")
Back to Journals » International Journal of Nanomedicine » Volume 15
PEG-Ceramide Nanomicelles Induce Autophagy and Degrade Tau Proteins in N2a Cells
Authors Gao J, Chen X, Ma T, He B, Li P, Zhao Y, Ma Y, Zhuang J, Yin Y
Received 15 April 2020
Accepted for publication 7 August 2020
Published 11 September 2020 Volume 2020:15 Pages 6779—6789
DOI https://doi.org/10.2147/IJN.S258311
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Jie Gao,1,2,* Xiaohan Chen,1,* Tianjun Ma,1,* Bin He,1 Peng Li,1 Yucheng Zhao,1 Yuejin Ma,1 Jianhua Zhuang,1 You Yin1
1Department of Neurology, Changzheng Hospital, Second Military Medical University, Shanghai 200003, People’s Republic of China; 2Institute of Translational Medicine, Shanghai University, Shanghai 200444, People’s Republic of China
*These authors contributed equally to this work
Correspondence: You Yin; Jianhua Zhuang
Department of Neurology, Changzheng Hospital, Second Military Medical University, Shanghai 200003, People’s Republic of China
Tel/Fax +86-21-63586818
Email [email protected]; [email protected]
Purpose: Alzheimer’s disease (AD) is a neurodegenerative disorder that manifests as abnormal behavior and a progressive decline in memory. Although the pathogenesis of AD is due to the excessive deposition of amyloid β protein (Aβ) outside the neurons in the brain, evidence suggests that tau proteins may be a better target for AD therapy. In neurodegenerative diseases, a decrease in autophagy results in the failure to eliminate abnormally deposited or misfolded proteins. Therefore, induction of autophagy may be an effective way to eliminate tau proteins in the treatment of AD. We investigated the effects of polyethylene glycol (PEG)-ceramide nanomicelles on autophagy and on tau proteins in N2a, a murine neuroblastoma metrocyte cell line.
Methods: Ceramide is a sphingolipid bioactive molecule that induces autophagy. PEG-ceramide is a polymer that is composed of the hydrophobic chain of ceramide and the hydrophilic chain of PEG-2000. In this study, we prepared PEG-ceramide nanomicelles that were 10– 20 nm in size and had nearly neutral zeta potential.
Results: The results show that PEG-ceramide nanomicelles caused an increase in the LC3-II/LC3-I ratio, while p62 protein levels decreased. Confocal microscopy revealed a significant increase in the number of dots corresponding to autophagosomes and autolysosomes, which indicated autophagic activation. Moreover, PEG-ceramide nanomicelles induced tau degradation in N2a cells through autophagy.
Conclusion: In summary, we have confirmed that PEG-ceramide nanomicelles enhanced autophagic flux and degraded overexpressed human tau proteins in N2a cells by regulating the autophagy pathway. Thus, PEG-ceramide nanomicelles show great promise as agents to induce autophagy and degrade tau proteins in the treatment of AD.
Keywords: Alzheimer’s disease, autophagosomes, lysosome, nano-carrier, autophagic flux
Introduction
With the aging of all populations worldwide, dementia has become the third leading cause of death in the world, after cardiovascular, cerebrovascular diseases, and cancer; Alzheimer’s disease (AD) accounts for 60–80% of all cases of dementia. The main clinical symptoms of AD patients are gradual memory loss and cognitive dysfunction. The pathogenesis of AD has been attributed to excessive deposition of amyloid β protein (Aβ) outside the neurons or to neurofibrillary tangles (NFTs) within the neurons, both of which can lead to necrosis and apoptosis of nerve cells.1–3 Although various molecules have demonstrated in vitro Aβ degradation, they have not caused any significant improvement in clinical symptoms or delayed the progression of AD.4,5
Recent reports have shown that cognitive impairment is closely associated with tau protein phosphorylation, NFTs, and the loss of synapses and neurons, but not with Aβ deposition. Tau proteins play roles in the maintenance of normal skeletal structure, brain maturation, regulation of axonal transport and neuronal signaling, the cellular response of heat shock, and generation of adult nerve impulses.6–8 Researchers in the Mayo Clinic have found that Aβ levels increase with NFT deposition, which is linked to the progression of AD and cognitive decline.9 However, positron emission tomography imaging of patients with early AD showed that the deposition of tau proteins in the temporal lobe region, which is responsible for memory, was more obvious than Aβ aggregation and was proportional to the impairment of memory.10 Current strategies to reduce the abnormal deposition of tau proteins mainly focus on inhibiting their aggregation and accelerating their clearance.11 Therefore, targeted clearance of tau proteins is a potentially effective strategy to treat AD.
Autophagy is a highly conserved catabolic process in mammalian cells. It is a process in which lysosomes degrade aged cells and misfolded proteins and is characterized by the formation of double membrane vesicles called autophagosomes.12 Homeostasis of neurons requires the maintenance of autophagy. There is evidence that decreased autophagy is positively correlated with neurodegenerative diseases, including the pathological progression of AD.13,14 Studies have shown an increase in the number of autophagic vesicles triggered by amyloid precursor protein and damaged organelles in the early stages of AD. However, due to the maturation and degradation dysfunction of these autophagic vesicles in the later stages of AD, the abnormal deposits of Aβ protein are not cleared, resulting in the formation of “senile plaques”.15 Besides, some studies have confirmed that inducing autophagy at the molecular or gene level could delay the neurodegenerative diseases caused by abnormal protein aggregation in animal models.16–21
Due to the development of nanotechnology and materials science, efforts are focused on the development of a brain-targeted nano-delivery system to deliver drugs effectively to the diseased parts of the brain.22 We had previously reviewed nanocarrier modifications usually linked with short peptides, monoclonal antibodies, or protein fragments for specific brain targeting through enhanced binding of the nanocarrier system to the surface receptors in the blood-brain barrier (BBB).6 Nano-carriers can also regulate autophagy by:23–26 (1) Inhibition of autophagy: The underlying mechanisms include inhibiting the formation of autophagosomes or inhibiting the fusion of autophagosomes and lysosomes by inducing lysosomal membrane permeability and inhibiting the activity of lysosomes. (2) Induction of autophagy: This is mainly caused by oxidative stress and a reduction in adenosine 5ʹ-triphosphate (ATP) levels. The nano-carriers enter cells and damage the mitochondria, leading to reactive oxygen species production and oxidative stress. Mitochondrial dysfunction also leads to a decrease in cellular ATP levels, which activates the 5ʹ-adenosine monophosphate (AMP)-mammalian target of rapamycin (mTOR) pathway and triggers autophagy. A polypeptide–polymer nanomaterial developed by the National Center for Nanoscience and Technology was injected intravenously to deliver the “nano-scavenger” to the lesion site and form a copolymer with Aβ through hydrophobic and hydrogen bond interactions. Large quantities of this “nano-scavenger” carrying the Aβ entered the cells and activated autophagy, which effectively reduced Aβ accumulation and neurotoxicity, thus improving the memory of the AD model mice.27 Thus, activation of the autophagy pathway by nano-carriers is a promising strategy for the treatment of AD.
