")
Back to Journals » International Journal of Nanomedicine » Volume 15
PEGylated PLGA Nanoparticle Delivery of Eggmanone for T Cell Modulation: Applications in Rheumatic Autoimmunity
Authors Haycook CP, Balsamo JA, Glass EB , Williams CH , Hong CC, Major AS, Giorgio TD
Received 16 October 2019
Accepted for publication 19 December 2019
Published 19 February 2020 Volume 2020:15 Pages 1215—1228
DOI https://doi.org/10.2147/IJN.S234850
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Christopher P Haycook,1 Joseph A Balsamo,2,3 Evan B Glass,1 Charles H Williams,4 Charles C Hong,5 Amy S Major,3,6 Todd D Giorgio1
1Department of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA; 2Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, TN, 37232, USA; 3Department of Medicine, Division of Rheumatology and Immunology, Vanderbilt Medical Center, Nashville, TN 37232, USA; 4Department of Medicine, Division of Physiology, University of Maryland School of Medicine, Baltimore, MD 21201, USA; 5Department of Medicine, Division of Cardiovascular Medicine, University of Maryland School of Medicine, Baltimore, MD, 21201, USA; 6U.S., Department of Veterans Affairs, Tennessee Valley Healthcare System, Nashville, TN 37212, USA
Correspondence: Todd D Giorgio
Department of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
Email [email protected]
Background: Helper T cell activity is dysregulated in a number of diseases including those associated with rheumatic autoimmunity. Treatment options are limited and usually consist of systemic immune suppression, resulting in undesirable consequences from compromised immunity. Hedgehog (Hh) signaling has been implicated in the activation of T cells and the formation of the immune synapse, but remains understudied in the context of autoimmunity. Modulation of Hh signaling has the potential to enable controlled immunosuppression but a potential therapy has not yet been developed to leverage this opportunity.
Methods: In this work, we developed biodegradable nanoparticles to enable targeted delivery of eggmanone (Egm), a specific Hh inhibitor, to CD4+ T cell subsets. We utilized two FDA-approved polymers, poly(lactic-co-glycolic acid) and polyethylene glycol, to generate hydrolytically degradable nanoparticles. Furthermore, we employed maleimide-thiol mediated conjugation chemistry to decorate nanoparticles with anti-CD4 F(ab’) antibody fragments to enable targeted delivery of Egm.
Results: Our novel delivery system achieved a highly specific association with the majority of CD4+ T cells present among a complex cell population. Additionally, we have demonstrated antigen-specific inhibition of CD4+ T cell responses mediated by nanoparticle-formulated Egm.
Conclusion: This work is the first characterization of Egm’s immunomodulatory potential. Importantly, this study also suggests the potential benefit of a biodegradable delivery vehicle that is rationally designed for preferential interaction with a specific immune cell subtype for targeted modulation of Hh signaling.
Keywords: advanced delivery systems, eggmanone, autoimmunity, controlled release
Introduction
Helper T cell activity is dysregulated in a variety of diseases for which rheumatic autoimmunity is a prime example. Rheumatic autoimmune diseases preferentially affect women and are characterized by general pathology characteristics including inappropriate activation of the immune system, resulting in systemic inflammation within connective tissues including cartilage, joint synovium, and the skin.1 With the exception of rheumatoid arthritis, targeted therapeutic options are limited, and treatment consists mainly of chronic, systemic delivery of immunosuppressive and anti–inflammatory agents that can result in compromised immunity, premature cardiovascular disease, and osteoporosis.1
Central to T cell and B cell cooperation is their physical interaction at the immune synapse (IS). The IS is an area of concentrated signaling at the point where the membranes of the T cell and antigen-presenting cell (APC) make physical contact. Formation of the IS between CD4+ T cells and B cells is critical for the production of autoantibodies that potentiate the systemic inflammation of connective tissues in rheumatic autoimmunity. IS formation involves intricate reorganization of the cytoskeleton facilitated by the polarization of the microtubule-organizing center (MTOC), as well as, actin partitioning and repositioning of the Golgi apparatus below the surface of the IS.2
MTOC reorganization and polarization to the IS is dependent on Hedgehog (Hh) signaling, a pathway that is traditionally associated with primary cilia in nonhematopoietic cells.3,4 De la Roche et al demonstrated that inhibitors of Hh signaling can disrupt the IS and the ability of CD8+ T cells to become activated and lyse antigen-presenting targets.3 Overactivation of Hh signaling in the thymus can lead to decreased negative selection and the escape of autoreactive T cell clones.5 Additionally, Hh signaling proteins are able to provide co-stimulatory effects to CD4+ T cells in the periphery that promote proliferation and cytokine production.6 Furthermore, others have demonstrated that the MTOC in CD4+ T cells is reoriented to face towards the IS junction with B cells in an antigen-dependent manner.7 Therefore, specific disruption of the IS via targeting the Hh-regulated MTOC may represent a potential new, specific therapeutic strategy to disrupt autoantibody production in rheumatic autoimmunity that could eliminate the need for chronic usage of immunosuppressants and glucocorticoids.
Eggmanone (Egm) is a small molecule inhibitor of the Hh signaling pathway that was discovered at Vanderbilt University.8 Unlike commercially available small molecule Hh inhibitors that inhibit the upstream G protein-coupled receptor Smoothened (SMO) and are susceptible to acquired resistance, Egm antagonizes phosphodiesterase 4 (PDE4), a downstream regulator of Hh gene transcription. Importantly, unlike other PDE4 inhibitors, Egm inhibits PDE4 by raising cyclic AMP locally at the basal body, instead of raising total cellular cyclic AMP content.8 If delivered to CD4+ T cells, Egm could potentially inhibit autoimmune lymphocyte activation through suppression of Hh mediated IS formation in CD4+ T cells. However, Egm is also extremely hydrophobic, leading to rapid excretion and ineffective intravenous administration if a rationally designed delivery vehicle is not utilized.9
Specific delivery of small molecule drugs to T cells is a challenging task due to their low phagocytic activity. Previous attempts to specifically deliver hydrophobic immunomodulatory cargo to CD4+ T cells have utilized several poly(lactic-co-glycolic acid) (PLGA) nanoparticle formulations to create localized drug delivery depots at the cell surface. McHugh et al conjugated biotin-labeled whole anti-CD4 antibodies to avidin-coated PLGA nanoparticles.10 Although they were able to achieve high CD4-targeting specificity ex vivo, avidin and streptavidin conjugation systems have previously been shown to be immunogenic, and could, therefore, exacerbate the inflammatory immune environment associated with autoimmunity.11 Additionally, conjugation of whole targeting antibodies that contain foreign fragment crystallizable (Fc) regions can lead to rapid clearance of nanoparticles in systemic circulation via Fc receptor-mediated recognition by the reticuloendothelial system (RES).12 Cao et al conjugated anti-CD4 antibody fragments to PLGA nanoparticles coated with polyethylene glycol (PEG) conjugated lipids. This formulation decreased the potential for opsonization-mediated clearance by the RES through incorporation of hydrophilic PEG,12 and also provided tunable control of nanoparticle surface charge via mixing of cationic and anionic lipids in the surface coating. Although this formulation incorporated several design elements to improve in vivo pharmacokinetics by bypassing RES mediated nanoparticle clearance mechanisms, the antibody fragmentation method utilized resulted in the conjugation of some non-functional fragments that reduced overall CD4-targeting specificity.13
In this work, we employ a therapeutic approach involving hydrolytically degradable nanoparticles that specifically bind to CD4+ helper T cell subsets to form membrane-localized drug delivery depots, bypassing their inability to perform professional phagocytosis. We utilized two FDA-approved polymers, PLGA and PEG, to generate hydrolytically degradable nanoparticles capable of providing rapid release rates of Egm and decreased non-specific delivery to off-target immune cells. Furthermore, we employed maleimide-thiol mediated conjugation of CD4-targeting F(ab’) antibody fragments to enable sustained, targeted delivery of Egm on the surface of CD4+ T cell membranes. This approach ensures the correct presentation of the antibody fragment for receptor binding, reduces bioconjugate chemistry-associated immunorecognition and clearance, and minimizes the overall size of the decorated delivery system relative to surface functionalization using a complete antibody. To the best of our knowledge, this work represents the first characterization of Egm’s immunomodulatory effects and its formulation as a potential nanomedicine for targeted immunomodulation of CD4+ T cells.
