")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 8
Peptide-Based Vaccines for Hepatocellular Carcinoma: A Review of Recent Advances
Authors Charneau J, Suzuki T, Shimomura M, Fujinami N, Nakatsura T
Received 14 May 2021
Accepted for publication 10 August 2021
Published 2 September 2021 Volume 2021:8 Pages 1035—1054
DOI https://doi.org/10.2147/JHC.S291558
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Manal Hassan
Jimmy Charneau,1 Toshihiro Suzuki,1,2 Manami Shimomura,1 Norihiro Fujinami,1 Tetsuya Nakatsura1
1Division of Cancer Immunotherapy, Exploratory Oncology Research and Clinical Trial Center, National Cancer Center, Kashiwa City, Japan; 2Department of Pharmacology, School of Medicine, Teikyo University, Tokyo, Japan
Correspondence: Tetsuya Nakatsura
Division of Cancer Immunotherapy, Exploratory Oncology Research and Clinical Trial Center, National Cancer Center, 6-5-1 Kashiwanoha, Kashiwa City, 277-8577, Japan
Tel +81-4-7131-5490
Fax +81-4-7133-6606
Email [email protected]
Abstract: Primary liver cancer is the sixth most commonly diagnosed cancer and the third leading cause of cancer-related deaths worldwide. After surgery, up to 70% of patients experience relapses. The current first-line therapy for advanced cases of hepatocellular carcinoma (HCC) comprises sorafenib and lenvatinib administered as single-drug therapies. Regorafenib, cabozantinib, and ramucirumab are administered as second-line therapies. Recently, it has been reported that using the immune checkpoint inhibitors atezolizumab (anti-PDL1 antibody) and bevacizumab (anti-VEGF antibody) leads to longer overall survival of unresectable cases, when compared with the use of sorafenib. The role of cancer immunity against HCC has attracted the attention of clinicians. In this review, we describe our phase I/II clinical trials of peptide vaccines targeting GPC3 in HCC and discuss the potential of peptide vaccines targeting common cancer antigens that are highly expressed in HCC, such as WT-I, AFP, ROBO1, and FOXM1. Further, we introduce recent cancer vaccines targeting neoantigens, which have attracted attention in recent times, as well as present our preclinical studies, the results of which might aid to initiate a neoantigen vaccine clinical trial, which would be the first of its kind in Japan.
Keywords: common cancer antigen, cancer vaccine, glypican-3, neoantigen, personalized peptide vaccine
Introduction
In 2020, there were 907,100 new cases of primary liver cancer, and the disease was the cause of death in 8.3% of the patients.1 These numbers make primary liver cancer the sixth most commonly diagnosed cancer and the third leading cause of cancer-related deaths worldwide. The prevalence of this disease is annually increasing owing to a higher rate of diagnosis as well as the longer life expectancy of the patient. Risk factors,2 leading in most of cases to a cirrhosis state, are hepatitis B and C type (which are viruses widely spread in Asia and Africa), fatty liver disease including nonalcoholic fatty liver diseases and NASH (nonalcoholic steatohepatitis) and alcoholic liver diseases. Cases of food contamination by the mycotoxin B1 aflatoxin produced by Aspergillus or industrial pollution have also been reported to contribute to the development of hepatocellular carcinoma (HCC).2 HCC has a high recurrence rate, ranging between 40% and 70%,3 and it is resistant against several standard therapies; therefore, it is classified as a refractory cancer. Early diagnosis leads to a 5-year survival rate higher than 70% when surgical resection or local radiofrequency ablation is applied.3,4 However, when the cancer is at a later advanced stage or is unresectable, the 5-year survival rate is lower than 16%. Sorafenib is the first line of treatment; it is very effective at early stages of the disease, but it efficacy reduces as the disease progresses.5 However, as sorafenib administration can lead to drug-resistance acquisition, favoring the growth of resistant tumor clone cells, the drug lenvatinib is also frequently used as the first-line treatment.6 Even though lenvatinib is not more efficient than sorafenib in terms of overall survival, lenvatinib is associated with a higher overall response rate and progression-free survival than sorafenib. In addition, new immunotherapy strategies are emerging as robust candidates for the treatment of HCC. Administration of small molecule inhibitors of multiple receptor tyrosine kinases in combination with immune checkpoint inhibitors (ICIs) has been reported to be effective in other cancers and is currently under investigation as a treatment option for HCC.
Immunotherapy approaches have been reported to be efficacious for various cancers.7 In addition, the idea of using of our own immune system to fight the disease is positively perceived and easily understood by the public. Immunotherapy is frequently used in combination with radiotherapy or chemotherapy or as the last resort if the two therapies have failed. The main immunotherapies already used in a clinical setting comprise ICIs.8 These compounds target immune checkpoint molecules present on the surface of cells, which physiologically restrict the risk of an auto-immune response after immune activity. However, cancer cells take advantage of these checkpoints and thus remain protected against tumor-specific T cells. PD1/PD-L19 and CTLA-410 are the most well-known checkpoint receptors and are highly expressed in the membrane of T cells and tumor cells. In cancer patients, T cells frequently remain in an “exhausted” state,11 which implies that they have been activated and stimulated at a low level by the tumor antigens but could not induce a strong immune response due to the “safe” control by Treg cells. Hence, as tumor cells express immune checkpoints in high quantities, they are easy targets for ICIs. A study showed that immunotherapy can be beneficial for HCC, as administering a combination of atezolizumab (anti-PDL-1) and bevacizumab (anti-VEGF) to unresectable HCC cases led to increased overall survival, longer median survival, and increased overall response rate in comparison with administering sorafenib.12
To further understand immunotherapy and its efficacy, it is necessary to consider the inter-individual differences in disease responses. Each person has their own “immunity ID” composed of the Human Leukocyte Antigen (HLA) system,13 a complex expressed at the surface of most molecules of MHC I, encoded by 6 different alleles, and MHC II, encoded by eight different alleles. The MHC system is the main guard against self-aggression of our bodies. Peptides that result from degradation of self or exogenous proteins are presented by MHC and recognized by some T cells, using a surface receptor system named T-cell receptor (TCR). In the case of a matched MHC-peptide-TCR combination, T cells are activated and start an expansion phase, which takes several days. CD4 T cells recognize MHC class II and help other immune cells by enhancing their immune response secretion factors, such as IFN-γ and interleukins.14 CD8 T cells present cytotoxic activity,15 which destroys the cells presenting the recognized peptides. Previous studies have reported the presence of antigens specifically expressed by cancer cells, termed “cancer antigens,” which have the potential to be the targets that are recognized by T cells, and used for cancer treatment. In this review, first, we summarize the “cancer antigens” expressed in HCC and then review the future development and potential of cancer vaccines targeting these cancer antigens based on our experience in clinical and preclinical studies.
Cancer Antigen Classification
The most commonly used cancer antigen classifications are listed in Table 1.16–25 To date, more than 200 cancer antigens have been identified. The antigens and their T cell epitope presented were obtained from TANTIGEN 2.0: Tumor T-cell Antigen Database (met-hilab.org) and Cancer antigen peptide database (https://caped.icp.ucl.ac.be/).
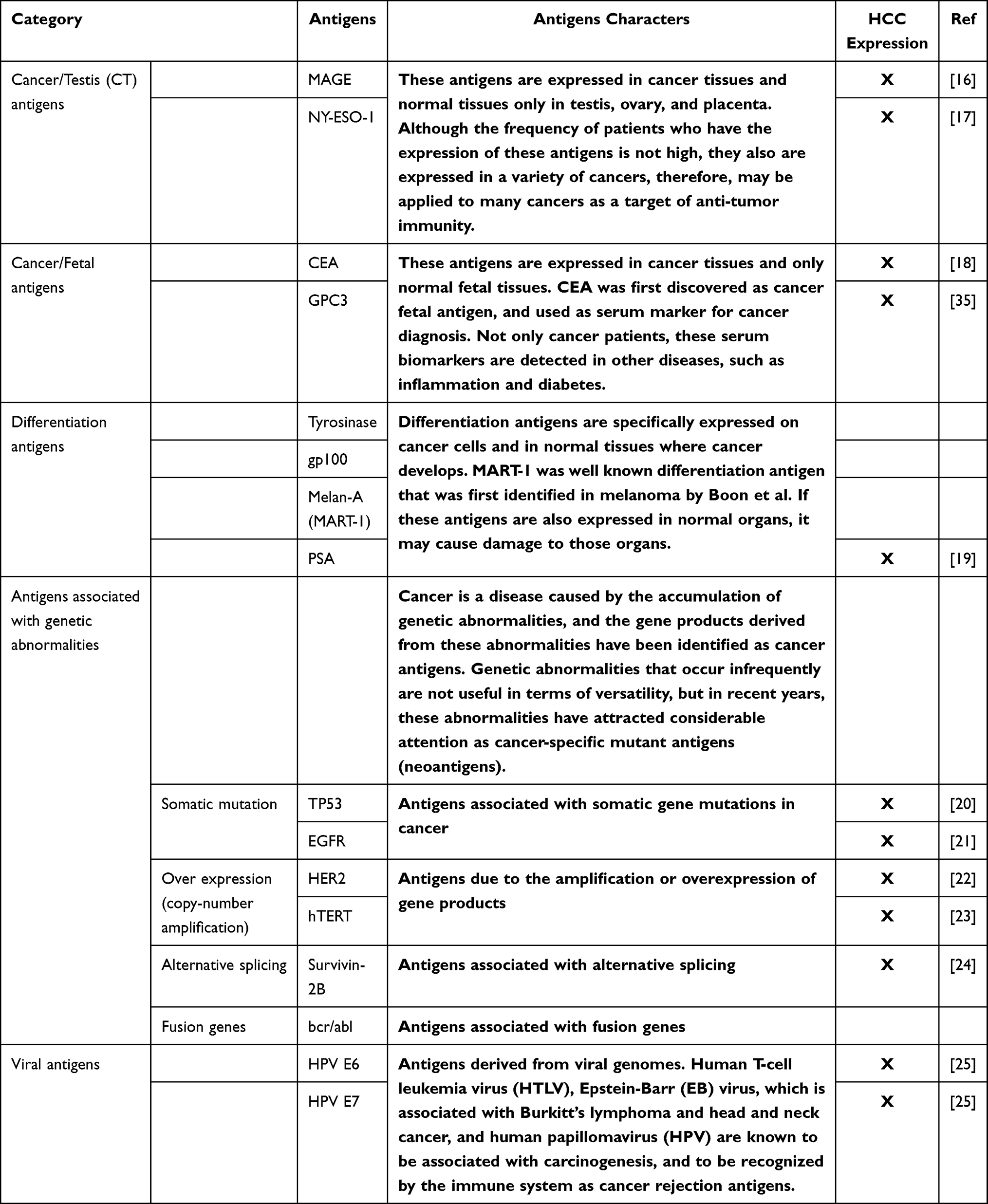 |
Table 1 The Classification of Cancer Associated Antigens and Their Expression in Cancers |
Common cancer antigens (CCAs) can usually be organized into three categories: tumor-associated antigens (TAAs), antigens derived from gene mutations in tumors, and virus-derived antigens.26 TAAs are usually overexpressed in tumors compared with those in normal tissues; therefore, they are also used as diagnostic markers. This is the case for prostate-specific antigen, carcinoembryonic antigen, and alpha-fetoprotein levels in serum. Since some TAAs are also expressed in normal cells at a lower level, targeting them with immunotherapy presents the risk of triggering an auto-immune response. Tumor-specific antigens (TSAs) originate from oncogenic drivers with non-synonymous mutations, leading to modified peptides or “neoantigens.” Therefore, TSAs are expressed exclusively in tumors and are not found in healthy tissues. TSAs serve as ideal targets for diagnosis and immunotherapy because of their tumor specificity and the absence of immune tolerance. The interest of using the mutated antigen to increase the immune response in comparison with the wild-type antigen for diagnosis or immunotherapy has been known for a long time.27 However, neoantigens are, in general, patient-specific as a result of the tumor heterogeneity and the inter-individual mutations observed in all cancers. Developing and using therapy approaches targeting neoantigens have several important requisites: 1) Neo-peptide sequences need to be predicted using sequencing technology and bioinformatics approaches; 2) The binding affinity for MHC molecules needs to be determined (increased or decreased); 3) The power of the immune response or antigenicity needs to be decreased. The disadvantages of this strategies are the high tumor mutation burden and the low number of neoantigens.