Ceramide is a bioactive sphingolipid that triggers autophagy and also induces apoptosis and other forms of cell death.28 Like amino acid starvation, ceramide triggers autophagy by interfering with the mTOR signaling pathway and by dissociating the Beclin 1:B-cell lymphoma 2 protein (Bcl-2) complex in a c-Jun N-terminal kinase1 (JNK1)-mediated, Bcl-2 phosphorylation-dependent manner.28 We had previously developed a novel system of nanoliposomes (Lip-ADR-Cer) for the co-delivery of adriamycin and ceramide to overcome cancer multidrug resistance, in which PEGylated C16-ceramide (PEG-ceramide) was employed as a liposomal component to increase the stability of the nanoliposomes.29 As the structure of PEG-ceramide is composed of the hydrophobic chain of C16-ceramide and the hydrophilic chain of PEG2000, we used PEG-ceramide to form nanomicelles to deliver salinomycin to treat liver cancer.28 To date, there has been no report on whether PEG-ceramide nanomicelles can accelerate the removal of abnormal proteins that mimic the pathology of AD by inducing autophagy. Hence, in this study, we prepared and characterized PEG-ceramide nanomicelles and examined their effects on tau proteins in mouse brain tumor cells, Neuro-2a (N2a).
Materials and Methods
Reagents
PEG-ceramide was purchased from Avanti Polar Lipids (AL, USA). Dimethyl sulfoxide, ammonium persulfate, and Earle’s Balanced Salt Solution were purchased from Sigma-Aldrich (MO, USA). Phosphate-buffered saline (PBS), Dulbecco’s Modified Eagle Medium (DMEM) with high glucose, 0.25% ethylene diaminetetraacetic acid (EDTA), pancreatic enzymes, penicillin-streptomycin solution, fetal bovine serum (FBS), and bicinchoninic acid assay kit were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Cell Counting Kit-8 was purchased from Dojindo Laboratories (Kumamoto, Japan). Rapamycin was purchased from MedChemExpress (MCE, USA). LC3 and p62 antibodies were purchased from Abcam (Cambridge, UK). FuGene HD Transfection Reagent was purchased from Promega (CA, USA). pRK5-EGFP-Tau plasmid and pRK5-EGFP-TauP301L plasmid were purchased from Addgene (MA, USA). Plasmid Miniprep Kit was purchased from Qiagen (MD, USA). mRFP-GFP-LC3 adenovirus was purchased from Hanbio Biotechnology Co. Ltd. (Shanghai, China). Western and IP cell lysis solution, phenylmethanesulfonyl fluoride, tetramethylethylenediamine, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer, and Ponceau S Staining Kit were purchased from Beyotine Biotechnology (Haimen, China). Tris(hydroxymethyl)aminomethane, glycine, and ampicillin were purchased from Bio-Light (Shanghai, China). Chloroform, ethyl alcohol, absolute ethyl alcohol, and polysorbate 20 were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
Cell Culture
Neuro-2a (N2a), a mouse-derived neuroblastoma cell line, was purchased from the Chinese Academy of Sciences Cell Bank and cultured in DMEM with high glucose containing 10% FBS and 1 mM sodium pyruvate solution in a humidified atmosphere of 5% CO2 at 37°C.
Preparation of PEG-Ceramide Nanomicelles
PEG-ceramide nanomicelles were prepared as described previously.30 Briefly, 5 mg PEG-ceramide in a test tube was mixed with 3 mL chloroform, followed by 1 mL methanol using a vortex mixer. The solution was transferred to a 250 mL round-bottom flask and evaporated under vacuum for 1 h using a rotary evaporator at 60 rpm and 37°C until a uniform film was distributed at the bottom of the round bottom flask. PBS (2 mL, pH=7.4) was subsequently added to the flask, and rotation was continued for 30 min at a speed of 60 rpm to dissolve the film. The resultant PEG-ceramide nano-micelles were stored at 4°C.
Characterization of PEG-Ceramide Nanomicelles
The particle size and zeta potential of the nanomicelles were analyzed using a Zetasizer Nano S (Malvern Instruments, Malvern, UK). Briefly, 0.05 mL of the PEG-ceramide nanomicelle solution was placed in a dish, diluted with 1 mL deionized water, and then placed on a Marvin laser particle size tester to determine the particle size distribution and zeta potential.
The morphological examination of nanomicelles was performed by transmission electron microscopy (TEM). Briefly, Briefly, samples were prepared by dropping one drop of nanomicelle dispersion onto a copper grid coated with a carbon membrane. Then the samples were dried, and were visualized under the Hitachi H-600 TEM (accelerating voltage of 200 kV).