Materials and Methods
Cell Culture
8- to 10-week old female mice (FVB (FVB/NJ stock no: 001800), C57BL/6J stock no: 000664, and OT-II (B6.Cg-Tg(TCRαTCRβ)425Cbn/J stock no: 004194), The Jackson Laboratory) were used for all experiments in order to reflect the higher incidence rates of rheumatic autoimmunity in females. All animal care was conducted in accordance with local and federal guidelines evaluated by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and an animal protocol (M1700021) approved by the Vanderbilt Institutional Animal Care and Use Committee. Whole splenocyte cultures were derived from spleens harvested from 8- to 10-week-old female FVB mice, for evaluation of nanoparticle toxicity, C57BL/6J mice, for evaluation of nanoparticle targeting specificity, and OT-II mice, for evaluation of therapeutic efficacy. Mice were euthanized and spleens were immediately harvested and placed in ice-cold T cell media (RPMI-1640 supplemented with 10% fetal bovine serum, penicillin/streptomycin, 55 mM 2-mercaptoethanol, 1 mM pyruvate, 2 mM glutamine and non-essential amino acids). Spleens were manually dissociated using 40-micron cell strainers (Fisher Scientific, nylon mesh) and the resulting cell suspension was centrifuged at 1500 rpm for 7 min at 4°C. The supernatant was decanted and residual cell pellets were broken up. Red blood cells in the resulting cell suspension were lysed by adding 900 uL of microbiology grade water (Corning) followed by 100 µL of 10× PBS (Sigma) while vortex mixing.
Evaluation of T Cell Activation and Cytokine Production
Whole splenocytes were seeded in standard 96-well plates at 100,000 cells/well and stimulated for 72 hrs with various concentrations of whole ovalbumin or ovalbumin peptide aa323-339 (OVA323-339). Interferon gamma (IFN-γ) production was measured in culture supernatants by specific ELISA (BD OptEIA, Catalogue # 551866) after incubation with either dimethyl sulfoxide (DMSO) or Egm dissolved in DMSO. T cell activation was evaluated by flow cytometry.
Nanoparticle Formulation
PEGylated PLGA nanoparticles were prepared using oil-in-water emulsion mediated by sonication. PLGA 50:50 lactic acid (LA):glycolic acid (GA) (10 kDa)-PEG(5 kDa)-maleimide (Nanosoft Polymers, lot number 27910051517) or PLGA 50:50 LA:GA(10 kDa)-PEG(5 kDa)-methyl (Nanosoft Polymers, lot number 275310050324) was added to PLGA 50:50 LA:GA (10 kDa, Durect corporation, lot number 902-82-1) at 25% (mass PLGA-PEG/mass PLGA) and dissolved in dichloromethane (DCM) at 25 mg polymer/mL DCM. Hydrophobic Egm and/or 1,1ʹ-dioctadecyl-3,3,3ʹ,3ʹ-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD)14 cargo was incorporated into the oil phase prior to sonication for encapsulation within PEGylated PLGA nanoparticles. Lyophilized Egm was dissolved directly in DCM at 4% (mass Egm/mass total polymer) prior to dissolving polymers. 2uL of DiD dissolved in DMSO at 2mg/mL was added after polymers were fully dissolved for fluorescently labeled materials. Resulting solutions were vortex mixed and transferred to ice-cold 0.25% (w/v poly(vinyl alcohol) in deionized water) surfactant solution. Emulsification was achieved using a Fisher Scientific Sonic Dismembrator (power level 3, 3 subsequent 10 s on, 20 s off cycles on ice). The resulting nanoparticle suspension was stirred for 3 hrs to evaporate residual DCM and allow nanoparticles to harden. Excess poly(vinyl-alcohol) and residual free Egm and DiD were removed via sonication in deionized water and centrifugation (20,000 g, 10 min). Recovered nanoparticles were suspended in either deionized water for chemical analysis, or 3% aqueous trehalose (Sigma) solutions for all other applications. Nanoparticles were filtered with 5 µm (Pall, acrodisc supor membrane) followed by 0.45 µm (ThermoFisher, PTFE) syringe filters prior to freezing (−80°C) overnight and lyophilization (−40°C, 0.2 mbar, 72 hrs) using a Labconco Freezone 4.5. All nanoparticle formulation variables are summarized below (Table 1).
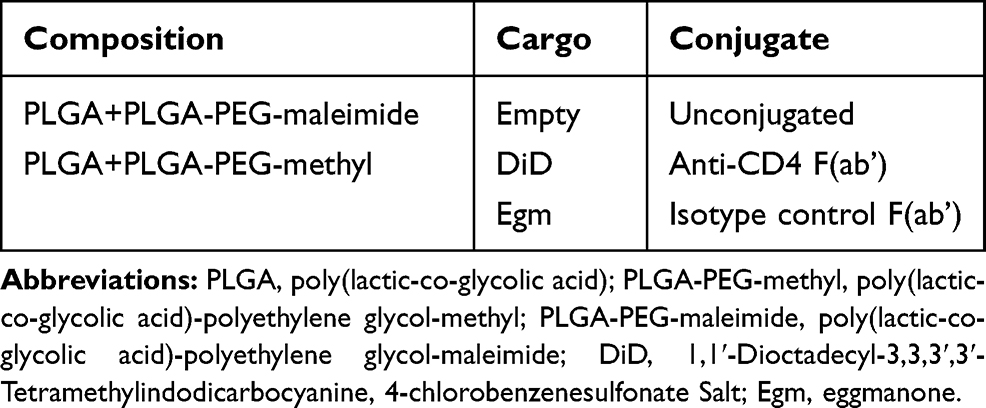 |
Table 1 Summary of Nanoparticle Formulation Variables |
Characterization of Egm-Loaded Nanoparticle Encapsulation Efficiency
Egm and Egm-loaded nanoparticle (Egm-NP) trehalose-free lyophilizate were dissolved in DMSO for evaluation of peak Egm absorbance at 323 nm by UV/VIS spectroscopy. At least three technical replicates of 180 uL volumes were prepared for each sample. Measurements were performed in UV-transparent 96-well plates (Nunc, 96-well UV microplates) using a Biotek M1000Pro plate reader. Measured Egm-NP absorbance values were adjusted to remove the contribution of PLGA by subtracting absorbance values of matched concentration empty particles. Encapsulation efficiency was calculated as the ratio of the loading capacity determined by UV/VIS spectroscopy to the theoretical loading capacity. Loading capacity was defined as the ratio of Egm mass to the total mass of polymer in the formulation.
Characterization of Nanoparticle Size, Zeta Potential, Reactive Chemistry, and Release Rate
Hydrated nanoparticle size and zeta potential were measured using nanoparticle tracking analysis (Malvern Panalytical, Nanosight NS 300) and laser doppler electrophoresis (Malvern Panalytcal, Zetasizer Nano ZS), respectively. Percent relative standard deviation (%RSD) of each formulation was calculated according to Malvern’s recommendations.15 Maleimide reactive end chemistry was verified using 1H nuclear magnetic resonance spectroscopy (Bruker, 400 MHz). At least 5 mg of trehalose-free nanoparticle lyophilizate was dissolved in 600 uL deuterated chloroform (Sigma) for NMR sample preparation. DiD release rate was measured using a Biotek M1000Pro plate reader. Nanoparticle lyophilizate was resuspended in 1× PBS and incubated at 37°C while shaking for up to 5 days. Resuspension times were staggered so that all release samples were collected at once. Following incubation, nanoparticles were centrifuged (20,000 g, 10 min), and supernatant was decanted. Unreleased DiD was measured after dissolving collected pellets in DMSO. Amount released was quantified by normalizing DiD fluorescence of release samples to 0 hr release controls.