Another type of TSAs are oncogenic viral antigens. Various viruses successfully infect human cells in a persistent manner and sometimes may integrate in the host DNA, such as the human papillomavirus or the Epstein–Barr virus, or remain in the cytoplasm as RNA forms, such as the hepatitis C virus. These viruses frequently inactivate the guardians of the genome p5328 and Rb,29 preventing apoptosis and causing the immortalization of cells. This could lead to the production of virus-induced tumor cells. These infected cells produce specific proteins and peptides that are recognized as foreign antigens, such as HBsAg (Hepatitis B),30 p24 (HIV),31 and pp65 (cytomegalovirus).32
Common Cancer Antigens in HCC
In the following section, we describe some well-studied CCAs, such as GPC3, AFP, WT-I, FOXM1, and ROBO1, which are overexpressed in HCC, which could be used for peptide vaccines using predicted or isolated peptides. CAAs in HCC are overexpressed, meaning that in comparison with healthy tissues, the tumor cells produce a lot of mRNA and proteins of these CAA. Consequently, it is considered that around 1 peptide for 10,000 degraded proteins is presented to MHC33 and that the production of MHC-I peptides is correlated to the mRNA quantity, it is likely that overexpressed CAAs is good to be considered as onco-antigen candidates for cancer vaccine.34
Glypican-3 (GPC3)
In physiological conditions, GPC3 regulates cell division and growth regulation through binding to growth factors via heparan sulfate chains.35,36 In addition, GPC3 is involved in pathological conditions, such as the Simpson-Golabi-Behmel syndrome, a genetic disease characterized by growth anomalies. Furthermore, GPC3 is highly expressed in some cancers, such as melanomas,37 ovarian clear cell carcinoma,38 lung squamous cell carcinoma,39 hepatoblastoma,40 nephroblastoma,41 yolk sac tumor,42 and hepatocellular carcinoma (HCC).35 Importantly, high GPC3 expression levels are associated with a poor prognosis in HCC.43 Because the expression of GPC3 is not observed in most normal tissues, except for the placenta, fetal liver, and fetal kidney, GPC3 is considered an ideal tumor-associated antigen to develop a cancer vaccine.44
Alpha Fetoprotein (AFP)
AFP is highly expressed in fetal tissues, but it is barely detected in adult tissues.45 AFP expression is commonly used for testicular cancer diagnosis and HCC, with levels higher than 400 μg/mL for an HCC diagnostic.46 As AFP is specifically overexpressed in HCC,47 targeting this antigen stands as a promising strategy. Recently, the interest of using high-avidity TCRs to induce an efficient anti-tumor response in HCC has been recently studied in a Japanese Phase I clinical trial.48 In this study, two AFP-derived peptides (AFP357 and AFP403) were injected into 15 patients with HCC, the toxicity of the treatment and the tumor evolution were monitored, and the high-avidity TCR of a patient who responded positively was cloned. Later on, the same team studied the potential of 14 predicted and AFP-derived peptides restricted to HLA class II, using the peripheral blood mononuclear cells from 47 patients with several types of liver cancer, including HCC.49 Four peptides showed interesting immune responses. Among them, AFP1 was the most active and presented a strong avidity for HLA-DR. Furthermore, patients expressing this peptide presented an increased survival. After HCC treatment, an increased frequency of peptide-specific T cells was observed in some patients with HLA-DRB1*1502, *0405, and *0901 alleles, demonstrating the interest of not only focus on class I but also research on the class II for a successful immunotherapy. Finally, it is important to mention that the AFP1 peptide is also presented by MHC class I, thus indicating the importance of developing immunotherapy considering the whole immune system.
Wilms’ Tumor-1 (WT-1)
Wilms’ tumor 1 (WT1) antigen, a part of a transcription factor encoded by a 50 kb gene that was discovered in 1990, is important for the development of the urogenital system.50 As a result of different mRNA splicing events, alternative start codons, and RNA editing, there are 24 isoforms of WT1, which have different functions. Similar to GPC3, WT1 is expressed in fetal tissues but not in healthy adult tissues. However, its expression is observed in chronic hepatitis and cirrhotic liver, both of which are associated with HCC. WT1 overexpression is oncogenic51 and is associated with poor prognosis in breast cancer52 and leukemia.53 In addition, a recent report detected the overexpression of the WT-1 factor in 95% of the tumors with worst prognostic analyzed.54 Furthermore, WT-1 overexpression is known to promote cell dedifferentiation and resistance to chemotherapy.55
Forkhead Box M1 (FOXM1)
FOXM1 is a proliferation-associated transcription factor that has four different isoforms. FOXM1 expression is a sign of active proliferation, such as that occurring in progenitor cells or regenerating tissues and also in several cancers.56 Overexpression of FOXM1 is associated with gene instability, cessation of senescence, and resistance to endocrine therapies in breast cancers.57 In addition, FOXM1 upregulates CCNB1, enhancing the proliferation of cells.58 FOXM1 is overexpressed in HCC when compared to that in non-tumor tissue, and its expression levels are correlated with tumor stage and tumor size.59 Hence, targeting FOXM1 is a suitable approach for a peptide vaccine. Some experiments have already been performed using pulsed dendritic cells with a cytoplasmic transduction peptide fusion protein of FOXM1 to evaluate the induced anti-tumor activity in HCC. In comparison with the control, a tumor regression with specific CTL activity was observed in the peptide condition.60 Furthermore, another study demonstrated that the use of FOXM1 predicted peptides to stimulate specific CTLs in vitro with human cells, confirming the potential for targeting this antigen.61
