")
Back to Journals » Adolescent Health, Medicine and Therapeutics » Volume 5
Patient considerations in the management of focal seizures in children and adolescents
Received 3 January 2014
Accepted for publication 30 January 2014
Published 9 April 2014 Volume 2014:5 Pages 49—65
DOI https://doi.org/10.2147/AHMT.S44316
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Video abstract presented by Daniel Kenney
Views: 3692
Daniel Kenney, Elaine Wirrell
Department of Neurology, Mayo Clinic, Rochester, MN, USA
Abstract: Focal epilepsy accounts for approximately one-half to two-thirds of new-onset epilepsy in children. Etiologies are diverse, and range from benign epilepsy syndromes with normal neuroimaging and almost certain remission to focal malformations of cortical development or hippocampal sclerosis with intractable seizures persisting lifelong. Other important etiologies in children include pre-, peri-, or postnatal brain injury, low-grade neoplasms, vascular lesions, and neuroimmunological disorders. Cognitive, behavioral, and psychiatric comorbidities are commonly seen and must be addressed in addition to seizure control. Given the diverse nature of focal epilepsies in children and adolescents, investigations and treatments must be individualized. First-line therapy consists of prophylactic antiepileptic drugs; however, prognosis is poor after failure of two to three drugs for lack of efficacy. Refractory cases should be referred for an epilepsy surgery workup. Dietary treatments and neurostimulation may be considered in refractory cases who are not good candidates for surgery.
Keywords: epilepsy, child, adolescent, treatment, surgery, ketogenic diet, epidemiology
Classification and epidemiology
The term “focal” has replaced “partial” or “localization-related” to describe a seizure that originates in networks limited to one hemisphere, either from subcortical structures or cortex.1 Focal seizures are further described as “dyscognitive” if there is associated alteration of awareness (formerly termed “partial complex”), and as “evolving to bilateral convulsive activity” if they spread to both hemispheres. The International League Against Epilepsy (ILAE)’s glossary of descriptive terminology for ictal semiology provides standard terminology that should be used to further describe specific features of focal seizures.2 Certain features are characteristic of seizures arising from a specific location and can aid in localization and lateralization of the epileptogenic focus. For example, a focal dyscognitive seizure with an epigastric aura and dysphasia is suggestive of a dominant temporal lobe focus, whereas a focal seizure with hyperkinetic pedaling automatisms most likely originates in the frontal lobe.
Focal seizures are the commonest mode of onset in children, accounting for 52%–68% of new-onset epilepsy in population-based pediatric studies.3,4 The incidence rate of focal epilepsy is highest in the first year of life (63.5 per 100,000 persons per year) and then declines steadily through adolescence.3,4
While the terms “generalized” and “focal” epilepsy remain useful in many cases of pediatric epilepsy, there are clinical scenarios in which such dichotomization is not supported by clinical, electrophysiological, or radiological data. Difficulty in dichotomizing epilepsy into generalized versus focal is a particular problem in many early-onset epileptic encephalopathies, such as Dravet syndrome, Lennox–Gastaut syndrome, or West syndrome, where children can have both focal and generalized seizures, or where focal pathology clearly leads to a generalized seizure.
Electroclinical syndrome and etiology
The underlying cause of focal epilepsy is one of the most important predictors of its natural history in children. Certain structural etiologies, such as cortical dysplasia or hippocampal sclerosis, are associated with markedly higher rates of pharmacoresistance.5,6 Conversely, other specific types of focal epilepsy, such as benign epilepsy with centrotemporal spikes or Panayiotopoulos syndrome, are likely to be self-limited with remission in childhood or adolescence. Recognition of specific etiologies is essential in choosing appropriate therapies and counseling families.
The most recent document on organization of the epilepsies, by the Commission on Classification and Terminology of the ILAE, suggests organizing epilepsies into two nonhierarchical, complementary domains: 1) electroclinical syndrome; and 2) etiology.1
An electroclinical syndrome is a “complex of clinical features, signs and symptoms that together defines a distinct disorder” and carries implications for treatment and prognosis.1 Syndromes are defined by age at onset, seizure type(s), specific electroencephalography (EEG) patterns, underlying causes, and comorbidities. Among children with new-onset epilepsy, an electroclinical syndrome can be identified in a significant minority of cases.