Evaluation of Nanoparticle Morphology and Egm Localization
Dehydrated nanoparticle morphology was evaluated using transmission electron microscopy (TEM) (Philips/FEI T-12). Freshly made nanoparticles suspended in deionized water were incubated at room temperature on poly-l-lysine-coated TEM grids (formvar coated stabilized with carbon film, Electron Microscopy Sciences) for 5 min before wicking away excess liquid with filter paper. No contrast agents were utilized for TEM imaging. Egm localization within nanoparticles was investigated using ImageJ analysis and energy-dispersive X-ray spectroscopy. Intensity profiles of equal magnification TEM images were generated using the line profile tool in ImageJ (magnification = 52000×, line width = 20 nm). Intensity profiles for each image were normalized to the average background intensity surrounding each nanoparticle. TEM imaging sample preparation was also utilized for elemental analysis of nanoparticles by scanning electron transmission energy-dispersive X-ray spectroscopy (STEM-EDS) (FEI Tecnai Osiris). Nanoscale X-ray element mappings generated from STEM-EDS spectral data were used to determine the spatial distribution of Egm within PEGylated PLGA nanoparticles. High angle annular dark-field images served as a reference for nanoparticle area among elemental mappings.
Evaluation of Egm-Loaded Nanoparticle Biocompatibility and Immunomodulatory Potential
Whole splenocytes were seeded in standard (for evaluation of therapeutic efficacy) or black-walled (for evaluation of biocompatibility) 96-well plates at 100,000 cells/well prior to adding nanoparticle treatments. Egm-NP and empty nanoparticle (empty-NP) lyophilizate was resuspended in T cell media (described above) via vortex mixing and water bath sonication immediately prior to use. Standard media was replaced with nanoparticle-containing media, and cell viability was evaluated 72 hrs later by Celltiter glo assay (Promega). IFN-γ production was evaluated 72 hrs after stimulation with 50 µg/mL ovalbumin and nanoparticles via ELISA (BD OptEIA, Catalogue # 551866).
Antibody Fragment Conjugation to Nanoparticles
Anti-CD4 (clone GK1.5, Bio X Cell) and isotype control (clone LTF-2, Bio X Cell) F(ab’)2 antibody fragments were generated using the Pierce F(ab’)2 Prep kit (ThermoFisher catalog # 44988). F(ab’) antibody fragments were generated from F(ab’)2 fragments according to previously published methods.16 Disulfide bonds between antibody fragments were reduced using 0.5 mM DTT in 1× PBS for 30 mins at room temperature. Excess DTT was removed from fragments using 7K MWCO zeba spin desalting columns (ThermoFisher catalog # 89882). Reduced antibody fragment concentration was quantified using nanodrop (Mettler Toledo UV5 nano) as recommended by ThermoFisher (280 nm absorbance, molar extinction coefficient = 1.4). Fragments were added to resuspended nanoparticles at a concentration of 12.5 ug antibody per 1.49 × 109 ± 1.89 × 108 resuspended nanoparticles (determined by Nanosight) in 1× PBS according to previously published methods.16 Resulting solutions were prepared with a final volume of 1 mL and shaken for 2 hrs in 2 mL microcentrifuge tubes at room temperature on a Fisher mini vortexer (speed 8) to initiate Maleimide-thiol antibody conjugation to nanoparticles.
Evaluation of Antibody-Conjugated Nanoparticle Targeting Efficacy
Targeting efficacy was evaluated using DiD (Invitrogen, catalog # D7757)-loaded fluorescent nanoparticles (DiD-NPs) with surface-conjugated antibody fragments. Whole splenocyte cultures were incubated for 30 mins with DiD-loaded, antibody fragment decorated nanoparticles suspended in PBS via flow cytometry (250,000 cells per 7.45 × 107 ± 9.45 × 106 NPs/mL) unless otherwise indicated.
Flow Cytometry
Product information for the fluorescently labeled antibodies used in the flow cytometry staining protocols within this work is provided in the following format: (surface marker-fluorochrome, clone, company). Flow cytometry staining probes used consisted of: (TCR-β-APC, H57-597, BD), (CD4-PE-Cy7, GK 1.5, BD), (CD4-PerCP, RM4-5, Tonbo), (CD8a-APC-Cy7, 53–6.7, Tonbo), (CD44-FITC, IM7, BD), (CD62L-PerCP, MEL-14, BD).
Cells were incubated with Fc block at 1:100 for 15 mins at room temp in FACS buffer containing HBSS, 1% BSA, 4.17 mM sodium bicarbonate, and 3.08 mM sodium azide. Cells were labeled with an antibody cocktail consisting of either (TCR-β-FITC, CD4-PE-Cy7, CD44-FITC, CD62L-PerCP) or (CD4-PerCP, CD8a-APC-Cy7) in FACS buffer for 30 mins on ice in the dark. Cells were washed and resuspended in 2% paraformaldehyde for analysis on a MACSQuant seven-color flow cytometer (Miltenyi Biotec); data were analyzed using FlowJo Single Cell Analysis Version 10.0.1.
Flow cytometry gating for T cell activation experiments involved selecting the lymphocyte population from forward scatter versus side scatter plots. TCR-β+CD4+ T cells were selected from TCR-β versus CD4 plots. Activated CD4+ T cells were selected from CD44 versus CD62L plots. Flow cytometry gating for nanoparticle targeting experiments involved selecting the lymphocyte population from forward versus side scatter plots. CD4+, CD8+, and non-T cells were selected from CD4 versus CD8 plots. Corresponding nanoparticle+ cell populations were then selected according to DiD staining intensity.
Statistical Analysis
All error bars represent standard error of the mean unless otherwise indicated. One or Two-way Analysis of variance followed by multiple comparison tests was performed for most data presented here as indicated, and statistical significance was defined as p < 0.05. Statistical analyses were performed using Prism 7.04 (GraphPad Software).
Results and Discussion
Egm Inhibits CD4+ T Cell Activation and Cytokine Responses
A central hypothesis of this study is that targeted inhibition of Hh signaling could significantly reduce CD4+ T cell activation in response to specific antigen. To test this hypothesis, OT-II whole splenocytes were incubated with increasing concentrations of whole OVA in the presence of unformulated Egm or an equivalent volume of DMSO (vehicle). At all OVA concentrations, Egm significantly inhibited CD4+ T cell production of IFN-γ (Figure 1A). Using OVA323-339, which eliminates the need for antigen processing, we demonstrated that Egm was a potent inhibitor of CD4+ T cell cytokine responses, and its effects were dose-dependent (Figure 1B). Significant reduction of IFN-γ was observed for Egm concentrations as low as 0.63 µM, and IFN-γ was undetectable at concentrations above 2.5 µM (Figure 1B). After observing that Egm treatment suppressed helper T cell cytokine responses, we investigated whether Egm also suppressed T cell activation. OT-II splenocyte experiments utilizing OVA323-339 were repeated and T cell activation was evaluated by flow cytometric analysis of T cell activation and memory markers (Figure 1C, D, and E). At concentrations of OVA323-339 above 25 µg/mL, Egm significantly reduced the percent of helper T cell populations expressing the activation/memory phenotype CD44hiCD62Llo (Figure 1D).17 Additionally, Egm treatment significantly reduced the expression of CD69, an additional T cell activation marker, for all concentrations of OVA tested when compared to DMSO controls (Figure 1E). Our results indicate that Egm treatment significantly inhibits CD4+ T cell activation in the presence of specific antigen. Thus, for the first time, we have demonstrated the immunomodulatory effects of Egm on CD4+ T cells and also highlighted its potential use as an immunosuppressive small molecule compound. However, Egm’s in vivo utility is limited by its poor water solubility that necessitates the use of a rationally designed carrier to enable further investigation of its immunomodulatory potential.