The critical micelle concentration (CMC) of PEG-ceramide was determined by using the pyrene fluorescence method, as described previously.31 Briefly, 100 mL of pyrene solution (6×10−5 M in acetone) was transferred into a series of volumetric flasks (10.0 mL), and the acetone was evaporated under a gentle stream of nitrogen gas for 4 h at room temperature. PEG-ceramide solutions were added into each flask to achieve a final pyrene concentration of 6×10−7 M and PEG-ceramide concentrations ranging from 0.02 μg/mL to 250 μg/mL. Then, the solutions were allowed to equilibrate for another 12 h at 37°C in a shaker (SHKE6000-8CE, Thermo Scientific, USA). A fluorescence spectrum was recorded using a fluorescent spectrometer F-7000 (Hitachi, Japan). The excitation wavelengths ranged from 300 to 360 nm, but the emission wavelength was fixed at 390 nm. Emission and excitation slit widths were both 2.5 nm, and the scanning speed was set at 240 nm/min.
Cytotoxicity Assay
The cytotoxicity of PEG-ceramide nanomicelles against N2a cells was measured using a CCK-8 kit. Briefly, N2a cells (dissociated into single cells) were seeded in 96-well plates at a density of 3×103 cells per well and incubated overnight. The cells were then incubated for 24 h with varying concentrations of PEG-ceramide nanomicelles (3.7–118.6 μM). Cytotoxicity was subsequently evaluated by adding the CCK-8 (10 μL) solution to each well of the plate. After incubation for 2 h, the absorbance was measured at 450 nm using an enzyme-labeled mini reader (Bio-Rad, CA, USA). Cell viability was calculated by using the formula: [(AE –AB)/(AC – AB)] × 100%, where AE, AC, and AB were defined as the absorbance of experimental samples, untreated controls, and blank controls, respectively. The IC50 value was calculated using GraphPad Prism version 6.04 for Windows (GraphPad Software, La Jolla, California, USA).
Western Blotting
Western blotting of LC3 and P62 was performed by using the standard instructions provided by Cell Signaling Technology (MA, USA). Briefly, extracted protein (40 μg) was subjected to SDS-PAGE and transferred onto nitrocellulose membranes. LC3 (final dilution 1:1000, Abcam), p62 (final dilution 1:1000, Abcam), and actin antibodies (final dilution 1:1000, Beyotine) were used as primary antibodies, and goat anti-rabbit (H+L) horseradish peroxidase conjugate (1:5000 dilution, Thermofisher) was used as the secondary antibody. The bands were detected by using an enhanced chemiluminescence (ECL) kit (GE Healthcare) and visualized with a ChemiDoc XRS system (Bio-Rad); protein quantitation was performed by normalizing relative to actin using Quantity One Version 4.6.2.
Autophagic Flux Analysis
An RFP-GFP-LC3 assay was performed to assess autophagic flux. Briefly, the cells were cultured overnight on Live-focus culture plates and transfected with the RFP-GFP-LC3 adenoviral vector, as described earlier.32 After treatment with 5 μM PEG-ceramide nanomicelles for 24 h, the cells were observed under a confocal laser scanning fluorescence microscope. Autophagic flux was measured by counting the cells with GFP+/mRFP+ (yellow) and GFP−/mRFP+ (red) puncta. A total of 20 cells were counted per sample in triplicate experiments.
Extraction of pRK5-EGFP-Tau and pRK5-EGFP-TauP301L Plasmid
The pRK5-EGFP-Tau and pRK5-EGFP-TauP301L plasmids were extracted from bacteria by using the following methods. Briefly, 1% plasmid-containing bacterium was cultured overnight in 2 mL LB medium at 37°C, and 1.5 mL of the bacterial suspension was transferred to an Eppendorf tube, centrifuged at 4000 rpm for 3 min, and the supernatant was discarded. Then, 0.1 mL of solution I (1% glucose, 50 mM EDTA pH 8.0, 25 mM Tris-HCl pH 8.0) and 0.2 mL of solution II (0.2 mM NaOH, 1% SDS) were mixed in an ice bath for 5 min, and 0.15 mL precooled solution III (5 M potassium acetate solution, pH 4.8) was added and mixed for 5 min in the ice bath. After centrifugation at 10,000 rpm for 20 min, the supernatant was transferred into another Eppendorf tube and mixed with an equal volume of isopropanol and incubated for 10 min. After centrifugation at 10,000 rpm for another 20 min, the supernatant was discarded. The pellet was washed with 0.5 mL of 70% (v/v) ethanol, and all the liquid was removed carefully to air-dry the pellet. The dried pellet was dissolved in 50 μL of Tris-EDTA buffer solution (or in deionized water at 60°C). The absorbance of the plasmid solution was determined at 260 nm using an ultraviolet spectrophotometer, and the plasmid concentration was calculated.
Analysis of Tau in N2a Cells After Treatment with PEG-Ceramide Nanomicelles
N2a cells were first transfected with pRK5-EGFP-Tau and pRK5-EGFP-TauP301L plasmids. Briefly, N2a cells were seeded in 6-well plates and incubated overnight at 37°C in a 5% CO2 incubator; 800 μL of serum-free, antibiotic-free medium was added to each well. Diluting plasmid with serum-free, antibiotic-free cell growth medium to a concentration of 0.02 mg/mL (2 μg of the plasmid in 100 μL of diluent). Then pipet the Fugene HD reagent (6 μL) directly into the medium containing the diluted DNA. The resultant mixture was incubated at room temperature for 12 min and then added to the 6-well plates, followed by incubation at 37°C in a 5% CO2 incubator for 2 h. Medium was added to the cells to achieve a final serum concentration of 1–4% in a volume of 1.5–2 mL. Fluorescent protein could be detected in the cells under a fluorescence microscope after culturing for 24 h. The cell suspensions were harvested and analyzed by flow cytometer to determine the transfection efficiency of pRK5-EGFP-Tau and pRK5-EGFP-TauP301L plasmids. Then, the transfected N2a cells were treated with PEG-ceramide micelles (5 μM) and the positive control, rapamycin (5 μM) for 24 h, the total protein was extracted for Western blotting analysis.