Over the last two decades, there have been significant advances in genetics and neuroimaging, which have greatly facilitated our understanding of the etiology of epilepsy in many patients. The Commission on Classification and Terminology divides etiology into six main categories, including genetic, structural, metabolic, immune, infectious, and unknown. Certain etiologies are best described by using a combination of two or more of these categories, such as tuberous sclerosis, which would be defined as a genetic–structural etiology.
Focal electroclinical syndromes in children and adolescents
Common syndromes
Benign epilepsy with centrotemporal spikes
Benign epilepsy with centrotemporal spikes, also known as benign rolandic epilepsy, is the commonest type of focal epilepsy. Typically, it begins in the early school-age years with hemifacial sensorimotor seizures, with drooling and dysarthria, which may spread to the ipsilateral upper extremity. A brief postictal Todd’s paresis is common. Seizures typically occur shortly before wakening in the morning or shortly after falling asleep at night, and nocturnal seizures may evolve to bilateral convulsive activity. The EEG shows characteristic centrotemporal spikes, which are activated by drowsiness and sleep, and have a characteristic dipole, with frontal positivity and temporal negativity. Neurological and developmental exams are usually normal, although, rarely, this condition can evolve to electrical status epilepticus in sleep and acquired auditory agnosia, consistent with Landau–Kleffner syndrome. Neuroimaging, if performed, shows no causative lesion. Many children have infrequent, focal seizures and may not require prophylactic antiepileptic medication. Those with more frequent seizures, particularly if convulsive, may require treatment; however, remission is the rule, usually in later childhood.
Panayiotopoulos syndrome
Panayiotopoulos syndrome predominantly affects neurologically normal, preschool-aged children and presents with prominent ictal autonomic symptoms such as retching, vomiting, pallor, and/or urinary incontinence, but preserved consciousness, which are often not recognized as ictal. Progression to gaze deviation, impairment of consciousness, and hemiconvulsive activity is common. Seizures are often prolonged with autonomic status epilepticus and frequently occur out of sleep. The EEG shows focal sharp waves, which may be multifocal, but commonly affect posterior head regions. Imaging is normal and remission typically occurs within 1–2 years of onset.
Less common syndromes
Late-onset benign occipital epilepsy (Gastaut syndrome)
Late-onset benign occipital epilepsy presents in later childhood or adolescence with brief, diurnal seizures, consisting of elementary visual hallucinations and ictal amaurosis, which may be followed by eye deviation and convulsive activity. Postictal headache, which is indistinguishable from migraine, is common. The EEG typically shows occipital spikes, which may show fixation-off sensitivity. The diagnosis of late-onset benign occipital epilepsy should be made cautiously and only after careful neuroimaging, as similar semiology can be seen with lesional occipital lobe epilepsy. Antiepileptic drug (AED) treatment is usually required, although most patients will remit by early adulthood.
Autosomal dominant frontal lobe epilepsy
This syndrome, due to mutations in the nicotinic acetylcholine receptor gene, presents in later childhood or early adolescence with brief, frequent, nocturnal events of dystonic posturing, hypermotor activity, or complex behaviors. Although autosomal dominant with 80% penetrance, affected family members may have been misdiagnosed as having parasomnias or behavior problems in sleep. Interictal EEG may be normal or show frontal discharges with unremarkable imaging. Genetic testing is commercially available. Response to antiepileptic medication is usually favorable; however, the long-term remission rate is not well studied.
Familial temporal lobe epilepsies
The familial temporal lobe epilepsies are divided into mesial and lateral subgroups and are relatively rare. Both types typically begin in adolescence or early adulthood.
The mesial subtype presents with seizures characterized by prominent déjà vu, fear, nausea, and altered awareness. Rarely, progression to bilateral convulsive activity is seen. Up to 10% of patients will have a history of prior febrile seizures. The interictal EEG is nonspecific, and may be normal or show temporal slowing or focal temporal discharges. While imaging is often normal, mesial temporal sclerosis may be seen. Antiepileptic medication usually provides effective seizure control; however, surgery can be considered in more refractory cases. The genetics of familial mesial temporal lobe epilepsy is not well understood, but complex inheritance is likely.
The lateral subgroup presents with focal seizures that are often triggered by external noises. Semiology consists of prominent elementary auditory auras, early ictal aphasia, and, at times, evolution to bilateral convulsive activity. Although of autosomal dominant inheritance, penetrance can be low. In approximately one-half of cases, mutations in the leucine-rich glioma-inactivated gene 1 (LGI1) on chromosome 10 can be documented. EEG shows nonspecific temporal discharges; magnetic resonance imaging (MRI) is normal; and seizures are generally pharmacoresponsive with favorable outcome.