Roundabout Homolog-1 (ROBO-1)
ROBO-1 was initially discovered in Drosophila,62 after which Robo homologs have been discovered in several species, including humans. Robo belongs to the immunoglobulin superfamily of cell adhesion molecules, and its ligand is SLIT. Physiologically, these receptors are important for axon guidance in the ventral midline of the neural tube during neural development. ROBO-1 has been described to have an antitumor effect in breast cancer and pancreatic ductal adenocarcinoma,63 but it is overexpressed in HCC. The overexpression of ROBO-1 on the cell surface in HCC makes it an interesting target for CAR-T cell therapy as well as peptide-based vaccines.64
Development of Cancer Vaccine for HCC
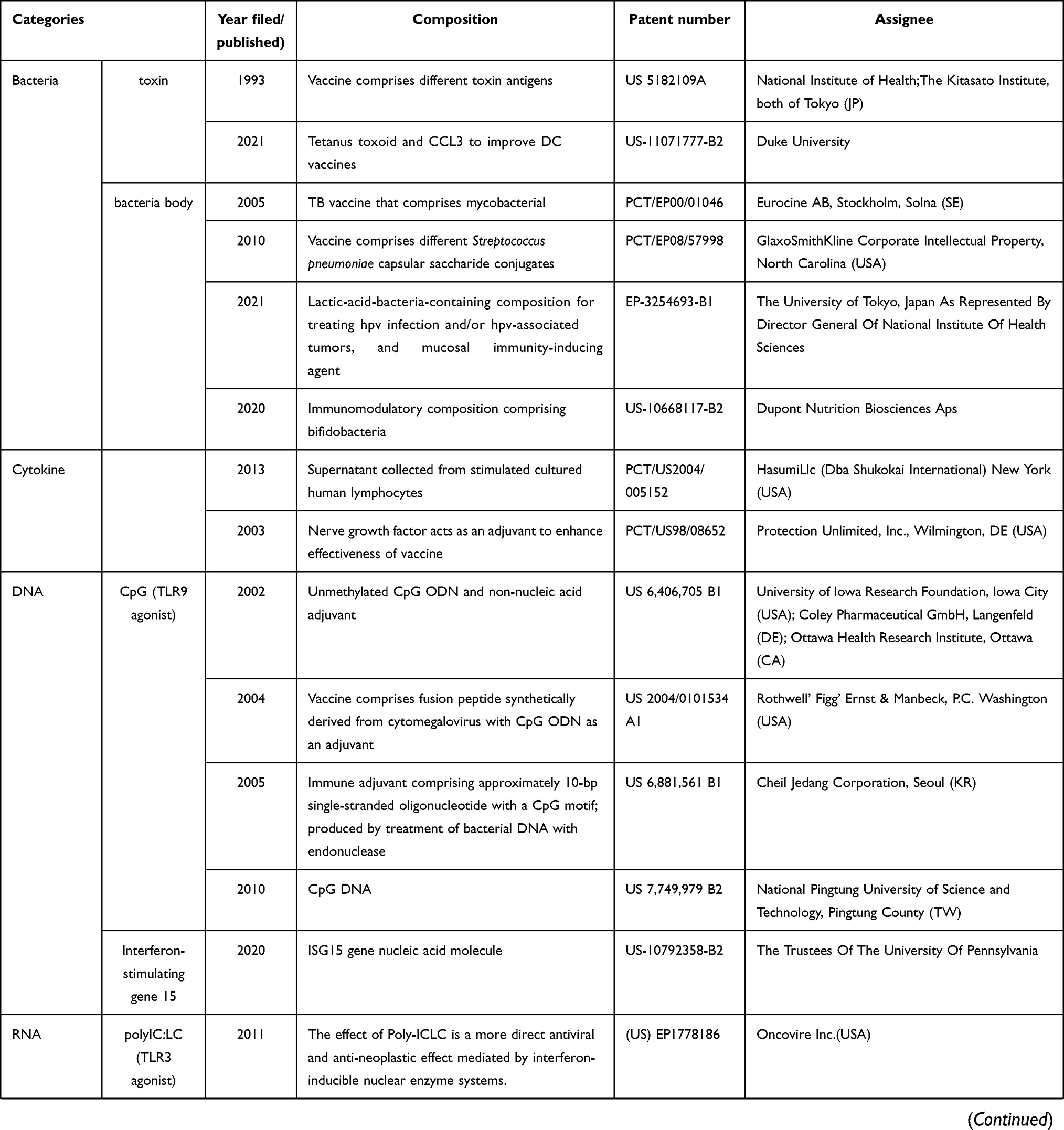
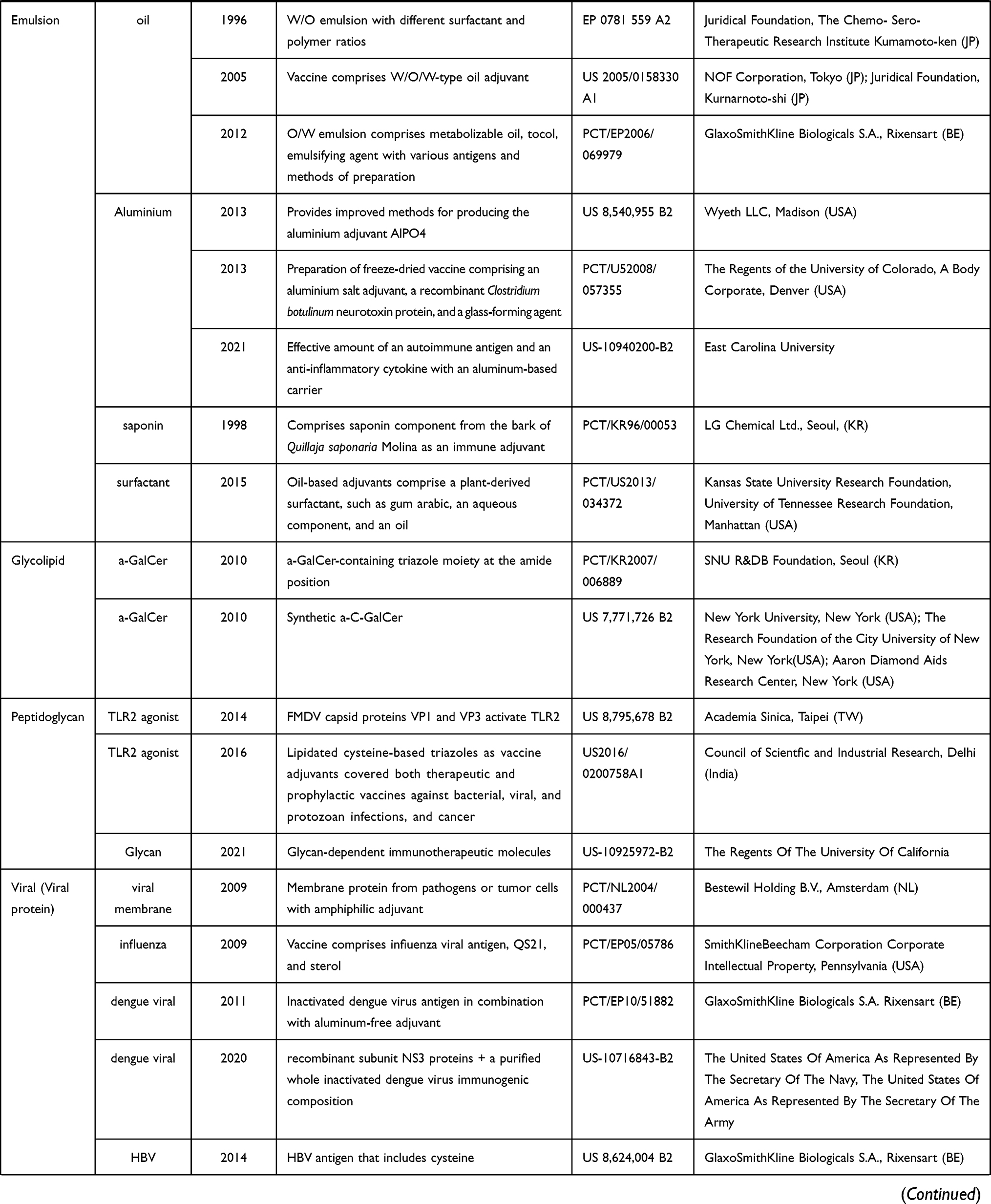
Historically, large-scale vaccination started after Edward Jenner’s experiment in the 18th century. Owing to vaccination, poliomyelitis, rinderpest and smallpox has been almost eradicated, and the number of people infected from diphtheria, tetanus, or pertussis has greatly diminished. The discovery of pathogens and a deep understanding of the functioning of the immunological system have helped unravel the mechanisms underlying protection immunity and the development of vaccines. How to efficiently establish a long-term and potent immunological memory against pathogens has been a critical research topic for a long time, including matters such as what kind of vectors and adjuvants, as well as the optimal type of vaccines, can best induce an effective immune response.65 DNA vaccines, consisting of a DNA plasmid encoding a certain antigen, have gained increasing attention over time. DNA vaccines can be directly injected into a specific site of the body, where APCs or other targeted cells take up the plasmid DNA and produce and expand the antigen. Another type of DNA vaccine that has been used for vaccination is the adenovirus, which allows the antigen genes to integrate into the host genome, and hence, the replication and expression of the gene at the same time as the cells do. However, these strategies involving the integration of a gene into the host genome convey the risk of inducing a mutation in the host genome. Hence, vaccines based on the transient expression of genes, inactivated pathogens, RNA, antigen proteins, or antigen peptides have been recently developed. Materials that are used as adjuvant for vaccine were summariz ed in Table 2, which is adapted from the review of Bonam et al,65 with new additions post-2015 made with the selection list using google patents and the terms, “adjuvant“, “vaccine“ and “cancer“, granted between 2015 to 2021 in US (USA) and EP (European).
 |
 |
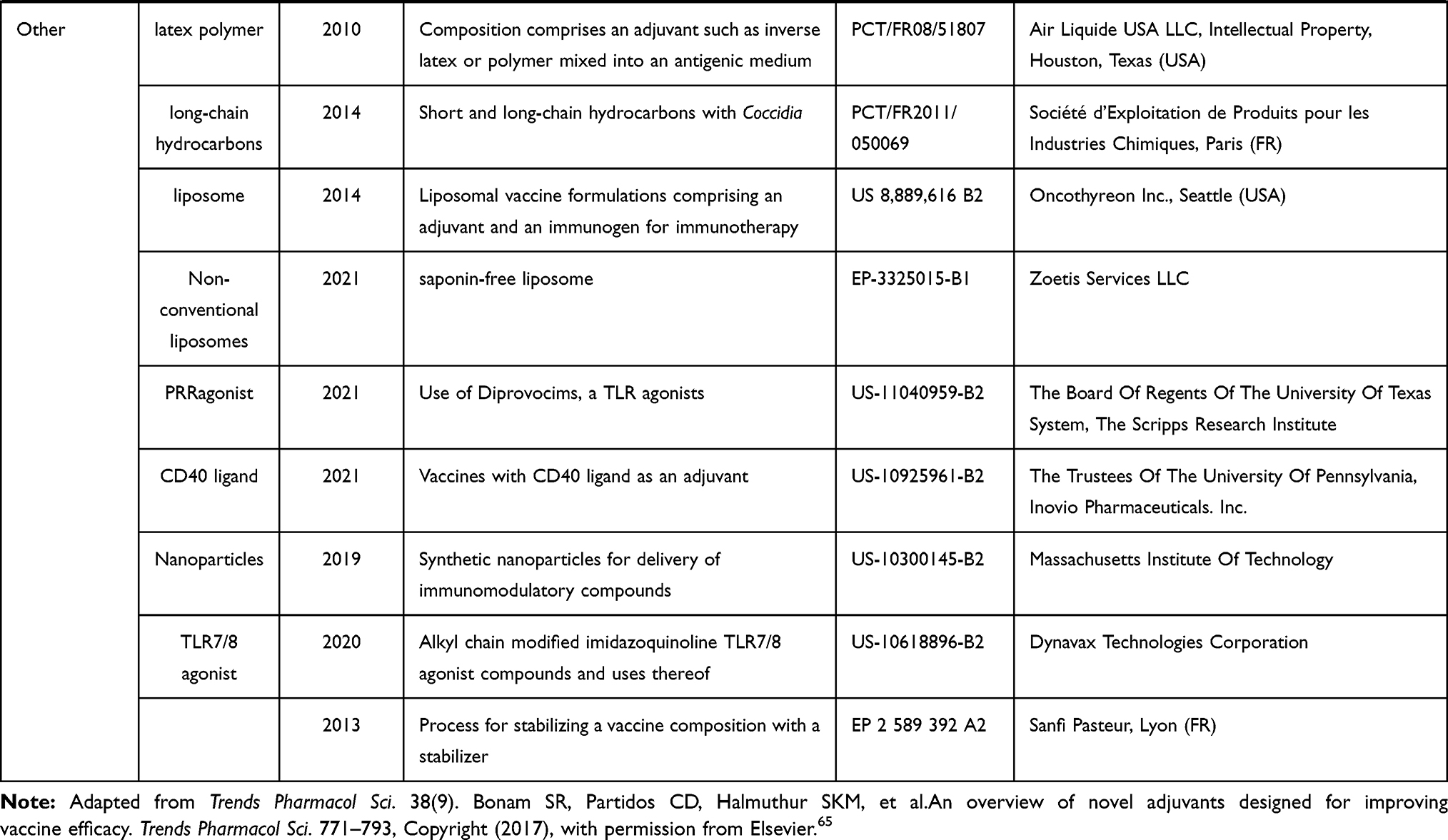 |
Table 2 Adjuvants Used for Vaccination |
In the 20th century, the identification of “cancer antigens” led to the development of vaccines to prevent, hopefully cure, cancer. Cancer vaccines need to induce a potent CTL response to regress tumor cells. In general, the antigenicity of tumor cells and their antigens is weaker than that of pathogens as a result of them originating from autologous cells. The development of cancer vaccines presents additional obstacles, including the chronic disease status of cancer patients, heterogeneity of tumor cells, and immunological tolerance against autologous tissues. A great variety of approaches have been developed to overcome these issues.66 Peptide vaccines are one of the most popular cancer vaccines. They consist of a peptide or a mix of linked or free peptides, combined with adjuvants that stabilize them and increase their efficacy. The specific peptide most suitable for developing a cancer vaccine is dependent on the specific type of cancer and the immunological characteristics of the patient. Despite this, peptide vaccines are frequently used for disease treatment because of their simplicity. Recent developments focused on various ways to target liver cancer. We can mention the use of a peptide vaccine cocktail67 with several antigens to increase the efficacy of antitumor response. The use of dendritic cells, an important platform for T cell activation, as vaccines has also demonstrated interesting results in several studies Table 3.68–74 Some other teams focused on the use of oncolytic viruses75 with results to be demonstrated in clinical trials in the future. Cold-inducible RNA binding protein73 or Tumor RNA-Loaded Lipid Nanoparticles74 have also recently been study and demonstrated potential, especially in use with ICI. At the time this review was written, a database search (https://clinicaltrials.gov/) using the key words “cancer” and “peptide vaccine” retrieved 282 completed clinical trials, 35 actives (not recruiting), and 73 currently recruiting clinical trials. A narrower search using the keyword “hepatocellular carcinoma” identified six studies, which are summarized in Table 4.