Statistical Analysis
Data in this study were analyzed using SPSS 13.0 software (SPSS, Inc., IL, USA). Student’s unpaired t-tests and one-way analysis of variance with Dunnett’s or Newman Keul’s post-tests were used. Differences with p values of less than 0.05 indicated significance. *P < 0.05; **P < 0.01; ***P < 0.001; n.s. represents not significant (P > 0.05).
Results
Characterization of PEG-Ceramide Nanomicelles
The PEG-ceramide nanomicelles showed a normal size distribution with a single peak and narrow peak width; the average diameter was 17.69 nm, and the zeta potential was 0.75 mv (Figure 1A and B). Theoretically, a small particle size facilitates better tissue permeability, while the neutral potential reduces binding to serum proteins and increases the penetration through cell membranes.33,34 Transmission electron microscopy revealed that PEG-ceramide nanomicelles were spherical and well-dispersed, with a size of ~20 nm (Figure 1C). To examine the amphiphilic property and self-assembling behavior of PEG-ceramide nanomicelles, the critical micelle concentration (CMC) of the copolymers was determined by using a pyrene fluorescence method described earlier.29 The shift of the excitation band at 334 nm was negligible at low concentrations of copolymers (Figure 1D). However, a redshift of the excitation band to 336 nm could be detected as the concentration of the copolymers increased. The fluorescence intensity ratio (I336/I334) was plotted against the concentrations of the copolymers, and the CMC values of the copolymers were defined as the intersection of the lines drawn through the points of flat regions of the curves at low concentrations and the rapidly rising parts of the curves at high concentrations. The CMC, an important characteristic of a surfactant, indicates the concentration above which the detergent forms micelle complexes. PEG-ceramide has a low CMC value (7.89 μg/mL), which might be beneficial in maintaining and stabilizing the original structure of PEG-ceramide nanomicelles.
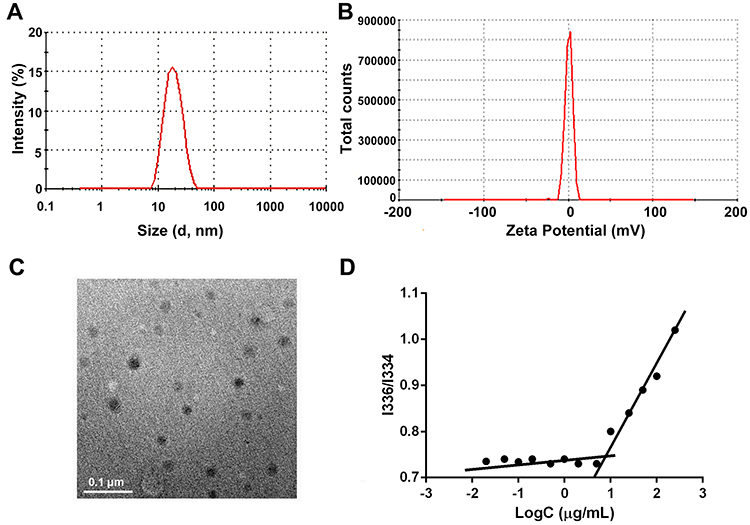 |
Figure 1 Characterization of nanomicelles. (A and B) Size distribution and zeta potential of PEG-ceramide nanomicelles. One representative image is shown. (C) TEM image of nanomicelles. Bars represent 100 nm. (D) CMC values of the PEG-ceramide copolymers were determined by a pyrene fluorescence method. The fluorescence intensity ratio (I336/I334) was plotted against the concentrations of the copolymers, and the CMC values of the copolymers were defined as the intersection of the lines drawn through the points of flat regions of the curves at low concentrations and the rapidly rising parts of the curves at high concentrations. Abbreviations: PEG, polyethylene glycol; TEM, transmission electron microscopy; CMC, critical micelle concentration. |
In vitro Cytotoxicity of PEG-Ceramide Nanomicelles
The toxic effects of PEG-ceramide nanomicelles on N2a cells were concentration-dependent, and the IC50 value was found to be 21.89 μM (Figure 2). Hence, concentrations below 20 μM (2.5 μM, 5.0 μM, 10.0 μM, and 20 μM) were selected for subsequent experiments.
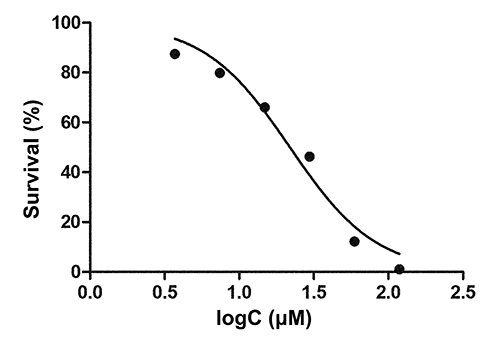 |
Figure 2 Dose-dependent cytotoxicity induced by nanomicelles in N2a cells. The cells were incubated for 24 h with varying concentrations of nanomicelles, and the cell viability was evaluated by the CCK-8 assay. The fitted curve of the average of three experiments is shown. |
Analysis of Autophagic Flux by LC3 and P62 Analysis
To elucidate the effects of PEG-ceramide nanomicelles on autophagic flux, we performed Western blotting to detect the expression of autophagy proteins, LC3 and p62, and immunofluorescence after transfection of an LC3 reporter protein in N2a cells after PEG-ceramide nanomicelle treatment. At the beginning of the autophagic process, LC3-I (cytosolic LC3) undergoes cleavage, and the remaining peptide is conjugated to phosphatidylethanolamine to form LC3-II (autophagosome-membrane-type LC3). As a consequence, LC3-I levels decrease while LC3-II levels increase; hence, the LC3-II/LC3-I ratio is usually used to estimate the level of autophagic activity. In the process of autophagy, the ubiquitin-binding protein, p62, binds to a ubiquitinated protein and forms a complex with the LC3-II protein on the membranes of the autophagosomes, which is eventually degraded in the lysosome. Hence, p62 is constantly degraded as a substrate when autophagy is activated.32
The LC3-II/LC3-I ratios of the experimental group and rapamycin group increased significantly (p < 0.01), whereas p62 protein levels decreased significantly (Figure 3, p < 0.01) except for 2.5 μM of PEG-ceramide nanomicelles. However, no concentration dependence was observed because higher concentrations of the PEG-ceramide nanomicelles were cytotoxic and inhibited autophagic activity. As the highest level of autophagic activity was induced by 5 μM of PEG-ceramide nanomicelles and rapamycin, and this concentration of the nanomicelles only caused low cytotoxicity in N2a cells, this concentration was selected for subsequent experiments.