Important etiologies associated with focal epilepsy in children and adolescents
Structural
Structural etiologies are commonly associated with medically intractable focal epilepsy in childhood. Various types of structural changes may be associated with epilepsy, some of which are more commonly associated with intractability.
Malformations of cortical development
Malformations of cortical development are one of the commonest pathologies reported in surgical series of pediatric epilepsy. They may be generalized and diffuse (ie, agyria-pachygyria), hemispheric (hemimegalencephaly), multifocal (periventricular nodular heterotopia), or focal (pachygyria, schizencephaly, or focal cortical dysplasia). The extent of involvement often predicts seizure type, severity, and coexisting neurologic disability. Diffuse lesions typically result in severe epileptic encephalopathy and profound developmental delay that begins very early in life, whereas more focal lesions may present with medically intractable epilepsy in otherwise well children and adults.
Seizure semiology depends on the location of the lesion; however, seizure frequency is usually high. MRI advances have been instrumental in improved detection of focal cortical dysplasias, with reported findings of increased cortical thickness, blurring of the cortical-white matter junction, increased T2 signal, transmantle sign, or localized brain atrophy. However, detection in young children prior to age 2–3 years is still challenging due to immature myelination. The focal cortical dysplasias are classified into three major types, with types I and II referring to isolated focal cortical dysplasia, and type III referring to dysplasia associated with other epileptogenic lesions, such as hippocampal sclerosis or tumors.7
Medical intractability occurs in up to 83% of children with such malformations, and such children should be referred expediently for surgical assessment if seizure control is not achieved with medical therapy.
Hippocampal sclerosis
Hippocampal sclerosis is also a common pathological finding in children undergoing temporal lobectomy for intractable focal epilepsy, and has generally been considered an acquired finding, secondary to postnatal injury such as prolonged febrile seizures. However, recent work has shown there is likely also a genetic predisposition, with higher rates of hippocampal developmental changes in children with febrile status epilepticus and smaller asymmetric hippocampi in relatives of patients with hippocampal sclerosis.8,9 Hippocampal sclerosis and dual pathology (hippocampal sclerosis and other lesion) were associated with only 11% and 3% seizure freedom at last follow-up in medically treated cases, respectively, suggesting that early referral for surgical evaluation should be considered.6,10
Low-grade neoplasms
Glioneuronal tumors, such as gangliogliomas and dysembryoplastic neuroepithelial tumors, account for up to 30% of intractable focal epilepsy.11 These tumors are usually low grade and carry a benign oncological prognosis. Both have a predilection for the temporal lobe and may be surrounded by a region of cortical dysplasia. Resection is often required to confirm benign pathology, and seizure outcome after surgery is usually favorable.
Hypothalamic hamartomas typically present with focal seizures, which may have a gelastic semiology. The ictal and interictal EEG may be normal or may show epileptiform discharge, and, on scalp recording, seizures may appear to originate from the temporal lobes. Many children also have a history of precocious puberty and significant behavioral difficulties. Seizures are typically medically intractable, and progressive cognitive and behavioral regression is seen. These cases should be referred expediently for surgery, with a goal to eradicate or completely disconnect the hamartoma.
Vascular lesions
Arteriovenous (AV) malformations are relatively uncommon causes of focal epilepsy, but are important to recognize as they carry a risk for hemorrhage. In the absence of obvious hemorrhage, unruptured AV malformations may result in seizures due to vascular steal and resultant gliosis of the intervening brain, development of aneurysms, and hemosiderin deposition from subclinical bleeding. Because of the risk of hemorrhage, which is approximately 3% per year, the goal of therapy should be obliteration of the malformation, if this can be safely achieved.
Cavernous malformations may present with various clinical symptoms; however, one-half of patients harboring these lesions present with seizures. Most often, cavernomas are solitary, but multiple lesions may exist, and, if present, should suggest a familial inheritance. The risk of hemorrhage is lower than with AV malformations, and the optimal management of cavernomas presenting with new-onset seizures is debated. Antiepileptic medications may be used first-line, but surgery should be considered in intractable cases.