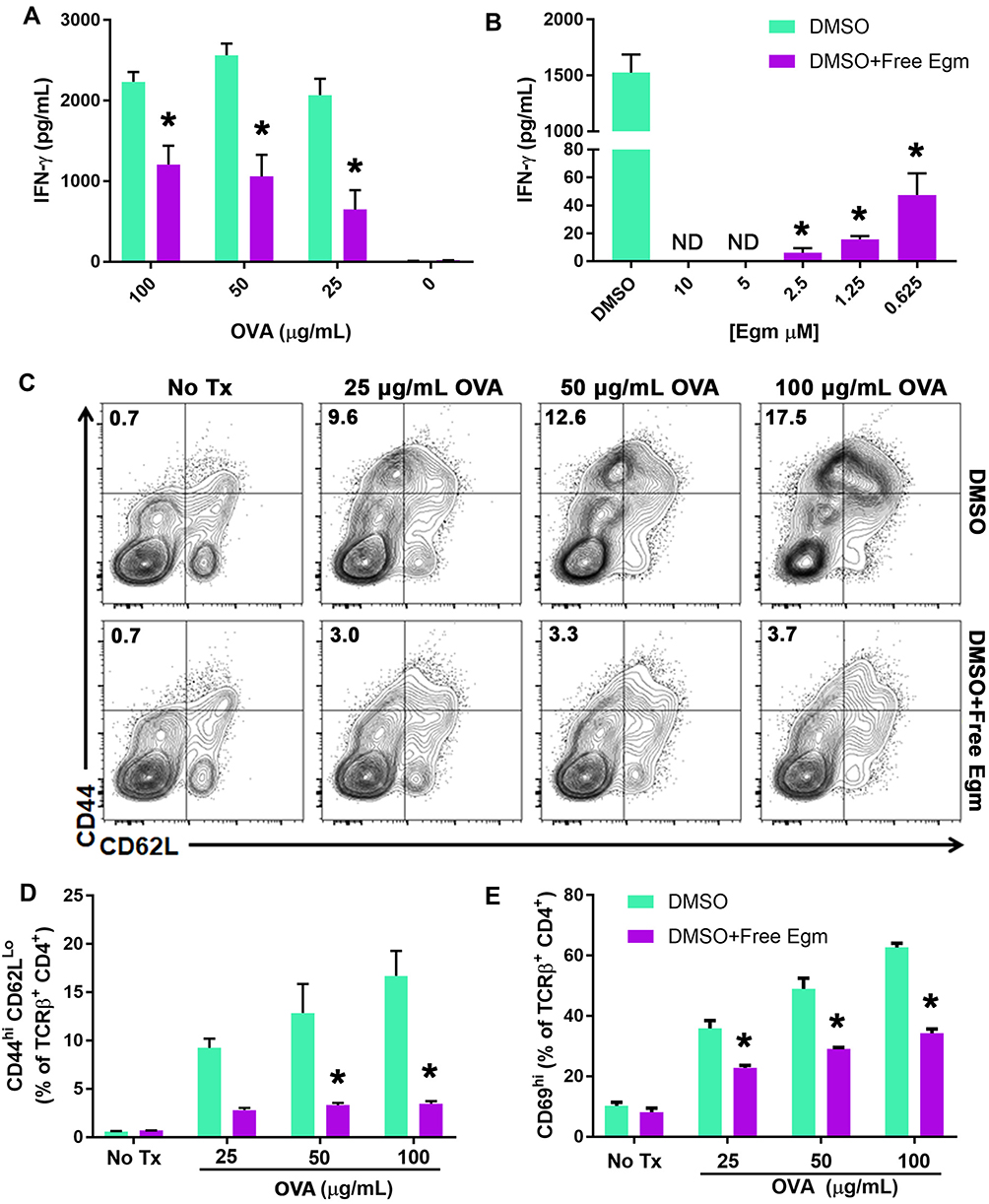 |
Figure 1 Egm inhibits CD4+ T cell activation and cytokine responses. Notes: (A) OT-II whole splenocytes were incubated with whole OVA protein at various concentrations for 72 hrs in the presence of DMSO (vehicle) or 10 µM Egm in DMSO. (B) Egm dose response of OT-II whole splenocytes incubated with OVA323-339 peptide (50 µg/mL) for 72 hrs. IFN-γ concentration was measured via ELISA. (C) Representative flow cytometric analysis of OT-II whole splenocyte cultures incubated with OVA323-339 peptide in the presence of DMSO or 10 µM Egm in DMSO for 72 hrs. T cell activation was evaluated by analysis of CD44hiCD62Llo T cell populations (plots gated from TCRβ+CD4+ T cells). (D) Quantification of CD44hiCD62Llo T cell populations from 3 biological replicates. (E) Quantification of CD69 expression in the presence of Egm or DMSO from 3 biological replicates. The significance of the data was evaluated via ordinary One-way ANOVA with multiple comparison test. (*P<0.05). Abbreviations: OVA, ovalbumin; DMSO, dimethyl sulfoxide; No Tx, no treatment. |
Synthesis and Characterization of PEGylated PLGA Nanoparticles Loaded with Egm and DiD
Single oil in water emulsion and solvent evaporation strategies were utilized to synthesize PEGylated PLGA nanoparticles as a potential delivery vehicle for in vivo Egm administration (Figure 2). Egm and a fluorescent surrogate compound, DiD, were encapsulated in nanoparticles in order to investigate whether Egm could be efficiently packaged and delivered to CD4+ T cells as a nanomedicine. Nanoparticle size and zeta potential were measured by nanoparticle tracking analysis and laser doppler electrophoresis, respectively. Because nanoparticle tracking analysis produces a population of single-particle size measurements, rather than hypothetical gaussian distributions of nanoparticle size,15 the polydispersity of each formulation was evaluated by calculating the %RSD. Egm encapsulation had no effect on particle size (Figure 2A–C), %RSD (Figure 2D), or zeta potential (Figure 2E). Loading of DiD increased mean particle size and SD compared to all formulations, as well as, %RSD compared to select formulations (Figure 2A, C and D). Differences in particle size and polydispersity measures associated with DiD loading may be an artifact caused by fluorescent excitation of the DiD inside nanoparticles by the 642 nm laser utilized in nanoparticle tracking analysis measures. Importantly, lyophilization did not significantly increase the particle size of any formulation, and the size of all lyophilized formulations was statistically similar. Lyophilization of nanoparticle formulations enabled prolonged shelf-life and “as-needed” usage of all formulations developed in this study. All formulations had zeta potentials near −42 mV, hydrodynamic diameters near 200 nm, and narrow size distributions with %RSD below 40. Therefore, particle suspensions were monodisperse, and the appropriate size to provide passive accumulation in the spleen18–20 in addition to T cell-mediated transport to the spleen and lymph nodes.16
Zeta potential and particle size are both known to be important factors that influence the circulation half-life of nanoparticles, and these factors were unaffected by Egm loading.21 Additionally, the zeta potential of nanoparticle targeting systems has also been shown to significantly affect the potential of nonspecific binding due to electrostatic interactions with the cell membrane.21 PLGA nanoparticles with cationic surface charges preferentially interact with negatively charged cell membranes irrespective of targeting ligand conjugation.22 Conversely, nanoparticles with anionic surface charges similar to our formulations possess elevated specific binding interaction to CD4+ T cells by reducing nonspecific electrostatic interactions with cell membranes, allowing conjugated targeting ligand binding affinity to dominate.13 Additionally, nanoparticle formulations with cationic zeta potentials can lead to pro-inflammatory immune responses while those with anionic zeta potentials are associated with reduced immunogenicity and increased circulation potential.23 Therefore, we have confidence that the measured zeta potentials of our three undecorated formulations (empty, Egm-loaded, and DiD-loaded), are well-suited for antibody fragment decoration and systemic CD4+ T cell targeting applications in rheumatic autoimmunity.
