, et al. Blood-brain barrier dysfunction in small vessel disease related intracerebral hemorrhage. Front Neurol. 2018;9:926. doi:10.3389/fneur.2018.00926
76. Kopf D, Frolich L. Risk of incident Alzheimer’s disease in diabetic patients: a systematic review of prospective trials. J Alzheimers Dis. 2009;16(4):677–685. doi:10.3233/JAD-2009-1011
77. Lalla E, Papapanou PN. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol. 2011;7(12):738–748. doi:10.1038/nrendo.2011.106
78. Luo B, Pang Q, Jiang Q. Tooth loss causes spatial cognitive impairment in rats through decreased cerebral blood flow and increased glutamate. Arch Oral Biol. 2019;102:225–230. doi:10.1016/j.archoralbio.2019.05.004
79. Cerutti-Kopplin D, Feine J, Padilha DM, et al. Tooth loss increases the risk of diminished cognitive function: a systematic review and meta-analysis. JDR Clin Trans Res. 2016;1(1):10–19. doi:10.1177/2380084416633102
80. Lockhart PB, Brennan MT, Sasser HC, Fox PC, Paster BJ, Bahrani-Mougeot FK. Bacteremia associated with toothbrushing and dental extraction. Circulation. 2008;117(24):3118–3125. doi:10.1161/CIRCULATIONAHA.107.758524
81. Forner L, Larsen T, Kilian M, Holmstrup P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J Clin Periodontol. 2006;33(6):401–407. doi:10.1111/j.1600-051X.2006.00924.x
82. Morris MC, Tangney CC. Dietary fat composition and dementia risk. Neurobiol Aging. 2014;35(Suppl 2):S59–64. doi:10.1016/j.neurobiolaging.2014.03.038
83. Gu Y, Scarmeas N. Dietary patterns in Alzheimer’s disease and cognitive aging. Curr Alzheimer Res. 2011;8(5):510–519. doi:10.2174/156720511796391836
84. Chianese R, Coccurello R, Viggiano A, et al. Impact of dietary fats on brain functions. Curr Neuropharmacol. 2018;16(7):1059–1085. doi:10.2174/1570159X15666171017102547
85. Murtaza N, Burke LM, Vlahovich N, et al. Analysis of the effects of dietary pattern on the oral microbiome of elite endurance athletes. Nutrients. 2019;11(3):614. doi:10.3390/nu11030614
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.