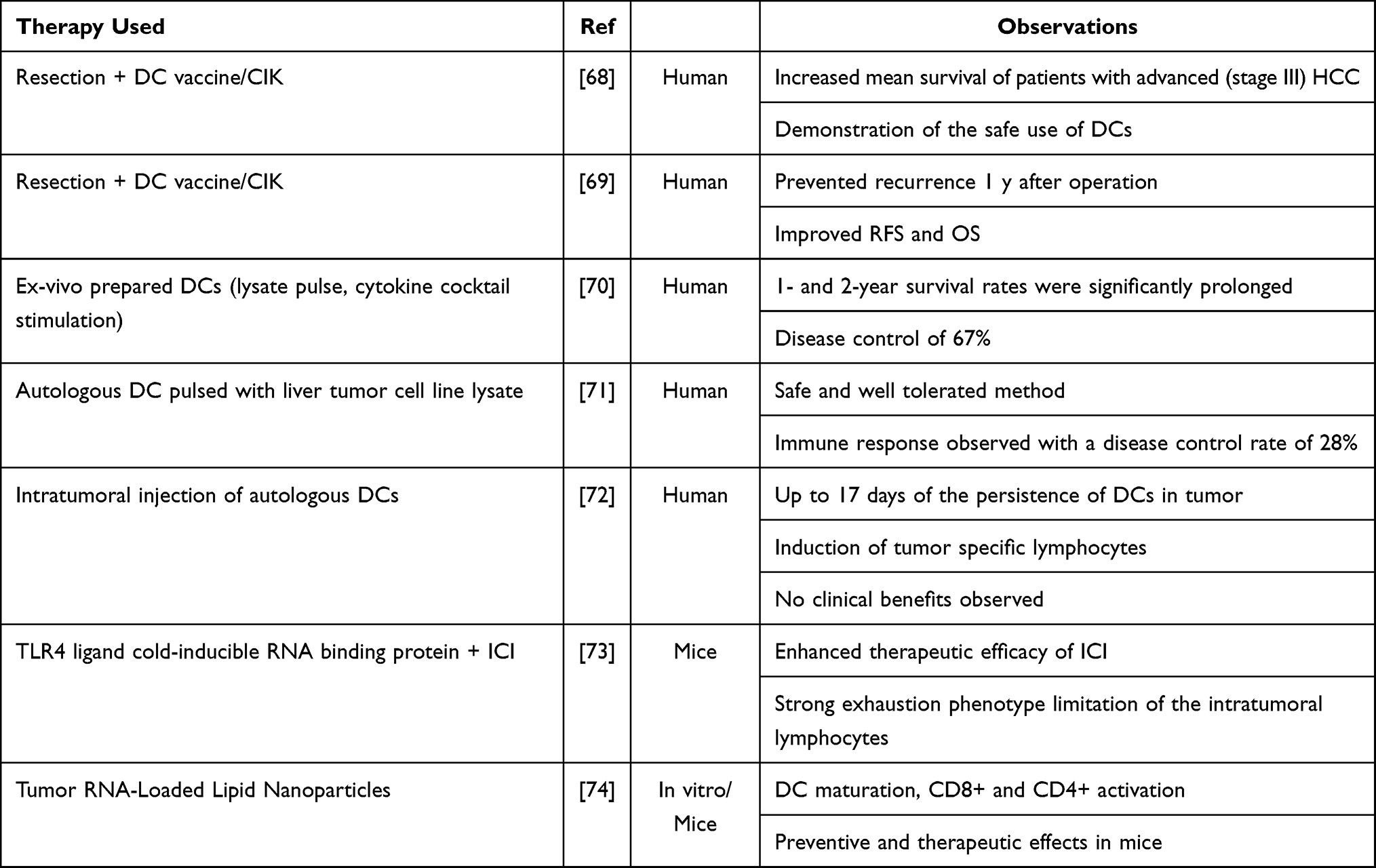 |
Table 3 Examples of Other Anti-Tumor Vaccine Methods |
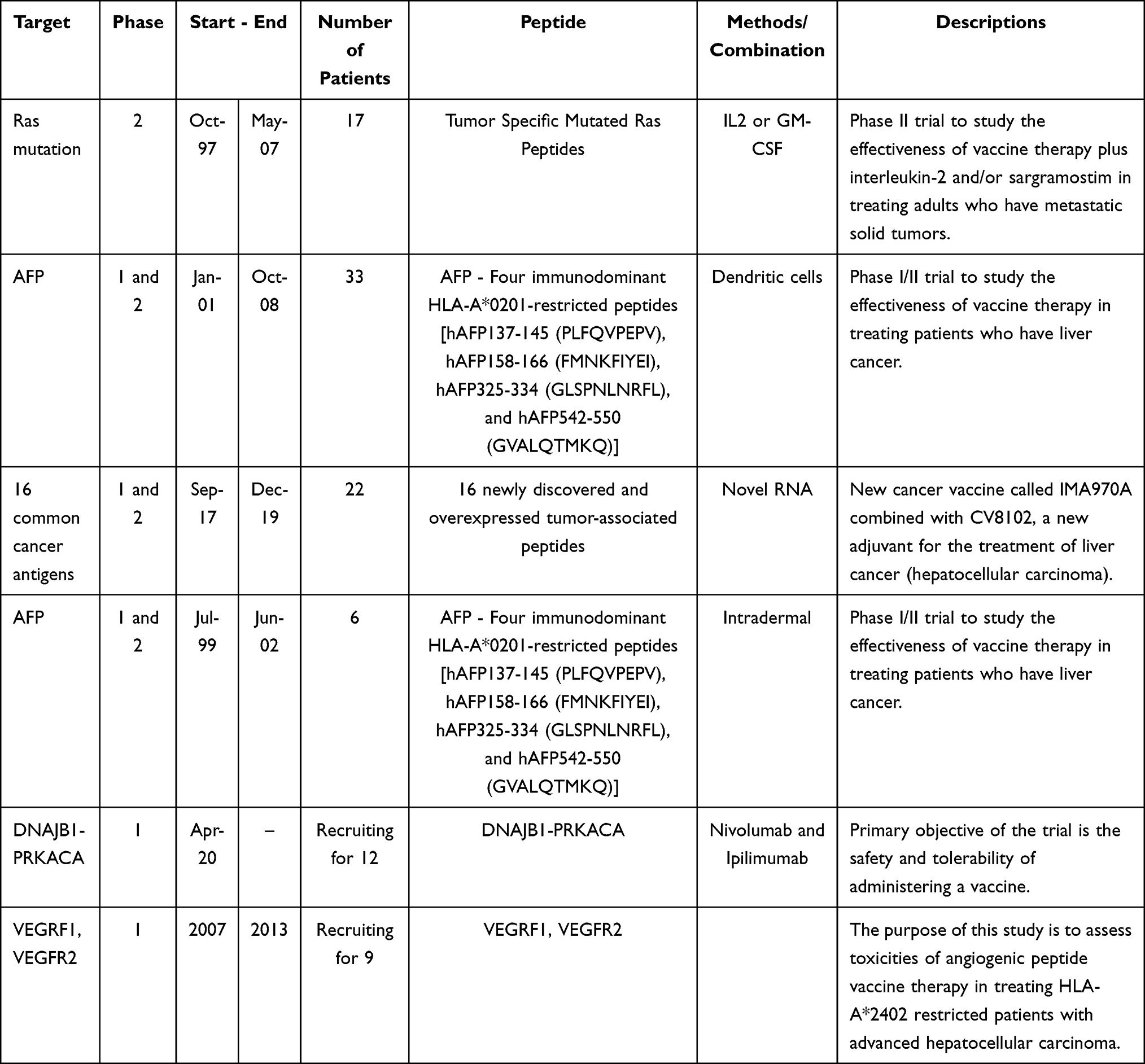 |
Table 4 Clinical Trial of Cancer Vaccines Targeting HCC |
Here, we summarize the results of our clinical trial targeting GPC3 and introduce our recent studies on the development of cancer vaccines targeting CCAs and neoantigens.
Phase I and II Clinical Trial of Peptide Vaccine Targeting Glypican-3
Several clinical trials have been performed and were simply summarized in the Figure 1. In 2011, we published the results of a peptide vaccine against glypican-3, which is frequently upregulated in HCC.76 From February 2007 to November 2009, 33 patients from the National Cancer Center Hospital East (Kashiwa, Japan) with advanced HCC were enrolled in a phase I trial to study the safety and immunogenic response of the peptide vaccine.77 Half of the patients with the allele HLA-A24 were vaccinated three times, every two weeks using an escalation method with the GPC3298–306 peptide (EYILSLEEL), and patients with the allele HLA-A02 vaccinated with the GPC3144–152 peptide (FVGEFFTDV). Both HLA types were chosen due to their high frequency in the population, with approximately 60% of the Japanese population bearing HLA-A24 and 40% of Caucasian population bearing HLA-A02. This study showed, for the first time, a correlation between the frequency of peptide-specific cytotoxic T lymphocytes and the overall survival of the patients as well as the safety of the use of the GPC3 peptide vaccine (Figure 2A).
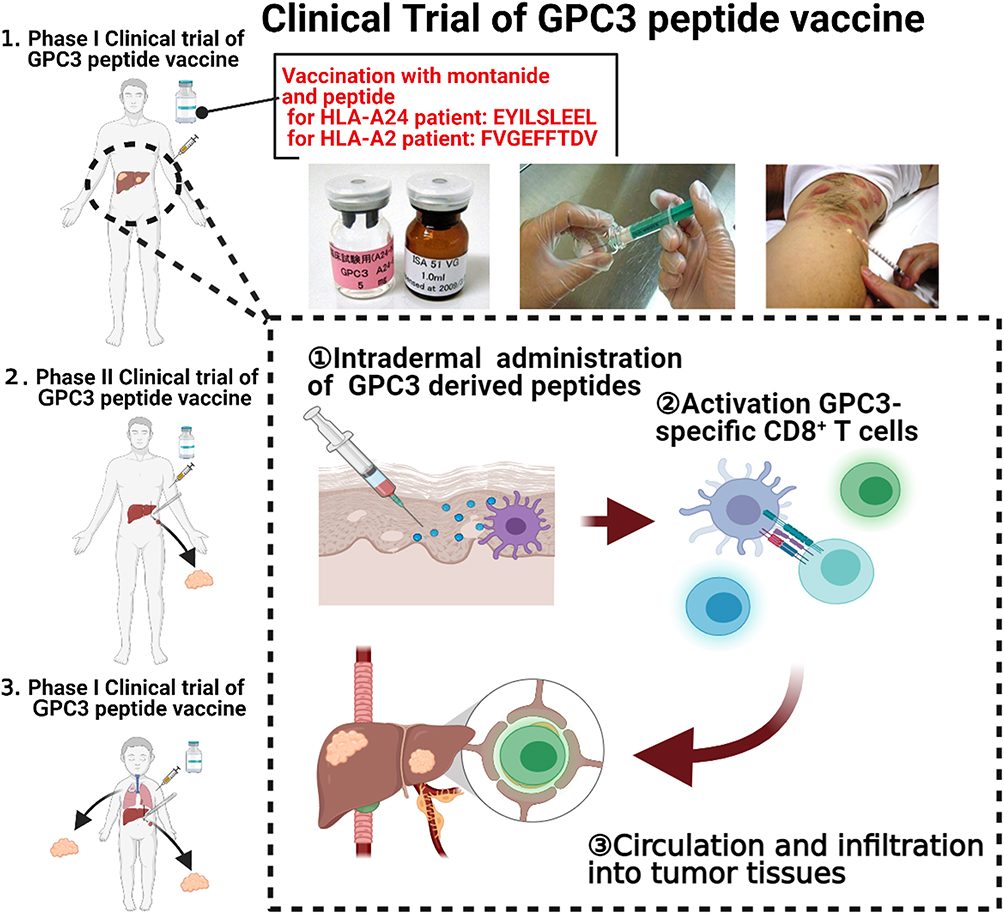 |
Figure 1 Scheme of our peptide vaccine trial targeting GPC3. Pictures show a series of steps to synthesize the vaccine. A24- or A2-restricted short peptides (bottle with pink label) were well mixed with montanide (bottle with white label) to prepare emulsions, and then administered to the patient intradermally. To induce a peptide-specific immune response, a series of physiological immune systems may occur in vivo (lower schemes). ① Antigen presenting cells (APCs), such as dendritic cells or Langerhans cells located in the intradermis, take up peptides of GPC3, then migrate to draining lymph nodes, where they mature. ② In draining lymph nodes, APCs present the peptides to GPC3 reactive CD8+ T cells and induce their activation and expansion. Activated CD8+ T cells differentiate into cytotoxic T lymphocytes (CTLs), infiltrate into the systemic circulation, and infiltrate into tumor tissues, resulting in tumor rejection. Using these peptide vaccines targeting GPC3, we performed phase I/II clinical trials (left figure): 1. Phase I clinical trial of GPC3 peptide vaccine in advanced HCC patients. 2. Phase II study of GPC3 peptide vaccine as an adjuvant therapy for HCC patients who underwent surgery or radiofrequency ablation. 3. Non-randomized, open-label, phase I clinical trial of GPC3 peptide vaccine in pediatric solid tumors. |
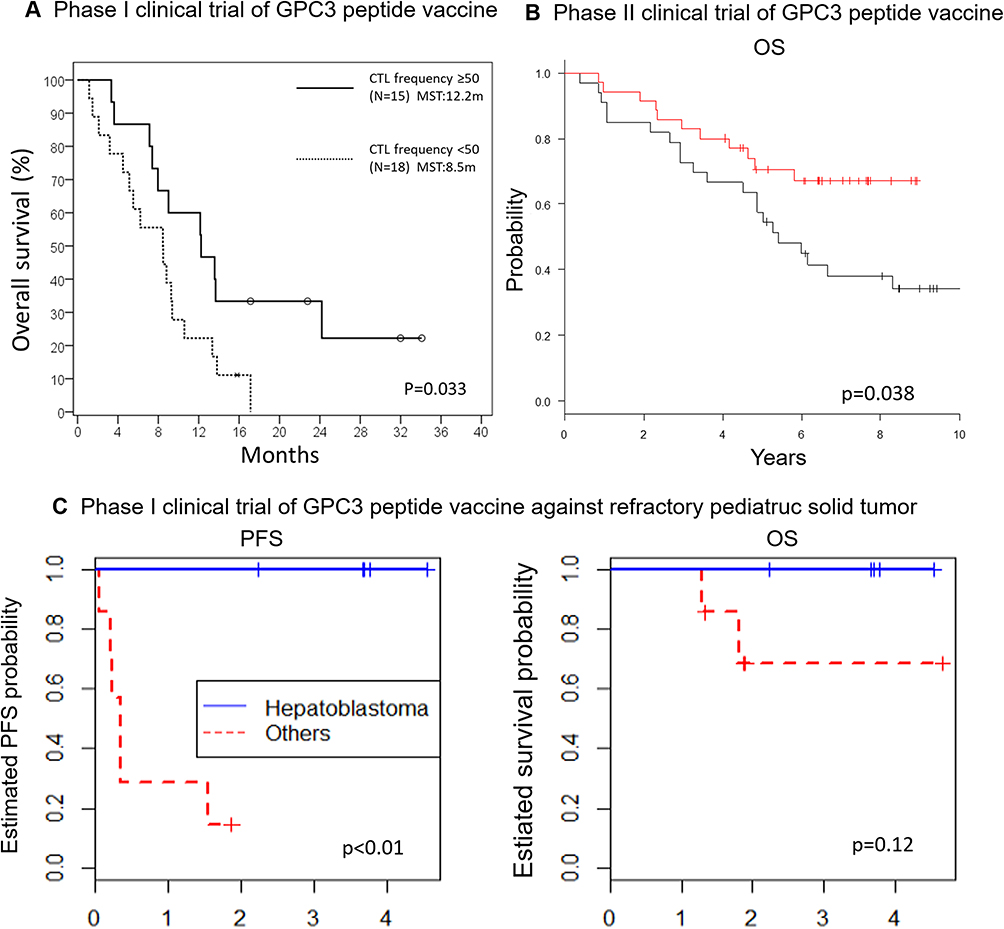 |
Figure 2 Overall survival (OS) or Progress free survival (PFS) of patients who had been experienced with vaccine in phase I/II clinical trials of GPC3 peptide vaccine. The details are shown in Figure 1. (A) Kaplan–Meier curves for OS of advanced HCC patients in a phase I clinical trial of GPC3 peptide vaccine.77 Patients with GPC3-specific CTL frequencies ≥50 (bold line) had a longer survival than those with GPC3-specific CTL frequencies <50 (dotted line) (P = 0.033). (B) Kaplan–Meier curves for OS in a phase II clinical trial of adjuvant GPC3 peptide vaccine.80 Among patients with GPC3-positive HCC, patients that underwent surgery and were vaccinated (red line) tended to have longer recurrence-free and overall survival rates than those who underwent surgery alone (black line). (C) Kaplan-Meier curves for PFS and OS in a phase I clinical trial of GPC3 peptide vaccine with pediatric solid tumors.93 Hepatoblastoma patients in the partial remission group (blue line) showed longer PFS and OS than those with other pediatric solid tumors (red line). Abbreviation: MST, median survival time. |
Since radiofrequency ablation leads to an enhancement of specific T cells against HCC-associated antigens or GPC3, the same team decided to perform a single-arm Phase II study, in which 41 patients were enrolled to receive a GPC3-derived peptide vaccine as adjuvant therapy.78 Although the primary endpoint was not successfully reached for 1- and 2-year recurrence rates, the presence of GPC3 peptide-specific CTL responses was detected in 35 of the 41 patients. It is interesting to note that two patients who presented GPC3-positive primary tumors and experienced relapses did not present GPC3 in the recurrent tumors. Indeed, the GPC3 peptide-specific CTL successfully killed the tumor cells expressing GPC3, leaving the GPC3-negative cells to expand by selective pressure. This observation highlights the necessity of using different antigen peptides simultaneously to increase the chance of tumor regression and reduce the risk of recurrence. Another important observation was the exhausted state of the specific CTLs post-vaccination, determined by PD-1 or CTLA-4 expression, which can be overcome by ICIs. Finally, another pilot study about the use of the GPC3 peptide vaccine against advanced HCC has been performed and it demonstrated the intra-tumoral infiltration of GPC3-specific peptides CTLs, the correlation between the CTLs frequency and the overall survival, the similar TCRs repertoire by comparing tumor infiltrated cells and PBMCs and they managed the isolation of GPC3 peptide-specific CTL clones.79