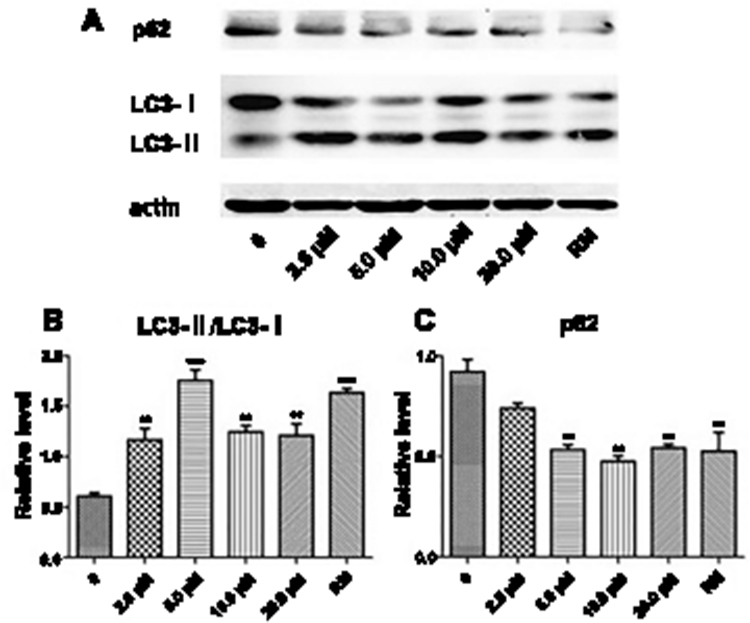 |
Figure 3 PEG-ceramide nanomicelles promote autophagy in N2a cells. (A) Western blots of LC3-I, LC3-II, and p62 in N2a cells treated with different concentrations of PEG-ceramide nanomicelles. RM (rapamycin, 5 μM) was used as positive control. (B and C) The graph of protein levels normalized to β-actin levels. All other groups were compared with the untreated control group by one-way analysis of variance with Dunnett’s post hoc test. Data are expressed as mean ± standard deviation (n = 3). **p < 0.01 and ***p < 0.001. Abbreviations: PEG, polyethylene glycol; LC3, light chain 3. |
Analysis of Autophagic Flux by the RFP-GFP-LC3 Assay
A novel tandem fluorescent-tagged reporter system, RFP-GFP-LC3, comprising the fusion of LC3 to an acid-resistant red fluorescent protein (RFP) and acid-sensitive green fluorescent protein (GFP), has been developed to analyze autophagosome maturation and degradation and monitor autophagic flux. The cells were transduced with a virus that expressed the RFP-GFP-LC3 fusion protein, which facilitated the tracking of labeled LC3. The fluorescence of acid-sensitive GFP is quenched by the fusion of autophagosomes and lysosomes. Thus, this reporter system can be used to identify autophagosomes (GFP+/RFP+; yellow dots) and autolysosomes (GFP−/RFP+; red dots). Consistent with the increase in LC3-II/LC3-I and the decrease in p62 protein, PEG-ceramide nanomicelles demonstrated a significant increase in the autophagic activity as determined by increased green/red fluorescence compared to controls (Figure 4A). The numbers of GFP and mRFP dots per cell were both significantly increased (p < 0.01) after treatment with 5 μM of PEG-ceramide nanomicelles for 24 h (Figure 4B). In the merged images, the numbers of yellow dots and free red dots were also significantly increased (p < 0.01), indicating significantly increased autolysosome and autolysosome formation, and suggesting that PEG-ceramide nanomicelles increases autophagic flux (Figure 4C).