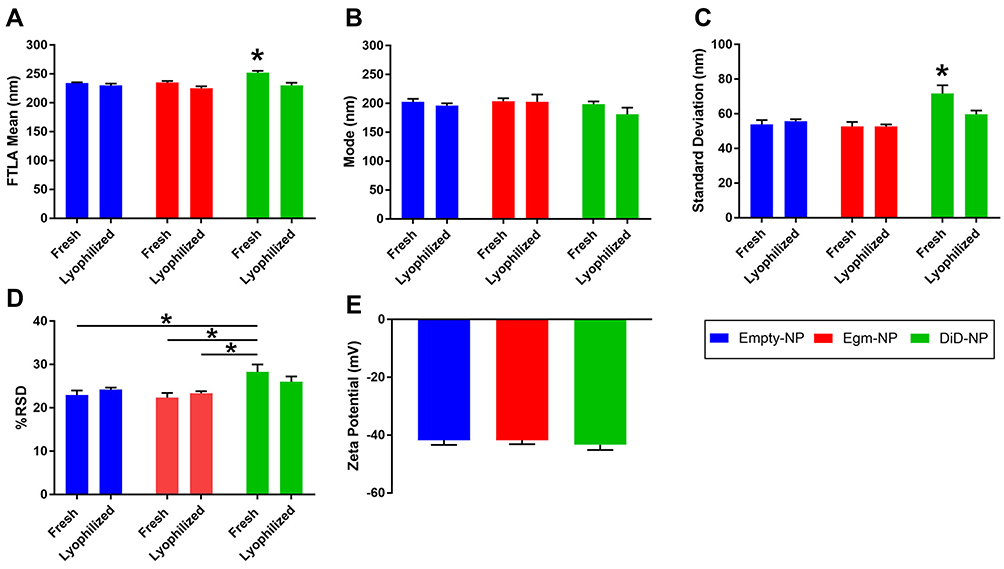 |
Figure 2 PEGylated PLGA nanoparticles retain physicochemical characteristics following Egm loading. Notes: (A) FTLA mean hydrodynamic diameter of nanoparticles. (B) Mode of nanoparticle diameter distributions. (C) Standard deviation of nanoparticle diameters. (D) Calculated percent relative standard deviation of nanoparticle distributions (E) Zeta potential measures of lyophilized formulations. All measurements were performed in deionized water. All data shown represent the mean ± SEM of at least three independent batches for each formulation. The significance of the data was evaluated via ordinary One-way or Two-way ANOVA with multiple comparison test. (*P<0.05). Abbreviations: FTLA, finite-track length adjusted; empty-NP, empty nanoparticle; Egm-NP, Eggmanone-loaded nanoparticle; DiD-NP, DiD-loaded nanoparticle; SEM, standard error of the mean. |
We verified that the presence of maleimide reactive end-chemistry on PEGylated PLGA nanoparticle coronas was unaffected by Egm loading through 1H nuclear magnetic resonance spectroscopy of nanoparticle formulations made with either PLGA-PEG-maleimide or PLGA-PEG-methyl. NMR spectra of nanoparticles made with PLGA-PEG-maleimide (Supplemental Figure 1B and C) exhibited a peak at 6.7 ppm, characteristic of maleimide,24 that was not present in the spectra of particles made with PLGA-PEG-methyl instead (Supplemental Figure 1A). Additionally, NMR spectra of nanoparticles loaded with Egm exhibited known characteristic peaks of Egm25 (Supplemental Figure 1C) that, in addition to UV/VIS spectroscopy, confirmed the loading of Egm within nanoparticles. Egm encapsulation efficiency was 76.7 ± 11.1% (calculated from UV/VIS measures).
Emulsion Mediated Fabrication Localizes Egm in the Core of Spherical Nanoparticles
Dehydrated nanoparticle size and morphology were investigated via TEM imaging of unstained empty- and Egm-NPs on positively charged polylysine-coated TEM grids (Figure 3A). Both nanoparticle formulations exhibited diameters in agreement with Nanosight measures (Figure 2A and B) and spherical morphology that is typical of emulsion-mediated fabrication strategies. Interestingly, TEM imaging of Egm-NPs revealed a region of increased electron density in nanoparticle cores when compared to empty-NPs (Supplemental Figure 2). This prompted further investigation of elemental distribution within nanoparticles via energy-dispersive X-ray spectroscopy.
The presence of sulfur, a unique element found only in the chemical structure of Egm in our formulations, was used to indicate the spatial distribution of Egm within nanoparticles. Analysis of STEM-EDS spectra revealed the presence of carbon, nitrogen, and oxygen peaks, corresponding to PLGA and PLGA-PEG-maleimide polymer components, in both particle formulations, while a sulfur peak was only present in Egm-NPs (Figure 3B).
Elemental mappings revealed a concentration of sulfur in the core of Egm-NPs, indicative of Egm loading, with little observable signal outside of the nanoparticle region (Figure 3C). In contrast, the sulfur signal observed in empty-NP elemental mappings was largely dispersed throughout the entire region of interest and, therefore, was presumed to be the result of shot-noise during signal collection (Figure 3D). Taken together with areas of increased contrast in TEM images of Egm-NPs, the concentration of sulfur within Egm-NPs provides evidence suggestive of Egm localization in the nanoparticle cores mediated by emulsion-mediated fabrication methods.
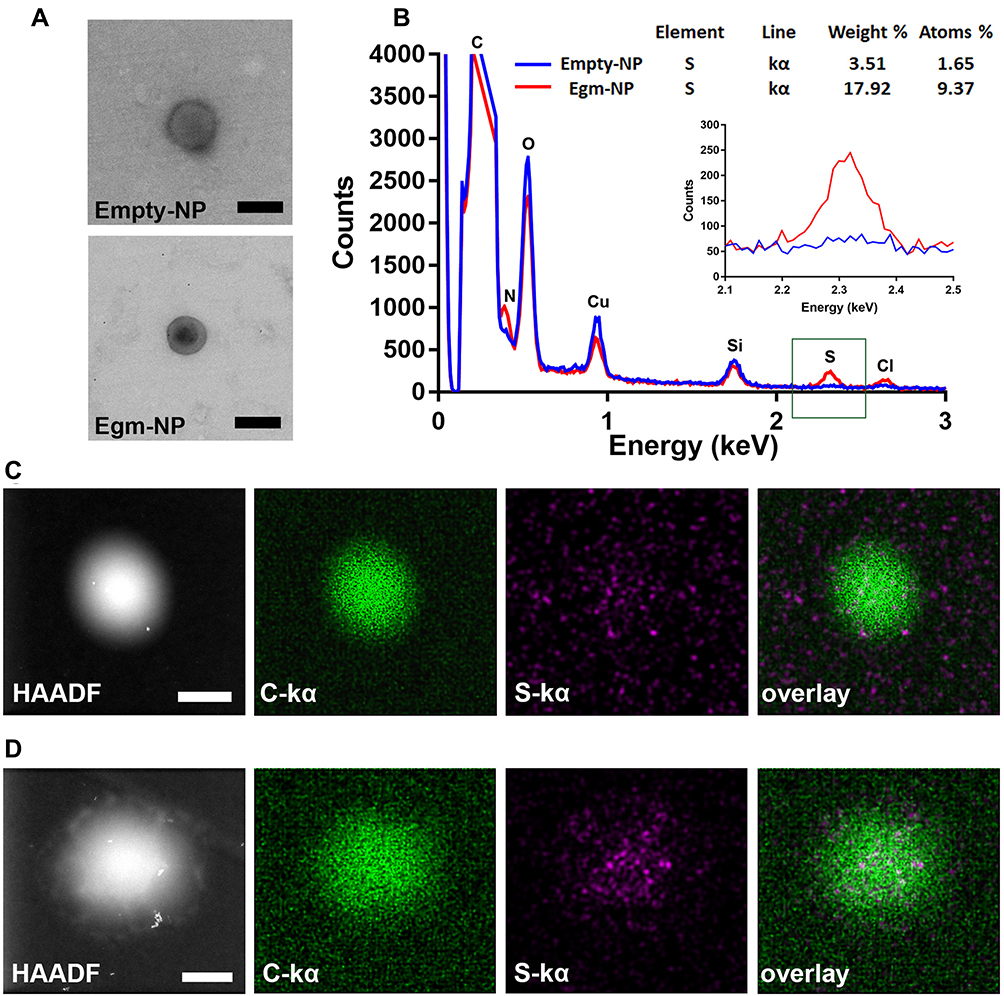 |
Figure 3 Emulsion mediated fabrication localizes Egm in nanoparticle cores. Notes: (A) Transmission electron micrographs of empty and Egm-loaded nanoparticles. No negative staining was utilized. (B) Elemental analysis and quantification of sulfur weight % and atoms % within empty and Egm-loaded nanoparticles. Inset corresponds to the energy region within the green box. (C) Nanoscale X-ray element mappings of empty nanoparticle. (D) Nanoscale X-ray element mappings of Egm-loaded nanoparticle. All images representative of corresponding nanoparticle populations. All scale bars represent 200 nm. Abbreviations: Empty-NP, empty nanoparticle; Egm-NP, Egm-loaded nanoparticle; HAADF, high-angle angular darkfield image; C, carbon; S, sulfur; O, oxygen; N, nitrogen, Cu, copper; Si, silicon; Cl, chlorine; kα, K-alpha emission line. |
Nanoparticle-Formulated Egm Inhibits Antigen-Specific CD4+ T Cell Cytokine Responses in a Therapeutically Relevant Timeframe
The use of low molecular weight PLGA with an equal molar ratio of lactic to glycolic acid in our formulation is intended to enable rapid release of Egm.26 The release of Egm from nanoparticle formulations was investigated using fluorescent DiD in order to enable sufficient signal strength during release measurements. Lyophilized nanoparticles that were resuspended and incubated in PBS at 37°C on an orbital shaker released 66.7% of DiD over the course of 5 days and exhibited a characteristic burst release associated with low molecular weight, 50:50 (lactic acid:glycolic acid) PLGA equal to 32.0% within the first 24 hrs (Figure 4A). The release kinetics we observed with DiD are expected to enable the efficacy of future potential therapeutic intervention strategies using Egm. The initial burst and controlled release periods appear suitable to address