Based on the positive results on this vaccine in the previous phase I clinical trial, we attempted to investigate the underlying immune mechanisms. For this purpose, five patients with HLA-A02 and six patients with HLA-A24 who were vaccinated with the same peptides previously described were selected. Most of the selected patients showed progressive disease and a poor response to sorafenib. Before- and after-vaccination tumor biopsies revealed the presence of GPC3-specific infiltrated CTLs, which was correlated with a better overall survival. The autopsy of a patient who received two injections showed a strong immune response with infiltrating CD8+ CTLs and tumor lysis. Although the disease continued to progress, the trial proved that an immune response was induced. A clone was successfully isolated for further analysis and experiments, including development of clinical TCR-engineered T cells (Figure 2B). Moreover, we have reported important observations in a phase I clinical trial of a GPC3 peptide vaccine against advanced pediatric solid tumors, including malignant hepatoblastoma. In this clinical trial, seven of 18 patients presented hepatoblastoma with uniform expression of GPC3. Of this cohort, two patients were not evaluated, and the rest showed complete responses (CR) and did not recur for more than six years (Figure 2C). These clinical trials stand as a proof of concept that peptide vaccines targeting GPC3 are an adequate therapy to prevent relapse and have contributed to a further understanding of the mechanisms of action of the vaccine.80 The number of patients who entered and received GPC3 peptide vaccine was however insufficient for the consideration of the efficacy of peptide vaccine against HCC. Finally, a proof-of-concept was successfully performed using glypican-3 and additional peptides, thus paving the way for treatment using various peptides and antigens.
Personalized Vaccine Targeting Common Cancer Antigens
The peptide vaccine using GPC3 was insufficient to induce a complete regression of the tumors. We are currently developing novel cocktail vaccines targeting various TAAs expressed in HCC, such as FOXM1, AFP, ROBO1, WT-1, and GPC3. As mentioned above, TAAs are frequently overexpressed in tumor tissues, but with their efficacy is debatable; as they have an autologous origin, they are not completely considered abnormal by the immune system. In addition, the risk of autoimmunity is increased. Hence, TAA-based peptide vaccines should be carefully developed, and after having performed clinical trials with GPC3, we wanted to explore other CAAs expressed in HCC to increase the efficacy spectra to kill tumor cells, therefore we have now started to research about. The scheme for the development of vaccines targeting common cancer antigens is shown in Figure 3.
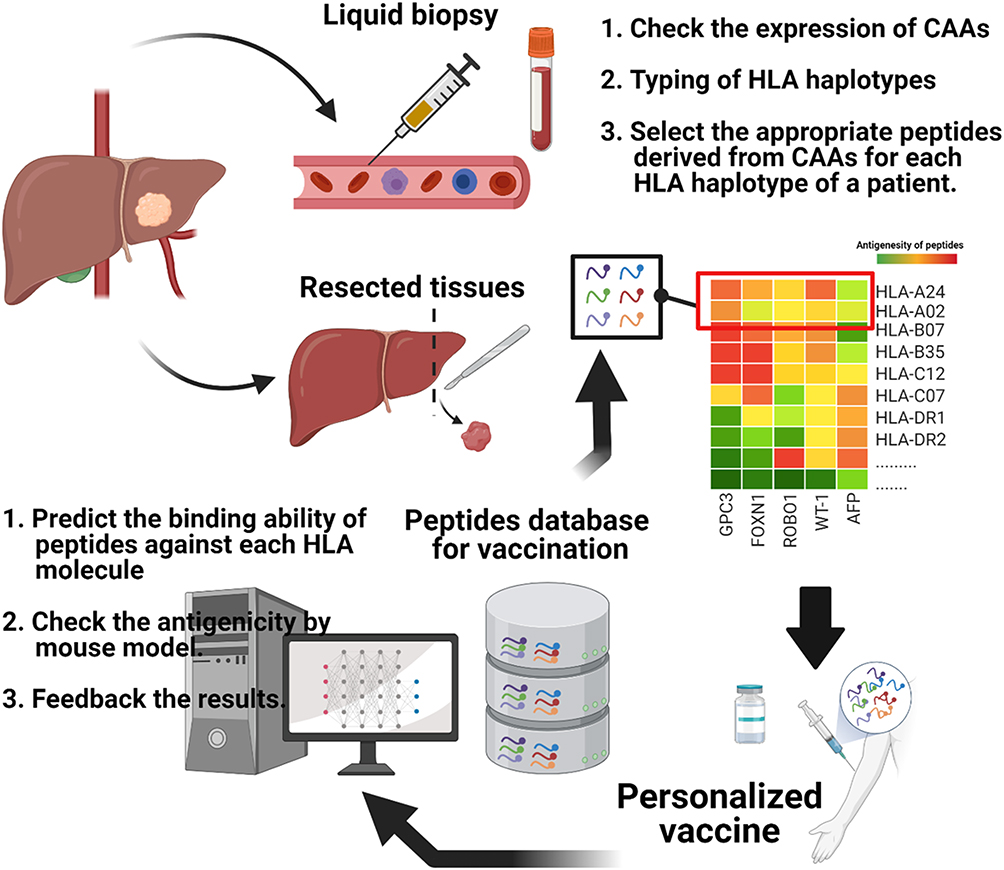 |
Figure 3 Scheme of personalized vaccination strategy targeting cancer associated antigens (CAAs). Personalized vaccines can be realized using a peptide prediction system for vaccines based on an off-the-shelf database of peptides derived from various types of CAAs. |
Personalized Vaccine Targeting Neoantigens
“Neoantigens” are tumor-specific antigens that appear as a result of novel mutations in tumor cells.81,82 These proteins are degraded by cells with the ubiquitin-proteasome system, resulting in the production and release of several peptides, including those that present the novel non-synonymous mutation, which are termed “neoantigens”. Tumors with high tumor mutational burden (TMB) present a higher number of neoantigens. However, HCC usually has low TMB,83 and hence, a deep analysis of the potential mutations and the development of personalized treatment for each individual should be performed to develop vaccines based on neoantigens, instead of solely relying on TMB.84–86 Furthermore, a deep understanding of the underlying mechanism is needed, as the neoepitope generated might not correspond to the HLA of the patient, HLA allele-specific expression loss frequently occurs in tumors, and only some neoepitopes that will successfully be presented on the MHC may trigger specific T-cells if there is no strong immunosuppression.87,88 What kind of neoantigen is the best for developing a potential peptide vaccine in HCC that is able to induce immune memory still needs to be determined.89,90
To evaluate the immunogenicity of neoepitopes derived from gene mutations in HCC, we examined whether neoepitopes could induce significant immune responses in vivo by repeatable vaccination of transgenic (Tg) mice expressing human HLA. Whole exome sequencing (WES) of resected HCC tissues in patients who have HLA-A24 or A02 and HLA typing were performed to find somatic mutations that occurred in the tumor cells. Using our original in silico prediction algorithm, neoantigens were predicted from nonsynonymous somatic mutations and frameshift mutations and subsequently synthesized. Then, peptides derived from genes conserved between mice and humans were selected and used for vaccination of HLA-A24 or A2-Tg mice using poly-ICLC as an adjuvant for a total of three times. Until now we used IFA for peptide vaccines, but it was a poor immunological adjuvant, this is why we used poly-ICLC, and in parallel, we are checking which other adjuvants would be the best (CpG, mRNA …). Splenocytes were harvested from vaccinated mice, and the response of T cells to predicted peptides was assayed by IFN-g ELISPOT assay (Figure 4A and B). Positive responses to peptide injection were observed in 43% of the A24-Tg mice, and in 41% of the A02-Tg mice. These responses were mutation-specific and did not occur against the wild-type peptide (Figure 4C). These results indicate the immunogenicity of the neoantigen epitope. Our prediction pipeline was shown to be generally accurate and capable of extracting antigenic neoantigen peptides, since many or the peptides that induced an immune response were selected by the algorithm (Figure 4D). However, some of the HLA-A02 peptides showed immune activity, including peptides with low scores predicted by the algorithm. It is necessary to improve the prediction accuracy of the algorithm by integrating new information and results from further analyses. Our results demonstrated the availability of HLA-Tg mouse models to validate prediction algorithms based on physiological immune response in vivo. Moreover, these vaccination models also are useful for the check any adverse effects and off target effect by vaccination. Now, we are preparing for publication of these results.