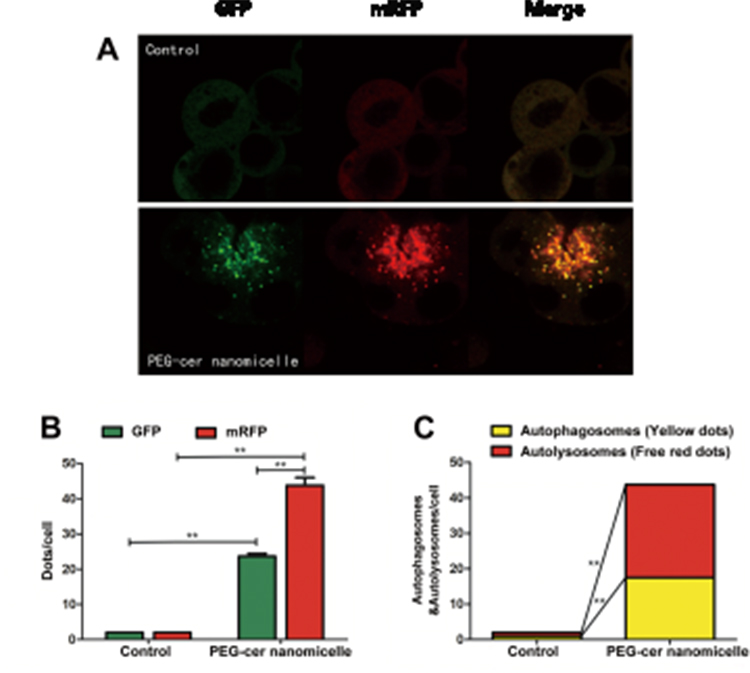 |
Figure 4 PEG-ceramide nanomicelles increase autophagic flux in the RFP-GFP-LC3 assay. The cells were transfected with the RFP-GFP-LC3 adenoviral vector. After treatment with PEG-ceramide nanomicelles, the cells were observed under a confocal microscope. (A) The cells under a confocal microscope. Scale bar: 20 μm. (B) The GFP+ and RFP+ LC3 puncta. (C) Autophagic flux was measured by counting the cells with GFP+/RFP+ (yellow) and GFP−/RFP+ (red) LC3 puncta. In all, 20 cells were counted per sample for each condition. Data (mean ± standard deviation, n = 20) are representative of three independent experiments. All the groups are compared to the control group by using non-paired Student’s t-test. **p < 0.01. Abbreviations: PEG, polyethylene glycol; RFP, red fluorescent protein; GFP, green fluorescent protein; LC3, light chain protein 3. |
Analysis of Tau in N2a Cells After Treatment with PEG-Ceramide Nanomicelles
As PEG-ceramide nanomicelles increased the autophagic flux in N2a cells, we hypothesized that the nanomicelles could degrade overexpressed Tau proteins by activating autophagy. N2a cells were transfected with plasmids to express the wild-type (WT) and mutant (P301L) human Tau proteins. After transfection, GFP was detected in the cells under the fluorescence microscope, which suggested that both wild-type Tau and mutant-type Tau were expressed in the cells (Figure 5A and B). Flow cytometric analysis suggested that the transfection efficiency of pRK5-EGFP-Tau and pRK5-EGFP-TauP301L plasmids were 60%-70% in N2a cells (Figure 5C). Western blotting revealed that, similar to 5 μM rapamycin, 5 μM PEG-ceramide nanomicelles significantly increased the LC3-II/LC3-I ratios, but significantly reduced the content of WT-Tau and P301L-Tau proteins in the cells (Figure 6, p < 0.001).
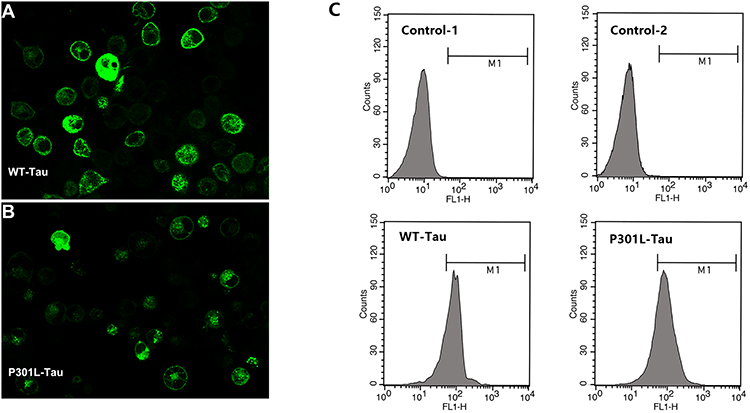 |
Figure 5 The transfection efficiency of N2a cells with pRK5-EGFP-Tau and pRK5-EGFP-TauP301L plasmids, as reflected by the GFP fluorescence. (A) Fluorescent image of N2a cells transfected with the pRK5-EGFP-Tau plasmid; (B) Fluorescent image of N2a cells transfected with the pRK5-EGFP-TauP301L plasmid. (C) The transfection efficiency of pRK5-EGFP-Tau and pRK5-EGFP-TauP301L plasmids were determined by flow cytometry, respectively. Abbreviations: EGFP, enhanced green fluorescent protein; GFP, green fluorescent protein. |
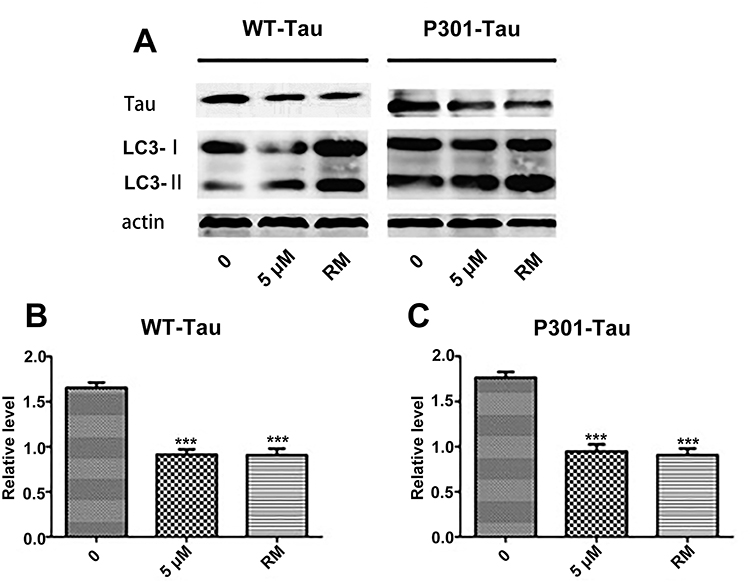 |
Figure 6 PEG-ceramide nanomicelles promote autophagy and degrade tau protein in N2a cells. (A) Western blotting of LC3-I, LC3-II, and tau protein in N2a cells treated with PEG-ceramide nanomicelles. RM (rapamycin, 5 μM) was used as positive control. (B and C) The graph of protein levels normalized to β-actin levels. All other groups were compared to the untreated control group by one-way analysis of variance with Dunnett’s post-hoc test. Data are expressed as mean ± standard deviation (n = 3). ***p < 0.001. Abbreviations: PEG, polyethylene glycol; LC3, light chain 3. |
Discussion
This study is the first to employ PEG-ceramide nanomicelles as an agent to induce autophagy to clear the tau protein in neuroblastoma cells. Targeted clearance of tau proteins is a potential therapeutic strategy for AD.21 Autophagy is a highly conserved catabolic process, and any decrease in autophagic activity can cause the abnormal deposition of tau proteins. We had previously shown that nanomicelles composed of amphiphilic block copolymers could be efficiently loaded with a hydrophobic anticancer drug.29,30 Ceramide has very small and poorly hydrated head groups, and the packing parameters (PP) are > 1.3.29 However, the PP of PEG-ceramide was significantly lower because of the hydrophilic tail of the PEG moiety, and its amphiphilic polymeric structure resulted in its ability to form nanomicelles. Our results show that the CMC of PEG-ceramide is very low (7.89 μg/mL), confirming that PEG-ceramide could form nanomicelles. Our data show that the PEG-ceramide nanomicelles are very small, ~10–20 nm, with a nearly neutral zeta potential (0.75 mV). The small size of the nanomicelles could facilitate cell penetration.33 Furthermore, nanoparticles with neutral zeta potential tend to have less harmful interactions with serum albumin compared to cationic nanoparticles.34
In the last 15 years, there have been no new drugs for AD, and the failure rate has been almost 100%, especially for drugs targeted at Aβ.4,5 Tau proteins have high solubility and are present in the central and peripheral nervous system neurons, astrocytes, and oligodendrocytes, especially in the axons of the central or peripheral nervous system neurons. Their physiological functions include promoting tubulin aggregation and microtubule stability, as well as playing an important role in the transmission of intracellular substances in neurons.6–8 In other words, Aβ is the key to pathological changes in the late stage of AD while abnormal deposition of tau proteins is the real source of cognitive decline and memory loss in early AD patients. Therefore, the clearance of Tau proteins is probably an effective means to relieve the symptoms of AD.