Encephalomalacia due to perinatal or other prior brain injury
Neonatal seizures are associated with an approximately 25% risk of postneonatal epilepsy, although the exact risk depends on underlying cause, with highest rates being seen with cerebral dysgenesis. Among children with cerebral palsy, those with quadriplegia or hemiplegia have higher rates of epilepsy (19%–36%) than those with diplegia (14%), dyskinetic/athetoid (8%–13%), or ataxic (13%–16%) groups.12
Seizures may also result from brain injury due to prior head injury. Risk factors for posttraumatic epilepsy include prolonged loss of consciousness, depressed skull fracture, multiple or hemorrhagic brain contusions, temporal or frontal contusion, and wound infection.
Epilepsy following central nervous system infections may be very problematic. Seizures are often refractory and multifocal in onset, which limits the ability to intervene surgically.
Neurocutaneous disorders
Tuberous sclerosis complex frequently presents with intractable epilepsy, which may begin with infantile spasms and progress to intractable generalized or focal seizures. Despite the presence of multiple tubers, seizures often arise from only one, or a small number of, tubers and, as such, resective surgery may be an option for such patients.
Seizures may result from neurofibromatosis; however, the literature is lacking data on this subject. Sturge–Weber syndrome typically presents with a port-wine stain involving the trigeminal region, glaucoma, and leptomeningeal angiomatosis, which is frequently unilateral and predominant posteriorly. While many patients may respond to antiepileptic medications, those with intractable epilepsy should be referred early for surgical assessment.
Immunological
Immunological etiologies for epilepsy are increasingly recognized in both adults and children; however, the exact proportion of children with epilepsy who harbor neuronal antibodies is not known. The following features, particularly if several are present, are suggestive of an underlying immunological cause:
- acute or subacute onset of epilepsy, with progressive course;
- multifocal neurological symptoms and signs, including intractable epilepsy, altered mental status or cognitive dysfunction, autonomic dysfunction, sleep disturbances, and movement disorders;
- investigations: slowing of the EEG background, fluid attenuated inversion recovery, or T2 signal changes on MRI or inflammatory changes in cerebrospinal fluid (CSF) with negative cultures; and
- personal or family history of autoimmunity.
Clinical subtypes
Autoimmune encephalopathies
N-methyl-D-aspartate (NMDA) receptor encephalitis: NMDA receptor encephalitis is due to antibodies directed against the NR1 subunit of the NMDA receptor and clinical symptoms are due to reduction of the NMDA receptor density on the cell surface, due to receptor internalization. Patients typically present with a prodrome of fever, headache, nausea, and/or vomiting, which is followed within a month by the typical psychiatric and behavioral symptoms, sleep disturbances, seizures, dyskinesias and choreoathetoid movements, and autonomic dysfunction. MRI is typically normal but may show T2 hyperintensities, and the EEG typically shows slowing or an extreme delta brush pattern. CSF is abnormal in 94% of cases,13 showing a lymphocytic pleocytosis, elevated protein, and/or positive oligoclonal bands. The NMDA receptor antibody is best detected in CSF, as serum may be falsely negative. While ovarian teratomas are present in nearly one-half of all adult women with this condition, underlying malignancies are uncommon in children. Treatment consists of tumor resection, if present, and initiation of immunotherapy (intravenous [IV] gamma globulin, IV steroids, or IV plasma exchange). Rituximab may be used as adjunctive therapy in more severe cases or if first-line therapies are suboptimal.
Voltage-gated potassium channel (VGKC) complex: The VGKC complex is critical for regulation of neuronal excitability, axonal conduction, and neurotransmitter release. This entity comprises a number of antibodies that are directed against the VGKC, including leucine-rich glioma-inactivated protein 1 (LGI1) and contactin-associated protein 2 (CASPR2). These antibodies often result in limbic encephalitis, with intractable epilepsy, hyponatremia, dysautonomia, sleep disturbances, and psychiatric symptoms. Leucine-rich glioma-inactivated protein 1 (LGI1) antibodies are characteristically associated with a unique seizure semiology – faciobrachial dystonic seizures, which are brief (<5–10-second) episodes of ipsilateral facial grimace and arm posturing. MRI is abnormal in nearly one-half of cases, showing mesial temporal T2 signal abnormality. The EEG typically shows background slowing, at times with temporal epileptiform discharges, and CSF is usually normal. VGKC antibodies are best detected in serum. Etiology is usually unknown in children, although a search for malignancy is recommended. Therapy is with immunosuppressive agents.