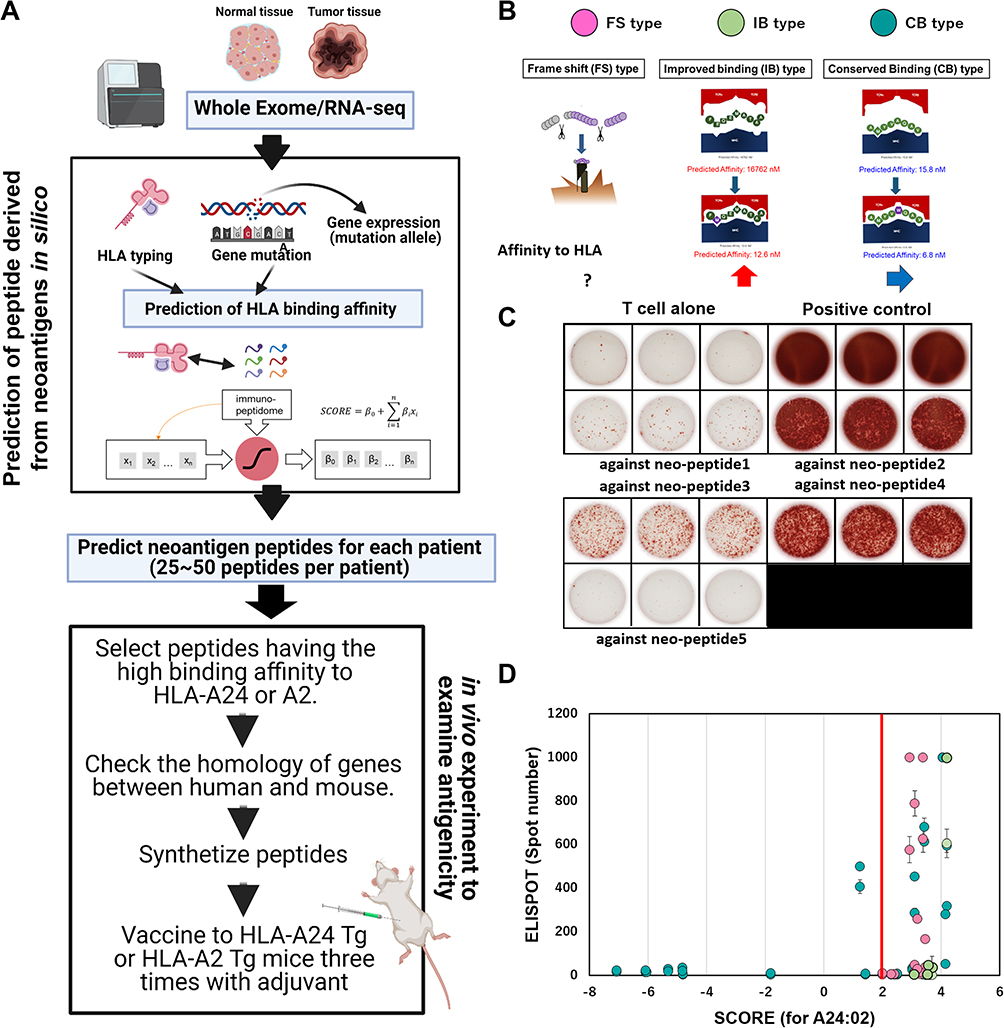 |
Figure 4 Scheme for the development of personalized neoantigen vaccines at the National Cancer Center. (A) In silico neoantigen prediction algorithm and its antigenicity validation DNA and RNA were extracted from the patient’s tumor tissue, and whole exon analysis and RNA-sequencing analysis were performed using a next-generation sequencer. From the omics analysis of gene mutations and their expression in each patient, neoantigen-derived peptides that can bind to the patient’s HLA were predicted in silico. We developed our own prediction algorithm in collaboration with BrightPath Biotechnology, Inc. A total of 25–50 predicted peptides were synthesized according to the prediction score per patient. For experiments using human HLA-transgenic mice (human HLA-Tg) experiments, peptides with wild-type sequences homologous to mice were selected and used for peptide vaccination with polyIC:LC as an adjuvant. After repeated vaccination with human HLA-Tg, spleen cells were collected, and immune responses to the peptides were evaluated using IFN -ELISPOT assay. (B) Classification of predicted neoantigen peptides by HLA binding affinity. Mutation-derived peptides were classified into three groups: frame shift (FS) type, improved binding (IB) type, and conserved binding (CB) type, based on the change in HLA-binding affinity depending on the location of the mutation in the peptide. (C) Validation of neoantigen peptide antigenicity using human HLA-Tg. Strong induction of immunity to neoantigens 02, 03, and 04 was confirmed. (C) Association between antigenicity prediction scores and immune responses to peptides (D). Correlation between immunogenicity in HLA-A24-Tg and the prediction score of our algorithms in HLA-A24. Each dot shows a peptide that was predicted and assessed for its immunogenicity in vivo. The color of the dots shows the neoantigen types in (B); red is FS type, blue is IB type, and green is CB type. |
To date, various clinical trials of cancer vaccines targeting neoantigens have been conducted, mainly by venture companies (Table 5). A vaccine with ICIs and peptides derived from DNAJB1-PRKACA fusion kinase is currently being tested for fibrolamellar hepatocellular carcinoma in Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins (NCT04248569). However, since the majority of these trials are still in Phase I, the therapeutic efficacy still remains to be determined. Some studies have reported the induction of CTL and helper T cells in patients responding to the administered neoantigen vaccines. The final goal of our study was to develop adjuvant vaccines targeting TSAs to prevent early recurrence and metastasis, resulting in a better prognosis for HCC.
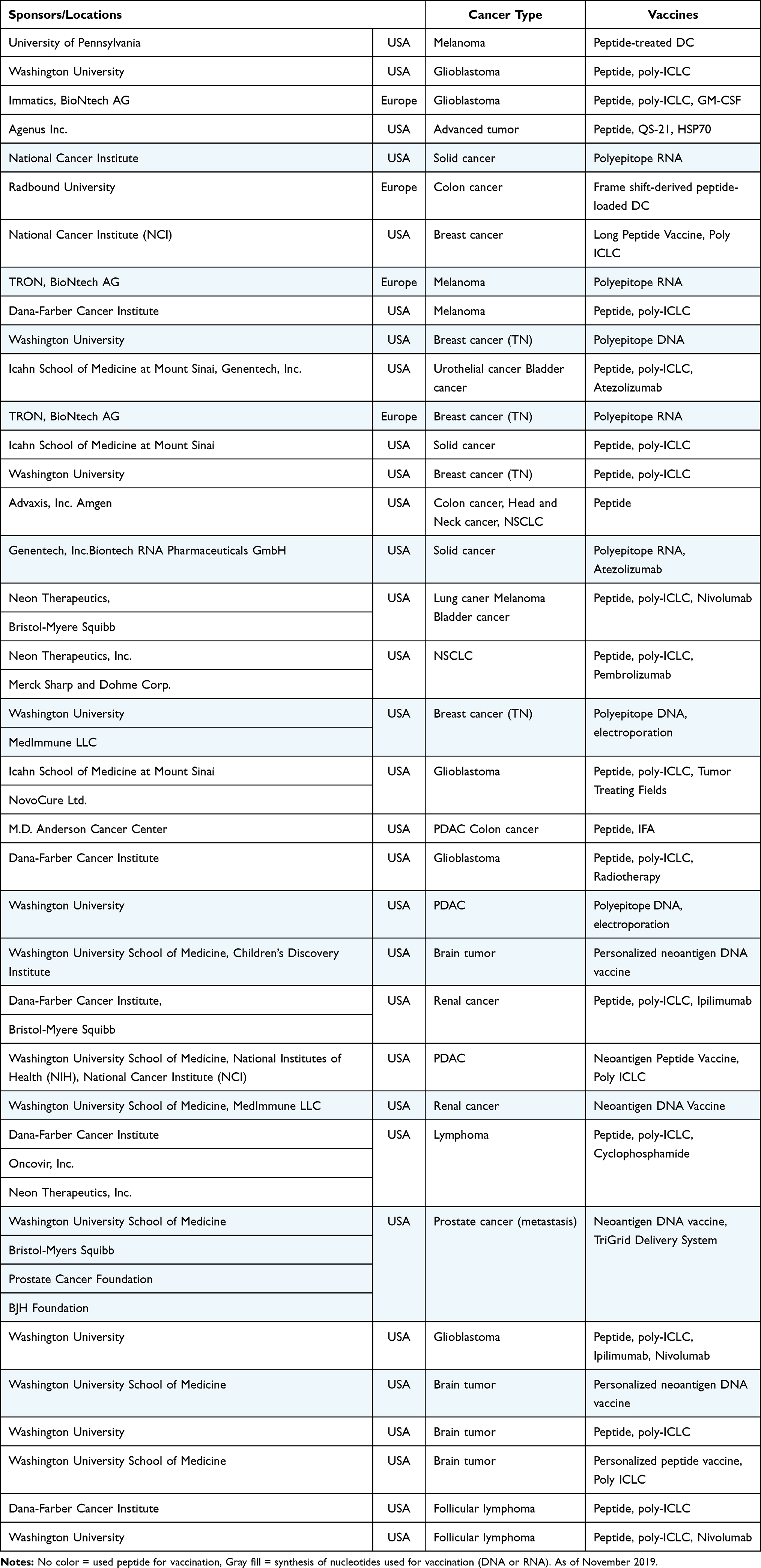 |
Table 5 The Development of Personalized Vaccines Targeting Neoantigens |
Summary
In summary, this review discusses several examples of positive results using peptide vaccines as a treatment for cancer. The induction of CTL in humans is promising. Furthermore, the technique is simple. We also described several common cancer antigens in HCC, which have been targeted in cancer treatment with peptides. Moreover, we discussed the potential for not focusing only on one cancer antigen or one peptide. Using previously reported studies, we concluded that some peptide vaccines against HCC antigens are efficacious and lead to potential tumor regression. Hence, there is a strong interest in peptide vaccine immunotherapy using a mix of common cancer antigens containing numerous and various epitopes for both HLA class I and II. However, the achieved efficacy is still partial, and so far, no total and definitive recovery has been observed. Cancers are complex from a physical molecular diversity perspective, with several mutations and adaptive mechanisms, challenging the innovation with the immunotherapies used. Advantages of TAAs-based vaccines are the density of peptides that are presented on HLA molecules by direct binding, the cost-effectiveness, the use of a peptide or a mix of peptides relevant for an immune response decreases the risk of contamination observed for inactivated or attenuated vaccines and reduces problems linked to autoimmunity and immune evasion. However, disadvantages are the necessity to use peptides having high immunogenicity, hence the interest of a higher in silico prediction or peptidome analysis, and the HLA haplotypes diversity which remains a strong challenge to design tailor-made vaccines with peptides against multiple antigens (including neoantigens). The need for strong antigens has led to intensive research on neoantigens specific to cancers, which are thought to be more immunogenic, leading to enhanced immunological responses. The use of strong adjuvants is also required for peptide vaccines. We have tried to use peptide vaccines that prevent recurrence as adjuvant therapy. For treatment of progressive cancers, peptide vaccine alone is not enough to induce regression, so it would require a peptide vaccine combined with powerful therapy such as ICI or radiotherapy. Our clinical trial suggests that the use of peptide vaccine as adjuvant therapy after surgery is good to prolong overall survival, especially for hepatoblastoma patients. Vaccine are usually used to prevent infections, so we want to use cancer vaccine to prevent recurrence or de novo tumor for patient with high risk factors (for HCC or other cancers). However, these points still need deeper investigation. The main aspect remaining to be resolved for the development of successful peptide vaccines is identifying which targets are the most effective. As previously mentioned, several antigens are overexpressed and mutated, but not all lead to a complete response. One reason why these peptide vaccines failed to induce tumor regression, despite the appearance of CTL by vaccination, may be that the CAA-derived peptides used for vaccination were not presented to HLA molecules on the tumor cell surface. HLA-ligandome analysis based on the identification of amino acid residues by mass spectrometry is an interesting, attractive and important approach to identified peptides that are presented on HLA therefore may be recognized by CTLs during tumor rejection. However, proteomics analysis has issues, only some peptides might likely be ionized during mass spectrometry analysis so it is difficult to find every peptide that may be presented on HLA. We used HLA ligandome analysis to determine neo-epitopes, but it is not enough and new techniques are required.91 Concerning neoantigen-based immunotherapy, challenges are the still low power prediction of algorithms to efficiently predict immunogenic peptide-derived neoantigens. Also, the poor research and prediction for CD4+ T cells needs research. However, concerning low frequency neoantigens in low mutational burden tumors, recent study demonstrated the use of radiotherapy to induce mutation, therefore neoantigens.92 Neoantigens belong to the intra-tumor heterogeneity, therefore, it is necessary to identify all neoantigens present in tumors to get an efficient treatment. These issues need further investigation in the future.