the lag phase of a secondary immune response to self-antigen generated by autoimmune memory cells. The lag phase of a secondary immune response is typically 2 to 5 days.27 During this time, antigen presentation and subsequent isotype switching and differentiation of B cells into autoantibody-producing plasma cells occur in the germinal centers of secondary lymphoid organs. Although rheumatic autoimmune diseases are mainly thought of as antibody-mediated diseases, the production of IgG antibodies, the isotype with the highest antigen affinity and plasma circulation potential, is a CD4+ T cell-dependent process that is regulated in germinal center responses. Thus, if a sufficient number of Egm-loaded nanoparticles with similar release kinetics could be targeted to the germinal centers during the lag phase, inhibition of plasma cell differentiation and autoantibody production could be achieved on a sufficient timescale to serve as a viable therapeutic intervention strategy for rheumatic autoimmune diseases. Although we did not directly measure the release of Egm from our formulation due to concentrations below the detection limits of appropriate analytical chemistry instrumentation, we expect a similar and/or faster release profile since Egm is similarly hydrophobic, and has a significantly lower molecular weight than DiD, 416 Da compared to 1052 Da, respectively.
After demonstrating that PEGylated PLGA nanoparticles could efficiently encapsulate and release Egm in a therapeutically relevant timeframe, we verified that the nanoparticle formulation of Egm was effective at inhibiting antigen-specific inflammatory cytokine responses of CD4+ T cells. IFN-γ is the hallmark cytokine produced by CD4+ T follicular helper cells (Tfh) in the germinal center response that enables B cell production of high-affinity, long-circulating IgG antibody isotypes.28 Inhibition of IFN-γ production by OT-II splenocytes stimulated with ovalbumin was measured 72 hrs after incubation with Egm loaded and empty, undecorated nanoparticle formulations. For all concentrations of Egm evaluated, both free and nanoparticle formulations of Egm significantly inhibited the production of IFN-γ compared to media controls and no statistical difference was observed between nanoparticle-formulated and free Egm. No significant changes were observed for DMSO and blank nanoparticle vehicle controls (Figure 4B).
Acute toxicity of lyophilized Egm- and empty-NPs was evaluated ex vivo 72 hrs after incubation of resuspended nanoparticles with whole splenocyte cultures derived from 8- to 10-week-old female FVB mice. No significant decreases in the viability of splenocytes during 72 hr incubations were observed for either formulation when compared to media controls (Figure 4C). Additionally, the lack of any change in IFN-γ production after treatment with empty nanoparticles is further evidence that our formulations are nontoxic and nonimmunogenic. Overall, neither formulation resulted in significant toxicity of whole splenocytes at any concentration investigated.
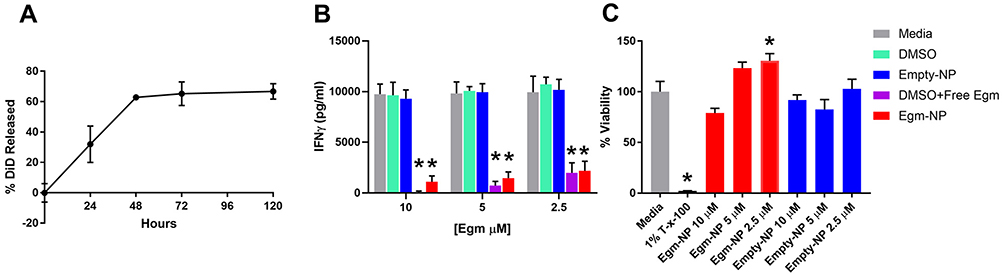 |
Figure 4 Nanoparticle-formulated Egm inhibits CD4+ T cell cytokine responses. Notes: (A) Normalized release profile of DiD from nanoparticles incubated in 37°C 1× PBS. (B) Interferon gamma (IFN-γ) production measured by ELISA from OT-II splenocytes stimulated with 50 µg/mL whole ovalbumin and incubated with vehicle controls, Egm in DMSO, and Egm-loaded nanoparticles for 72 hrs (n=3 biological replicates). (C) Average viability normalized to media controls of whole FVB splenocytes incubated for 72 hrs with Egm- and empty-NPs (n=3 biological replicates). No acute toxicity was observed for either particle formulation. Final Egm concentration of particle suspensions is listed. Empty particle concentrations correspond to matched polymer doses. The significance of the data was evaluated via ordinary One-way ANOVA with multiple comparison test (*p<0.05). Abbreviations: DiD, 1,1ʹ-dioctadecyl-3,3,3ʹ,3ʹ-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt; T-x-100, triton-x-100; empty-NP, empty nanoparticle; Egm-NP, Egm-loaded nanoparticle; DMSO, dimethyl sulfoxide. |
In combination with the cargo release profile and lack of acute toxicity observed from nanoparticle treatments within 72 hrs (Figure 4A and C), IFN-γ inhibition was inferred to be the result of Egm released from nanoparticles that inhibited the activation of CD4+ T cells undergoing antigen presentation. These studies cannot distinguish between inhibition due to Egm delivery to CD4+ T cells or to antigen-presenting cells in the mixed cell culture. Nevertheless, we have demonstrated the first characterization of antigen-specific CD4+ T cell inhibition mediated by nanoparticle-formulated Egm.
Maleimide-Thiol Chemistry-Conjugated Anti-CD4 f(ab’) Antibody Fragments Efficiently Target CD4+ T Cells in Heterogeneous Cell Suspensions
Successful encapsulation of fluorescent DiD within PEGylated PLGA nanoparticles enabled the characterization of CD4+ T cell targeting specificity afforded by maleimide-thiol antibody fragment decoration. Anti-CD4 and isotype control F(ab’) antibody fragments were conjugated to the surface of DiD-NPs and incubated for 30 mins with whole splenocyte cultures derived from immunocompetent mice. Evaluation of DiD+, nanoparticle-bound immune cell subtypes was performed via flow cytometry (Figure 5A).