In this study, we investigated the effects of PEG-ceramide nanomicelles on autophagic flux by determining LC3 and p62 levels by Western blotting and measuring RFP-GFP-LC3 fluorescence by confocal microscopic analysis, both standard protocols for evaluating autophagic flux.32 PEG-ceramide nanomicelles induced autophagic flux, which was reflected in the increased LC3-II/LC3-I ratios and decrease in p62 levels. Furthermore, analysis of the distribution of the RFP-GFP-LC3 fusion protein shows that the number of yellow and red dots significantly increased in N2a cells, indicating an increase in autophagic flux. Current studies have shown that autophagosomes can be seen in the pathologic anatomy of brain tissues of a considerable number of AD patients after death. These findings have drawn attention to the study of the pathological process of autophagy in AD.35 Autophagy promotes the degradation of “waste” by lysosomal proteases, and the digested material is reused to make the necessary components for cells.36,37 With aging, the levels of autophagy tend to decrease;38 therefore, autophagy defects may contribute to the progression of many neurodegenerative diseases.39–41
Current therapeutic strategies focus on inhibiting the aggregation of tau proteins.42,43 Methylene blue directly affects the formation of tau polymers in AD; however, due to its high hydrophilicity, its access to the brain is restricted.43,44 Some studies have used nano-carriers to activate autophagy to accelerate the degradation of Aβ and Huntingtin proteins for the treatment of AD.24,27 However, this is the first study to examine induction of autophagy by nanomicelles to accelerate the clearance of tau proteins for the treatment of AD. We have confirmed that PEG-ceramide nanomicelles could degrade tau proteins by inducing autophagy in N2a cells, but its exact mechanism and in vivo effects still need further study, mainly including: (1) The pathway of PEG-ceramide nanomicelles in regulating autophagy; (2) The permeability and toxicity of PEG-ceramide nanomicelles to the blood-brain barrier in vivo; (3) Demonstrates that PEG-ceramide nanomicelles induce autophagy to degrade tau protein in vivo. In conclusion, nanomedicine induced autophagy is expected to be a new strategy for the treatment of AD.
Conclusion
This study demonstrates that these novel PEG-ceramide nanomicelles could degrade overexpressed human tau proteins by activating autophagy in N2a cells; however, the mechanism still needs to be elucidated. Future studies will explore the efficiency of removal of tau proteins through autophagy induced by PEG-ceramide nanomicelles. These materials could become a potential nano-drug for the treatment of AD.
Abbreviations
AD, Alzheimer’s disease; Aβ, amyloid beta protein; PEG, polyethylene glycol; LC3, light chain 3 protein; NFT, neurofibrillary tangle; BBB, blood brain barrier; ATP, adenosine 5ʹ-triphosphate; EGFP, enhanced green fluorescent protein; GFP, green fluorescent protein; RFP, red fluorescent protein; IP, immunoprecipitation.
Funding
This work was supported by Science and Technology Support Projects in Biomedicine Field of Shanghai Science and Technology Commission, No.19441907500; Naval Medical University Military Medical Innovation, No.2017JS07; Science and Technology Action Innovation Program by Science and Technology Commission of Shanghai, No.17411950104.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Prince M, Wimo A, Guerchet M, et al. World alzheimer report 2015. The global impact of dementia. An analysis of prevalence, incidence, cost and trends. Alzheimers Dis Int. 2015.
2. Ramirez-Bermudez J. Alzheimer’s disease: critical notes on the history of a medical concept. Arch Med Res. 2012;43(8):595–599.
3. Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S. Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience. 2002;115(1):201–211.
4. Holmes C, Bocheetal D, Wikinson D, et al. Long-term effects of Aβ 42 immunisation in alzheimer’s disease: follow-up of a randomised, placebo-controlled Phase I trial. Lancet. 2008;372(9634):216–223.
5. Salloway S, Sperling R, Gilman S, et al. A Phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate alzheimer disease. Neurology. 2009;73(24):2061–2070.
6. Ma TJ, Gao J, Liu Y, et al. Nanomedicine strategies for sustained, controlled and targeted treatment of alzheimer’s disease. Mini Rev Med Chem. 2018;18(12):1035–1046.
7. Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33(1):95–130.