Data Sharing Statement
The results that support the findings of Figure 4 are not publicly available due to our preliminary data. Now, we are going to submission for any public journal.
Acknowledgments
We specially thank Takashi Yamada, Kazushi Hiranuka, Noriko Watanabe, Yuji Mishima, Norihiro Nakamura, and the members of Brightpath Bio Co. Ltd (Tokyo, Japan) for the development of our prediction pipelines for neoantigen vaccines (shown in Figure 4), omics analysis with NGS, prediction of peptides, and synthesis of peptides.
Funding
This study was funded by the collaboration fee with the corresponding author and Brightpath Bio Co. Ltd (Tokyo, Japan). Although the funding body mainly contributed to the results of Figure 4, Brightpath Bio Co. Ltd had no control over the writing, or publication in this review.
Disclosure
Tetsuya Nakatsura is currently receiving royalties from Onco Therapy Science, Inc. and fundamental research funding support from Thyas Co., Ltd, BrightPath Biotherapeutics Co. Ltd. and Takara Bio Inc. Other authors declare that they have no commercial or financial relationships that could be construed as a potential conflict of interest. No potential conflict of interest was reported by the other authors.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Galle PR, Forner A, Llovet JM. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236.
3. Tampaki M, Papatheodoridis GV, Cholongitas E. Intrahepatic recurrence of hepatocellular carcinoma after resection: an update. Clin J Gastroenterol. 2021;1–5. doi:10.1007/s12328-021-01394-7
4. Forner A, Reig M, Bruix PJ. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314.
5. Xia S, Pan Y, Liang Y, et al. The microenvironmental and metabolic aspects of sorafenib resistance in hepatocellular carcinoma. EBioMedicine. 2020;51:102610. doi:10.1016/j.ebiom.2019.102610
6. Al-Salama ZT, Syed YY, Scott LJ. Lenvatinib: a review in hepatocellular carcinoma. Drugs. 2019;79(6):665–674. doi:10.1007/s40265-019-01116-x
7. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–668.
8. Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun. 2020;11(1):3801. doi:10.1038/s41467-020-17670-y
9. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. 2020;10(3):727–742.
10. Salama AKS, Hodi FS. Cytotoxic T-lymphocyte–associated antigen-4. Clin Cancer Res. 2011;17(14):4622. doi:10.1158/1078-0432.CCR-10-2232
11. Zhang Z, Liu S, Zhang B, et al. T cell dysfunction and exhaustion in cancer. Front Cell Dev Biol. 2020;8:17. doi:10.3389/fcell.2020.00017
12. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894–1905. doi:10.1056/NEJMoa1915745
13. Dendrou CA, Petersen J, Rossjohn J, et al. HLA variation and disease. Nat Rev Immunol. 2018;18(5):325–339. doi:10.1038/nri.2017.143
14. Rishi VL, Rui Z, Asha DV, Bing X. CD4+T cells: differentiation and functions. Clin Dev Immunol. 2012;2012:925135. doi:10.1155/2012/925135
15. Basu R, Whitlock BM, Husson J, et al. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell. 2016;165(1):100–110. doi:10.1016/j.cell.2016.01.021
16. Gu X, Fu M, Ge Z, et al. High expression of MAGE-A9 correlates with unfavorable survival in hepatocellular carcinoma. Sci Rep. 2014;4(1):6625. doi:10.1038/srep06625
17. Raza A, Merhi M, Inchakalody VP, et al. Unleashing the immune response to NY-ESO-1 cancer testis antigen as a potential target for cancer immunotherapy. J Transl Med. 2020;18(1):140. doi:10.1186/s12967-020-02306-y
18. Edoo MIA, Chutturghoon VK, Wusu-Ansah GK, et al. Serum biomarkers AFP, CEA and CA19-9 combined detection for early diagnosis of hepatocellular carcinoma. Iran J Public Health. 2019;48(2):314–322.
19. Malavaud B, Miédougé M, Payen J-L, et al. Prostate-specific antigen in acute hepatitis and hepatocellular carcinoma. Prostate. 1999;41(4):258–262. doi:10.1002/(SICI)1097-0045(19991201)41:4<258::AID-PROS6>3.0.CO;2-1
20. Villanueva A, Hoshida Y. Depicting the role of TP53 in hepatocellular carcinoma progression. J Hepatol. 2011;55(3):724–725. doi:10.1016/j.jhep.2011.03.018
21. Sueangoen N, Tantiwetrueangdet A, Panvichian R. HCC-derived EGFR mutants are functioning, EGF-dependent, and erlotinib-resistant. Cell Biosci. 2020;10(1):41. doi:10.1186/s13578-020-00407-1
22. Shi J-H, Guo W-Z, Jin Y, et al. Recognition of HER2 expression in hepatocellular carcinoma and its significance in postoperative tumor recurrence. Cancer Med. 2019;8(3):1269–1278. doi:10.1002/cam4.2006
23. Zhou XU, Lu J, Zhu H. Correlation between the expression of hTERT gene and the clinicopathological characteristics of hepatocellular carcinoma. Oncol Lett. 2016;11(1):111–115. doi:10.3892/ol.2015.3892
24. Kannangai R, Wang J, Liu QZ, et al. Survivin overexpression in hepatocellular carcinoma is associated with p53 dysregulation. Int J Gastrointest Cancer. 2005;35(1):53–60. doi:10.1385/IJGC:35:1:053
25. Ma T, Su Z, Chen L, et al. Human papillomavirus type 18 E6 and E7 genes integrate into human hepatoma derived cell line hep G2. PLoS One. 2012;7(5):e37964. doi:10.1371/journal.pone.0037964
26. Vigneron N. Human tumor antigens and cancer immunotherapy. BioMed Res Int. 2015;2015:948501. doi:10.1155/2015/948501
27. Monach PA, Meredith SC, Siegel CT, et al. A unique tumor antigen produced by a single amino acid substitution. Immunity. 1995;2(1):45–59. doi:10.1016/1074-7613(95)90078-0
28. Levine AJ. P53 and the immune response: 40 years of exploration-A plan for the future. Int J Mol Sci. 2020;21(2):541. doi:10.3390/ijms21020541
29. Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25(38):5220–5227. doi:10.1038/sj.onc.1209615
30. Rodella A, Galli C, Terlenghi L, et al. Quantitative analysis of HBsAg, IgM anti-HBc and anti-HBc avidity in acute and chronic hepatitis B. J Clin Virol. 2006;37(3):206–212. doi:10.1016/j.jcv.2006.06.011
31. Sutthent R, Gaudart N, Chokpaibulkit K, et al. p24 antigen detection assay modified with a booster step for diagnosis and monitoring of human immunodeficiency virus Type 1 infection. J Clin Microbiol. 2003;41(3):1016. doi:10.1128/JCM.41.3.1016-1022.2003
32. Allice T, Cerutti F, Pittaluga F, et al. Evaluation of a novel real-time PCR system for cytomegalovirus DNA quantitation on whole blood and correlation with pp65-antigen test in guiding pre-emptive antiviral treatment. J Virol Methods. 2008;148(1–2):9–16. doi:10.1016/j.jviromet.2007.10.006
33. Yewdell JW, Reits E, Neefjes J. Making sense of mass destruction: quantitating MHC class I antigen presentation. Nat Rev Immunol. 2003;3(12):952–961. doi:10.1038/nri1250
34. Fortier M-H, Caron É, Hardy M-P, et al. The MHC class I peptide repertoire is molded by the transcriptome. J Exp Med. 2008;205(3):595–610. doi:10.1084/jem.20071985
35. Nakatsura T, Yoshitake Y, Senju S, et al. Glypican-3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem Biophys Res Commun. 2003;306(1):16–25. doi:10.1016/S0006-291X(03)00908-2
36. Nakatsura T, Komori H, Kubo T, et al. Mouse homologue of a novel human oncofetal antigen, glypican-3, evokes T-cell–mediated tumor rejection without autoimmune reactions in mice. Clin Cancer Res. 2004;10(24):8630. doi:10.1158/1078-0432.CCR-04-1177
37. Nakatsura T, Kageshita T, Ito S, et al. Identification of glypican-3 as a novel tumor marker for melanoma. Clin Cancer Res. 2004;10(19):6612. doi:10.1158/1078-0432.CCR-04-0348
38. Maeda D, Ota S, Takazawa Y, et al. Glypican-3 expression in clear cell adenocarcinoma of the ovary. Mod Pathol. 2009;22(6):824–832. doi:10.1038/modpathol.2009.40
39. Aviel-Ronen S, Lau SK, Pintilie M, et al. Glypican-3 is overexpressed in lung squamous cell carcinoma, but not in adenocarcinoma. Mod Pathol. 2008;21(7):817–825. doi:10.1038/modpathol.2008.37
40. Zynger DL, Gupta A, Luan C, et al. Expression of glypican 3 in hepatoblastoma: an immunohistochemical study of 65 cases. Hum Pathol. 2008;39(2):224–230. doi:10.1016/j.humpath.2007.06.006
41. Toretsky JA, Zitomersky NL, Eskenazi AE, et al. Glypican-3 expression in wilms tumor and hepatoblastoma. J Pediatr Hematol Oncol. 2001;23(8):496–499. doi:10.1097/00043426-200111000-00006
42. Esheba GE, Pate LL, Longacre TA. Oncofetal protein glypican-3 distinguishes yolk sac tumor from clear cell carcinoma of the ovary. Am J Surg Pathol. 2008;32(4):600–607. doi:10.1097/PAS.0b013e31815a565a
43. Shirakawa H, Suzuki H, Shimomura M, et al. Glypican-3 expression is correlated with poor prognosis in hepatocellular carcinoma. Cancer Sci. 2009;100(8):1403–1407. doi:10.1111/j.1349-7006.2009.01206.x
44. Haruyama Y, Kataoka H. Glypican-3 is a prognostic factor and an immunotherapeutic target in hepatocellular carcinoma. World J Gastroenterol. 2016;22(1):275–283. doi:10.3748/wjg.v22.i1.275