We analyzed DiD+CD4+ T cells, CD8+ T cells, and non-T cells (B cells, dendritic cells, and macrophages) to determine nanoparticle CD4-targeting specificity. Anti-CD4 decorated particles achieved (~83%) CD4+ T cell staining, and significantly increased targeting specificity for CD4+ T cells compared to isotype (~5%) and undecorated controls (~2%). Additionally, anti-CD4 decorated particles also targeted CD4+ T cells significantly more than CD8+ T cells (~8%) and non-T cells (~3%) (Figure 5B). We further validated nanoparticle targeting specificity by incubating increasing concentrations of nanoparticles with whole splenocyte cultures. For anti-CD4 decorated particles, the degree of CD4+ T cell targeting was dependent on particle concentration, while particle concentration had no effect on CD8+ T cell targeting (Figure 5C). In all cases, undecorated and untargeted isotype-decorated particles exhibited low levels of nonspecific binding that was presumably due to electrostatic repulsion of negatively charged cell membranes mediated by sufficiently negative zeta potentials achieved in our formulations (Figure 2D). Therefore, the high degree of targeting specificity achieved by anti-CD4 decorated particles was almost certainly due to zeta potential mediated electrostatic repulsion from nonspecific cell types that was overcome by antibody fragment affinity for CD4+ T cells.
The nanoparticle formulation designed and fabricated in this study yielded low levels of non-specific binding, and strong CD4-targeting specificity. More importantly, we were able to achieve these results in a physiologically relevant, heterogenous whole splenocyte population, rather than purified T cell populations.16
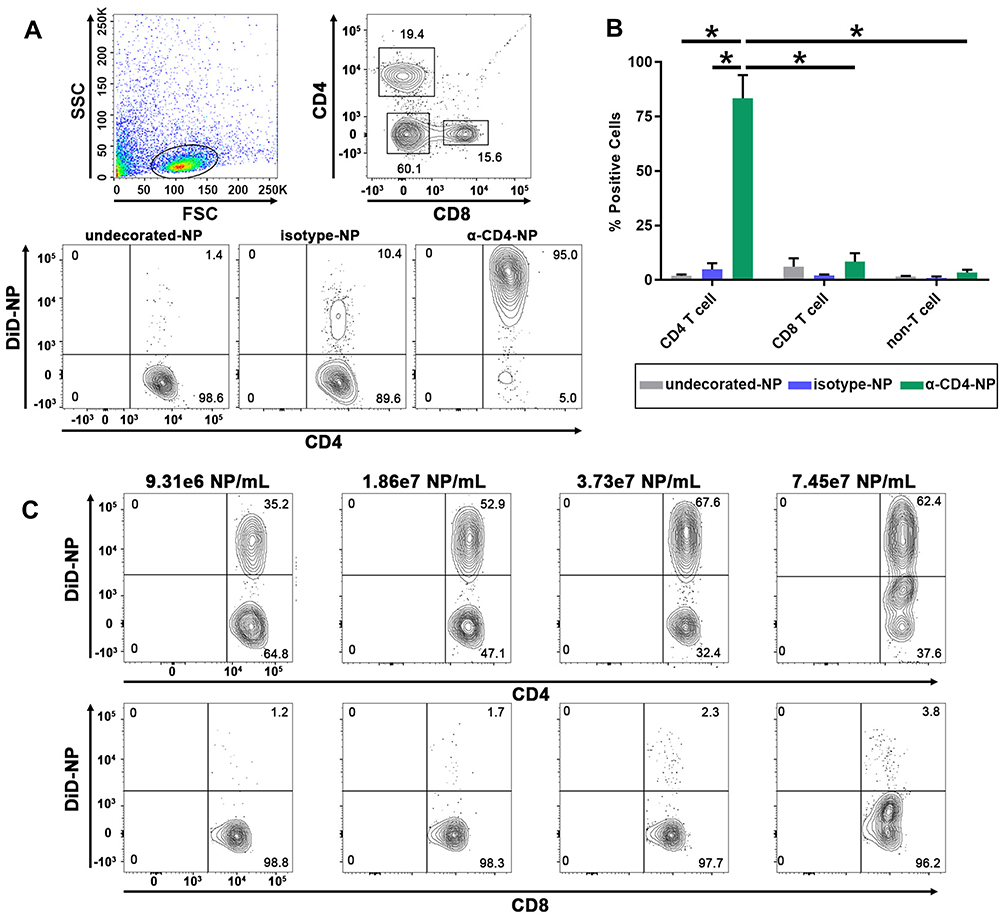 |
Figure 5 F(ab’) fragment conjugated PEGylated PLGA nanoparticles target CD4+ T cells. Notes: (A) Flow cytometry gating strategy used to analyze the targeting specificity of anti-CD4 decorated DiD-NPs and representative staining results from 1 of 3 experiments. Full flow cytometry gating strategy for targeting experiments shown in Supplemental Figure 3. (B) Quantification of DiD positive splenocytes incubated with either undecorated, isotype control decorated, or anti-CD4 decorated fluorescent nanoparticles suspended in 1× PBS. The flow cytometry probe for CD4 staining utilized an antibody clone that recognized a different epitope of CD4 than those used to decorate nanoparticles. (C) Analysis of CD4+ T cell targeting specificity after incubation with increasing concentrations of anti-CD4-decorated DiD-NPs. Nanoparticle concentrations were determined by nanoparticle tracking analysis. The percentage of DiD+CD4+ T cells and DiD+CD8+ T cells is indicated in the upper right-hand quadrants of the flow cytometry plots. Splenocytes were derived from female C57BL/6J mice. The significance of the data was evaluated via ordinary Two-way ANOVA with multiple comparison test (*p < 0.05). Abbreviations: SSC, side scatter; FSC, forward scatter; undecorated-NP, undecorated DiD-NP; isotype-NP, isotype control antibody fragment decorated DiD-NP; α-CD4-NP, anti-CD4 antibody fragment decorated DiD-NP. |
Conclusion
In this work, we synthesized and characterized anti-CD4 decorated PEGylated PLGA nanoparticle formulations for specific delivery of Egm to CD4+ T cells. Encapsulation of Egm did not affect the physical or chemical characteristics of the vehicle believed to be important for in vivo administration and cell-targeted delivery of cargo. Nanoscale elemental analysis and analytical chemistry methods supported the notion that emulsion mediated fabrication localized Egm within nanoparticle cores. We verified that nanoparticle formulations of Egm made from the FDA-approved polymers PLGA and PEG were biocompatible and capable of releasing the majority of their payload in a therapeutically relevant timeframe. Using maleimide-thiol chemistry conjugation of targeting antibody fragments, we were able to achieve high levels of CD4+ T cell-targeting specificity ex vivo with a minimal degree of non-specific particle binding. Furthermore, we have demonstrated antigen-specific inhibition of CD4+ T cell cytokine responses mediated by nanoparticle-formulated Egm. Collectively, this work represents the first characterization of the immunomodulatory effects of Egm, as well as the research and development of a rationally designed nanoparticle delivery vehicle intended for systemic Egm administration for the treatment of CD4+ T cell hyperactivity in rheumatic autoimmunity.
Although we limited the investigation of our formulation to CD4+ T cells, others have recently validated a similar formulation for delivery to CD8+ T cells.16 In fact, the conjugation mechanism of our formulation could presumably enable preferential delivery of hydrophobic cargo to any specific cell type possessing a unique cell surface marker with a corresponding antibody that can be decorated on the surface of biomaterial-based particles. We highlighted the clinical potential of Egm delivery for the treatment of autoimmune rheumatic diseases such as rheumatoid arthritis, systemic lupus erythematosus, and scleroderma, however, this formulation could also have broader applications in the treatment of human cancers associated with dysfunctional Hh signaling. Additionally, numerous FDA-approved hydrophobic immunosuppressants are currently used to broadly suppress the immune systems of patients with autoimmune diseases so that disease symptoms can be managed. The modular encapsulation mechanism employed by our formulation could enable focused delivery of these agents to improve the therapeutic index by limiting the necessary dosage, reducing off-target toxicities, and mitigating opportunistic infection. Additionally, several commercially available hedgehog inhibitors are currently approved for the treatment of human cancers while others are undergoing clinical trials for use in rheumatoid arthritis. Investigation of hedgehog signaling as a new therapeutic target with Egm-loaded nanoparticles may enable off-label use of existing FDA-approved Hh inhibitors in rheumatic autoimmunity.