8. Mitchison T, Kirschner M. Cytoskeletal dynamics and nerve growth. Neuron. 1988;1(9):761–772.
9. Murray ME, Lowe VJ, Graffradford NR, et al. Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain. 2015;138(Pt 5):1370–1381.
10. Brier MR, Gordon B, Friedrichsen K, et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci Transl Med. 2016;8(338):338ra66.
11. Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010;7(8):656–664.
12. Li L, Zhang X, Le W. Autophagy dysfunction in alzheimer’s disease. Neurodegener Dis. 2010;7(4):265–271.
13. Li Q, Liu Y, Sun M. Autophagy and alzheimer’s disease. Cell Mol Neurobiol. 2017;37(3):377–388.
14. Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by alzheimer-related PS1 mutations. Cell. 2010;141(7):1146–1158.
15. Zhu XC, Yu JT, Jiang T, Tan L. Autophagy modulation for Alzheimer’s disease therapy. Mol Neurobiol. 2013;48(3):702–714.
16. Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983–997.
17. Ling D, Salvaterra PM. A central role for autophagy in Alzheimer-type neurodegeneration. Autophagy. 2009;5(5):738–740.
18. Zare-Shahabadi A, Masliah E, Johnson GV, Rezaei N. Autophagy in alzheimer’s disease. Rev Neurosci. 2015;26(4):385–395.
19. Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4(7):949–951.
20. Guenther GG, Peralta ER, Rosales KR, Wong SY, Siskind LJ, Edinger AL. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc Natl Acad Sci U S A. 2008;105(45):17402–17407.
21. Wang Y, Mandelkow E. Degradation of tau protein by autophagy and proteasomal pathways. Biochem Soc Trans. 2012;40(4):644–652.
22. Celia C, Cosco D, Paolino D, Fresta M. Nanoparticulate devices for brain drug delivery. Med Res Rev. 2011;31(5):716–756.
23. Mei L, Zhang X, Feng SS. Autophagy inhibition strategy for advanced nanomedicine. Nanomedicine (Lond). 2014;9(3):377–380.
24. Wei PF, Zhang L, Nethi SK, et al. Accelerating the clearance of mutant huntingtin protein aggregates through autophagy induction by europium hydroxide nanorods. Biomaterials. 2014;35(3):899–907.
25. Mittal S, Sharma PK, Tiwari R, et al. Impaired lysosomal activity mediated autophagic flux disruption by graphite carbon nanofibers induce apoptosis in human lung epithelial cells through oxidative stress and energetic impairment. Part Fibre Toxicol. 2017;14(1):15.
26. Arya BD, Mittal S, Joshi P, Pandey AK, Ramirez-Vick JE, Singh SP. Graphene oxide–chloroquine nanoconjugate induce necroptotic death in A549 cancer cells through autophagy modulation. Nanomedicine (Lond). 2018;13(18):2261–2282.
27. Luo Q, Lin YX, Yang PP, et al. A self-destructive nanosweeper that captures and clears amyloid β-peptides. Nat Commun. 2018;9(1):1802.
28. Pattingre S, Bauvy C, Levade T, Levine B, Codogno P. Ceramide-induced autophagy: to junk or to protect cells? Autophagy. 2009;5(4):558–560.
29. Su X, Song H, Niu F, et al. Co-delivery of doxorubicin and PEGylated C16-ceramide by nanoliposomes for enhanced therapy against multi-drug resistance. Nanomedicine (Lond). 2015;10(13):2033–2050.
30. Wang M, Xie F, Wen X, et al. Therapeutic PEG-ceramide nanomicelles synergize with salinomycin to target both liver cancer cells and cancer stem cells. Nanomedicine (Lond). 2017;12(9):1025–1042.
31. Huang J, Zhang H, Yu Y, et al. Biodegradable self-assembled nanoparticles of poly(D,L-lactide-co-glycolide)/hyaluronic acid block copolymers for target delivery of docetaxel to breast cancer. Biomaterials. 2014;35(1):550–566.
32. Yang K, Lu Y, Xie F, et al. Cationic liposomes induce cell necrosis through lysosomal dysfunction and late-stage autophagic flux inhibition. Nanomedicine (Lond). 2016;11(23):3117–3137.
33. Shang L, Nienhaus K, Nienhaus GU. Engineered nanoparticles interacting with cells: size matters. J Nanobiotechnology. 2014;12:5.
34. Elsana H, Olusanya TOB, Carr-Wilkinson J, Darby S, Faheem A, Elkordy AA. Evaluation of novel cationic gene based liposomes with cyclodextrin prepared by thin film hydration and microfluidic systems. Sci Rep. 2019;9(1):15120.
35. Lee YJ, Han SB, Nam SY, Oh KW, Hong JT. Inflammation and alzheimer’s disease. Arch Pharm Res. 2010;33(10):1539–1556.
36. Tung YT, Wang BJ, Hu MK, et al. Autophagy: a double-edged sword in Alzheimer’s disease. J Biosci. 2012;37(1):157–165.
37. Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441(2):523–540.
38. Ulamek-Koziol M, Furmaga-Jablonska W, Januszewski S, et al. Neuronal autophagy: self-eating or self-cannibalism in Alzheimer’s disease. Neurochem Res. 2013;38(9):1769–1773.
39. Orr ME, Oddo S. Autophagic/lysosomal dysfunction in alzheimer’s disease. Alzheimers Res Ther. 2013;5(5):53.
40. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–695.
41. Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–884.
42. Bulic B, Pickhardt M, Schmidt B, Mandelkow EM, Waldmann H, Mandelkow E. Development of tau aggregation inhibitors for alzheimer’s disease. Angew Chem Int Ed Engl. 2009;48(10):1740–1752.
43. Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93(20):11213–11218.
44. Harrington C, Rickard JE, Horsley D, et al. Methylthioninium chloride (MTC) acts as a Tau aggregation inhibitor (TAI) in a cellular model and reverses Tau pathology in transgenic mouse models of Alzheimer’s disease. Alzheimers Dement. 2008;4(4):T120–T121.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.