45. Adinolfi A, Adinolfi M. Alpha-feto-protein during development and in disease. J Med Genet. 1975;12(2):138.
46. Zhang J, Chen G, Zhang P, et al. The threshold of alpha-fetoprotein (AFP) for the diagnosis of hepatocellular carcinoma: a systematic review and meta-analysis. PLoS One. 2020;15(2):e0228857. doi:10.1371/journal.pone.0228857
47. Chen T, Dai X, Dai J, et al. AFP promotes HCC progression by suppressing the HuR-mediated Fas/FADD apoptotic pathway. Cell Death Dis. 2020;11(10):822. doi:10.1038/s41419-020-03030-7
48. Nakagawa H, Mizukoshi E, Kobayashi E, et al. Association between high-avidity T-cell receptors, induced by α-fetoprotein−derived peptides, and anti-tumor effects in patients with hepatocellular carcinoma. Gastroenterology. 2017;152(6):1395–1406.e10. doi:10.1053/j.gastro.2017.02.001
49. Tamai T, Mizukoshi E, Kumagai M, et al. A novel α-fetoprotein-derived helper T-lymphocyte epitope with strong immunogenicity in patients with hepatocellular carcinoma. Sci Rep. 2020;10(1):4021. doi:10.1038/s41598-020-60843-4
50. Hastie ND. Wilms’ tumour 1 (WT1) in development, homeostasis and disease. Dev Camb Engl. 2017;144(16):2862–2872.
51. Qi X, Zhang F, Wu H, et al. Wilms’ tumor 1 (WT1) expression and prognosis in solid cancer patients: a systematic review and meta-analysis. Sci Rep. 2015;5(1):8924. doi:10.1038/srep08924
52. Zhang Y, Yan W-T, Yang Z-Y, et al. The role of WT1 in breast cancer: clinical implications, biological effects and molecular mechanism. Int J Biol Sci. 2020;16(8):1474–1480. doi:10.7150/ijbs.39958
53. Rosenfeld C, Cheever MA, Gaiger A. WT1 in acute leukemia, chronic myelogenous leukemia and myelodysplastic syndrome: therapeutic potential of WT1 targeted therapies. Leukemia. 2003;17(7):1301–1312. doi:10.1038/sj.leu.2402988
54. Sera T, Hiasa Y, Mashiba T, et al. Wilms’ tumour 1 gene expression is increased in hepatocellular carcinoma and associated with poor prognosis. Eur J Cancer. 2008;44(4):600–608. doi:10.1016/j.ejca.2008.01.008
55. Perugorria MJ, Castillo J, Latasa M, et al. Wilms’ tumor 1 gene expression in hepatocellular carcinoma promotes cell dedifferentiation and resistance to chemotherapy. Cancer Res. 2009;69(4):1358–1367. doi:10.1158/0008-5472.CAN-08-2545
56. Raychaudhuri P, Park HJ. FoxM1: a master regulator of tumor metastasis. Cancer Res. 2011;71(13):4329. doi:10.1158/0008-5472.CAN-11-0640
57. Bergamaschi A, Madak-Erdogan Z, Kim YJ, et al. The forkhead transcription factor FOXM1 promotes endocrine resistance and invasiveness in estrogen receptor-positive breast cancer by expansion of stem-like cancer cells. Breast Cancer Res. 2014;16(5):436. doi:10.1186/s13058-014-0436-4
58. Chai N, Xie H, Yin J, et al. FOXM1 promotes proliferation in human hepatocellular carcinoma cells by transcriptional activation of CCNB1. Biochem Biophys Res Commun. 2018;500(4):924–929. doi:10.1016/j.bbrc.2018.04.201
59. Hu G, Yan Z, Zhang C, et al. FOXM1 promotes hepatocellular carcinoma progression by regulating KIF4A expression. J Exp Clin Cancer Res. 2019;38(1):188. doi:10.1186/s13046-019-1202-3
60. Su H, Li B, Zheng L, et al. Immunotherapy based on dendritic cells pulsed with CTPFoxM1 fusion protein protects against the development of hepatocellular carcinoma. Oncotarget. 2016;7(30):48401–48411. doi:10.18632/oncotarget.10269
61. Yokomine K, Senju S, Nakatsura T, et al. The forkhead box M1 transcription factor as a candidate of target for anti-cancer immunotherapy. Int J Cancer. 2010;126(9):2153–2163.
62. Bisiak F, McCarthy AA. Structure and function of roundabout receptors. In: Harris J, Marles-Wright J, editors. Macromolecular Protein Complexes II: Structure and Function. Subcellular Biochemistry. Springer International Publishing. 2019;93:291–319.
63. Yuan M, Guo H, Li J, et al. Slit2 and Robo1 induce opposing effects on metastasis of hepatocellular carcinoma Sk-hep-1 cells. Int J Oncol. 2016;49(1):305–315. doi:10.3892/ijo.2016.3506
64. Ito H, Funahashi S, Yamauchi N, et al. Identification of ROBO1 as a novel hepatocellular carcinoma antigen and a potential therapeutic and diagnostic target. Clin Cancer Res. 2006;12(11):3257. doi:10.1158/1078-0432.CCR-05-2787
65. Bonam SR, Partidos CD, Halmuthur SKM, et al. An overview of novel adjuvants designed for improving vaccine efficacy. Trends Pharmacol Sci. 2017;38(9):771–793. doi:10.1016/j.tips.2017.06.002
66. Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. Npj Vaccines. 2019;4(1):7. doi:10.1038/s41541-019-0103-y
67. Ikeda M, Okusaka T, Ohno I, et al. Phase I studies of peptide vaccine cocktails derived from GPC3, WDRPUH and NEIL3 for advanced hepatocellular carcinoma. Immunotherapy. 2021;13(5):371–385. doi:10.2217/imt-2020-0278
68. Qiu Y, Xu M-B, Yun MM, et al. Hepatocellular carcinoma-specific immunotherapy with synthesized α1,3- galactosyl epitope-pulsed dendritic cells and cytokine-induced killer cells. World J Gastroenterol. 2011;17(48):5260–5266. doi:10.3748/wjg.v17.i48.5260
69. Shimizu K, Kotera Y, Aruga A, et al. Postoperative dendritic cell vaccine plus activated T-cell transfer improves the survival of patients with invasive hepatocellular carcinoma. Hum Vaccines Immunother. 2014;10(4):970–976. doi:10.4161/hv.27678
70. Palmer DH, Midgley RS, Mirza N, et al. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology. 2009;49(1):124–132. doi:10.1002/hep.22626
71. Lee W-C, Wang H-C, Hung C-F, et al. Vaccination of advanced hepatocellular carcinoma patients with tumor lysate-pulsed dendritic cells: a Clinical Trial. J Immunother. 2005;28(5):496–504. doi:10.1097/01.cji.0000171291.72039.e2
72. Nakamoto Y, Mizukoshi E, Tsuji H, et al. Combined therapy of transcatheter hepatic arterial embolization with intratumoral dendritic cell infusion for hepatocellular carcinoma: clinical safety. Clin Exp Immunol. 2007;147(2):296–305. doi:10.1111/j.1365-2249.2006.03290.x
73. Silva L, Egea J, Villanueva L, et al. Cold-inducible RNA binding protein as a vaccination platform to enhance immunotherapeutic responses against hepatocellular carcinoma. Cancers. 2020;12(11):3397. doi:10.3390/cancers12113397
74. Zhang Y, Xie F, Yin Y, et al. Immunotherapy of tumor RNA-loaded lipid nanoparticles against hepatocellular carcinoma. Int J Nanomedicine. 2021;16:1553–1564. doi:10.2147/IJN.S291421
75. Abudoureyimu M, Lai Y, Tian C, et al. Oncolytic adenovirus-A nova for gene-targeted oncolytic viral therapy in HCC. Front Oncol. 2019;9:1182. doi:10.3389/fonc.2019.01182
76. Yoshikawa T, Nakatsugawa M, Suzuki S, et al. HLA-A2-restricted glypican-3 peptide-specific CTL clones induced by peptide vaccine show high avidity and antigen-specific killing activity against tumor cells. Cancer Sci. 2011;102(5):918–925. doi:10.1111/j.1349-7006.2011.01896.x
77. Sawada Y, Yoshikawa T, Nobuoka D, et al. Phase I Trial of a glypican-3–derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res. 2012;18(13):3686. doi:10.1158/1078-0432.CCR-11-3044
78. Sawada Y, Yoshikawa T, Ofuji K, et al. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. OncoImmunology. 2016;5(5):e1129483. doi:10.1080/2162402X.2015.1129483
79. Tsuchiya N, Yoshikawa T, Fujinami N, et al. Immunological efficacy of glypican-3 peptide vaccine in patients with advanced hepatocellular carcinoma. OncoImmunology. 2017;6(10):e1346764. doi:10.1080/2162402X.2017.1346764
80. Taniguchi M, Mizuno S, Yoshikawa T, et al. Peptide vaccine as an adjuvant therapy for glypican-3-positive hepatocellular carcinoma induces peptide-specific CTLs and improves long prognosis. Cancer Sci. 2020;111(8):2747–2759. doi:10.1111/cas.14497
81. Turajlic S, Litchfield K, Xu H, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 2017;18(8):1009–1021. doi:10.1016/S1470-2045(17)30516-8
82. Yang W, Lee K-W, Srivastava RM, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. 2019;25(5):767–775. doi:10.1038/s41591-019-0434-2
83. Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi:10.1038/nature12477
84. Martin SD, Brown SD, Wick DA, et al. Low mutation burden in ovarian cancer may limit the utility of neoantigen-targeted vaccines. PLoS One. 2016;11(5):e0155189. doi:10.1371/journal.pone.0155189
85. Subudhi SK, Vence L, Zhao H, et al. Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer. Sci Transl Med. 2020;12(537):eaaz3577. doi:10.1126/scitranslmed.aaz3577
86. Keenan TE, Burke KP, Van Allen EM. Genomic correlates of response to immune checkpoint blockade. Nat Med. 2019;25(3):389–402. doi:10.1038/s41591-019-0382-x
87. Rosenthal R, Cadieux EL, Salgado R, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567(7749):479–485. doi:10.1038/s41586-019-1032-7
88. Mauriello A, Zeuli R, Cavalluzzo B, et al. High somatic mutation and neoantigen burden do not correlate with decreased progression-free survival in hcc patients not undergoing immunotherapy. Cancers. 2019;11(12):1824. doi:10.3390/cancers11121824
89. Lu L, Jiang J, Zhan M, et al. Targeting neoantigens in hepatocellular carcinoma for immunotherapy: a futile strategy? Hepatology. 2021;73(1):414–421. doi:10.1002/hep.31279
90. Hu Z, Leet DE, Allesøe RL, et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat Med. 2021;27(3):515–525. doi:10.1038/s41591-020-01206-4
91. Timp W, Timp G. Beyond mass spectrometry, the next step in proteomics. Sci Adv. 2020;6(2):eaax8978. doi:10.1126/sciadv.aax8978
92. Lussier DM, Alspach E, Ward JP, et al. Radiation-induced neoantigens broaden the immunotherapeutic window of cancers with low mutational loads. Proc Natl Acad Sci. 2021;118(24):e2102611118. doi:10.1073/pnas.2102611118
93. Tsuchiya N, Hosono A, Yoshikawa T, et al. Phase I study of glypican-3-derived peptide vaccine therapy for patients with refractory pediatric solid tumors. Oncoimmunology. 2017;7(1):e1377872–e1377872. doi:10.1080/2162402X.2017.1377872
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.