Future experiments will focus on characterizing nanoparticle biodistribution over time following intravenous administration to determine optimal therapeutic administration strategies. Optimization of the current formulation components will be performed to evaluate circulation half-life, in vivo targeting efficiency, and suppression of T follicular helper cell activation in germinal centers. Additionally, studies utilizing prophylactic and therapeutic administration of CD4-targeted, Egm-NPs still need to be performed in order to elucidate the translational potential of Hh signaling inhibition for the restoration of peripheral tolerance mechanisms that are deficient in autoimmunity.
Acknowledgments
The authors would like to thank the laboratory of Dr. Charles C. Hong, MD, PhD, for providing the eggmanone used in these experiments. The authors also thank the staff of the Vanderbilt Cell Imaging Shared Resource and the Vanderbilt Institute for Nanoscale Science and Engineering for technical support with electron microscopy imaging instrumentation and analysis. Transmission electron microscopy was conducted at the Vanderbilt Cell Imaging Shared Resource and electron dispersive X-ray spectroscopy was conducted at the Vanderbilt Institute for Nanoscale Science and Engineering. This research was supported by NIAID award number: R03AI124190 (ASM and TDG), NHLBI award number: T32HL144446 (CPH), and METAvivor Research and Support, Inc. (TDG). This paper was presented at the 2018 BMES annual meeting as a poster presentation with interim findings. The poster’s abstract was published in the “annual meeting abstract archive” found here: https://www.bmes.org/abstractarchive.
Disclosure
Professor Charles C Hong and Dr Charles H Williams report a US Patent #: US 10,329,304 B2 issued. Professor Todd D Giorgio reports patent materials and methods for the treatment of immune cells pending. The authors report no other conflicts of interest in this work.
References
1. Marder W, Vinet É, Somers EC. Rheumatic autoimmune diseases in women and midlife health. Women’s Midlife Heal. 2015;1:1–8.
2. Vaughan S, Dawe HR. Common themes in centriole and centrosome movements. Trends Cell Biol. 2011;21:57–66. doi:10.1016/j.tcb.2010.09.004
3. De La Roche M, Ritter AT, Angus KL, et al. Hedgehog signaling controls T cell killing at the immunological synapse. Science. 2013;342:1247–1250. doi:10.1126/science.1244689
4. Smelkinson M. The hedgehog signaling pathway emerges as a pathogenic target. J Dev Biol. 2017;5:14. doi:10.3390/jdb5040014
5. Crompton T, Outram SV, Hager-Theodorides AL. Sonic hedgehog signalling in T-cell development and activation. Nat Rev Immunol. 2007;7:726–735. doi:10.1038/nri2151
6. Chan VS, Chau SY, Tian L, et al. Sonic hedgehog promotes CD4+T lymphocyte proliferation and modulates the expression of a subset of CD28-targeted genes. Int Immunol. 2006;18:1627–1636. doi:10.1093/intimm/dxl096
7. Kupfer A, Singer SJ. The specific interaction of helper T cells and antigen-presenting B cells. IV. Membrane and cytoskeletal reorganizations in the bound T cell as a function of antigen dose. J Exp Med. 1989;170:1697–1713. doi:10.1084/jem.170.5.1697
8. Williams CH, Hempel J, Hao J, et al. An in vivo chemical genetic screen identifies phosphodiesterase 4 as a pharmacological target for hedgehog signaling inhibition. Cell Rep. 2015;11:43–50. doi:10.1016/j.celrep.2015.03.001
9. Xie C, Ramirez A, Wang Z, Chow MSS, Hao J. A simple and sensitive HPLC–MS/MS method for quantification of eggmanone in rat plasma and its application to pharmacokinetics. J Pharm Biomed Anal. 2018. doi:10.1016/j.jpba.2018.01.009
10. McHugh MD, Park J, Uhrich R, et al. Paracrine co-delivery of TGF-β and IL-2 using CD4-targeted nanoparticles for induction and maintenance of regulatory T cells. Biomaterials. 2015;59:172–181. doi:10.1016/j.biomaterials.2015.04.003
11. Chinol M, Casalini P, Maggiolo M, et al. Biochemical modifications of avidin improve pharmacokinetics and biodistribution, and reduce immunogenicity. Br J Cancer. 1998;78:189–197. doi:10.1038/bjc.1998.463
12. Song G, Petschauer J, Madden A, Zamboni W. Nanoparticles and the mononuclear phagocyte system: pharmacokinetics and applications for inflammatory diseases. Curr Rheumatol Rev. 2014;10:22–34. doi:10.2174/1573403X10666140914160554
13. Cao S, Jiang Y, Levy CN, et al. Optimization and comparison of CD4-targeting lipid–polymer hybrid nanoparticles using different binding ligands. J Biomed Mater Res Part A. 2018;106:1177–1188. doi:10.1002/jbm.a.v106.5
14. ThermoFisher. Lipophilic tracers. 1–6. 2008. Available from: https://www.thermofisher.com/order/catalog/product/D7757.
15. Malvern Instruments Limited. Comparison of statistical measures reported by NTA and DLS techniques. 2015. Available from: www.malvern.com.
16. Schmid D, Park CG, Hartl CA, et al. T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat Commun. 2017;8:1–11. doi:10.1038/s41467-017-01830-8
17. Gerberick GF, Cruse LW, Miller CM, Sikorski EE, Ridder GM. Selective modulation of t cell memory markers CD62L and CD44 on murine draining lymph node cells following allergen and irritant treatment. Toxicol Appl Pharmacol. 1997;146:1–10. doi:10.1006/taap.1997.8218
18. Li S-D, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5:496–504. doi:10.1021/mp800049w
19. Moghimi SM, Hunter AC, Andresen TL. Factors controlling nanoparticle pharmacokinetics: an integrated analysis and perspective. Annu Rev Pharmacol Toxicol. 2012;52:481–503. doi:10.1146/annurev-pharmtox-010611-134623
20. Hoshyar N, Gray S, Han H, Bao G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine. 2016;11:673–692. doi:10.2217/nnm.16.5
21. Honary S, Zahir F. Effect of zeta potential on the properties of nano-drug delivery systems - a review (Part 1). Trop J Pharm Res. 2013;12:255–264.
22. Fröhlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomed. 2012;7:5577–5591. doi:10.2147/IJN.S36111
23. Kishimoto TK, Maldonado RA. Nanoparticles for the induction of antigen-specific immunological tolerance. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.00230
24. Olivier JC, Huertas R, Hwa JL, Calon F, Pardridge WM. Synthesis of pegylated immunonanoparticles. Pharm Res. 2002;19:1137–1143. doi:10.1023/A:1019842024814
25. Hong C, et al. Compounds and Methods for Inhibition of Hedgehog Signaling and Phosphodiesterase. 2016. Avalable from: https://patentimages.storage.googleapis.com/56/95/27/735383ea5acde1/US10329304.pdf.
26. Makadia HK, Siegel SJ. Poly Lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polym. 2011;3:1377–1397. doi:10.3390/polym3031377
27. Ademokun AA, Dunn-Walters D. Immune responses: primary and secondary. Encycl Life Sci. 2010. doi:10.1038/npg.els.0000947
28. Jackson S, Jacobs HM, Arkatkar T, et al. B cell IFN-γ receptor signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. J Exp Med. 2016;213:733–750. doi:10.1084/jem.20151724
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.