Rasmussen encephalitis
Rasmussen encephalitis is typically a unihemispheric, presumed immunological disorder that begins in a previously healthy child or teenager with focal seizures, which progressively worsen over time, resulting in epilepsia partialis continua in approximately one-quarter of cases. Progressive focal neurological deficits such as hemiparesis, hemianopsia, and cognitive decline correlate with atrophy of the affected hemisphere. Pathology is suggestive of cytotoxic T-cell injury; however, no consistent antibodies have been found. While various immunotherapies may slow progression of this disorder, functional hemispherectomy is usually ultimately required.
Febrile infection-related epilepsy syndrome
This devastating disorder begins in school-aged children following a mild viral prodrome. Seizures rapidly evolve to multifocal, refractory status epilepticus and mortality and morbidity are high. The pathogenesis is unknown – extensive antibody testing on blood and CSF has been unrewarding, and there has been no consistent response to various immunotherapies.
Cognitive, behavioral, and psychiatric comorbidities
Although there is a great deal of interpatient variability, children with focal epilepsy may show dysfunction across multiple cognitive domains, including executive function, language use, working memory, and numeracy. Risk factors for cognitive impairment include the presence of anatomic abnormalities, multifocal (particularly bilateral) ictal foci, specific location of seizure focus, medical intractability, and earlier age at onset. Behavioral symptoms are also quite common among children with focal epilepsies. These can range from hyperactivity all the way to psychosis.
Impact of localization of the epileptic focus
The location of the epileptogenic focus may predict the types of cognitive and behavioral comorbidities. There is overlap between the deficits seen in patients with nonepileptogenic lesions and those with focal epilepsy in a given lobe. For example, a patient with focal epilepsy localized to the frontal lobes may have a similar dysexecutive picture to another patient with a destructive frontal lobe lesion. However, it is common for the spectrum of disability to be wider than might have been expected strictly on the location of seizure focus. For example, while children with temporal lobe epilepsy are at greater risk of memory impairment, executive function (more typically a frontal lobe function) is also commonly impaired. This may happen due to disruption of networks important for cognitive function, to seizure propagation through areas relatively remote to the ictal focus, or to other factors.
Frontal lobe epilepsy
The frontal lobe is a large and complex brain region that subserves many functions, and deficits from a lesion in this area may be protean. Most epileptogenic lesions of the frontal lobe are structural, and static deficits secondary to these lesions can be difficult to tease apart from those that are secondary to abnormal electrical activity. Patients with frontal lobe epilepsy have frequent impairments in attention, planning, and impulse control, difficulties in proverb recognition, and problematic social cognition, with faux pas, humor, or facial expressions not being readily recognized.14–16 Children with frontal lobe epilepsy may also show problems in motor planning, coordination, and dexterity, with possible linguistic problems in those with left-sided lesions.17,18
Psychiatric effects of frontal lobe seizure foci are also well documented, although these are less prominent than with temporal lobe epilepsy. Mood disorders and even ictal psychosis may be found.
Temporal lobe epilepsy
The cognitive and behavioral effects of temporal lobe seizure foci range from subtle problems with memory and psychomotor speed to severe impairments in intelligence and psychiatric disorders. Memory can be severely impaired, with episodic memory more impaired than semantic. Patients with left-sided foci typically have greater verbal memory problems, whereas those with right-sided foci manifest visuospatial deficits. Learning, naming, and word-finding problems are also well documented. Intelligence is frequently but variably impaired. Attentional problems can also be present and severe.
Psychiatric symptoms are common, often manifesting as anxiety and depression, although other psychiatric disorders (from psychosis to personality disorders) may be seen.19–21
Occipital and parietal lobe epilepsy
Relatively little attention has been given to neuropsychological deficits in focal epilepsies involving the parietal or occipital cortices. Because of the varied connections of the parietal or occipital cortices with other brain regions, seizures may propagate to motor areas, sensory association cortices, or temporolimbic structures, with correspondingly different manifestations and, one might predict, different interictal deficits. Focal lesions in these areas correlate with deficits of sensory processing, particularly visual, as well as problems with numeracy and language function.22 Involvement of the occipital cortex can result in visual field deficits corresponding to the size and location of the lesion.
Impact of electroclinical syndrome
Focal electroclinical syndromes
Electroclinical syndrome is an important predictor of the risks and types of cognitive and behavioral comorbidities (Table 1).
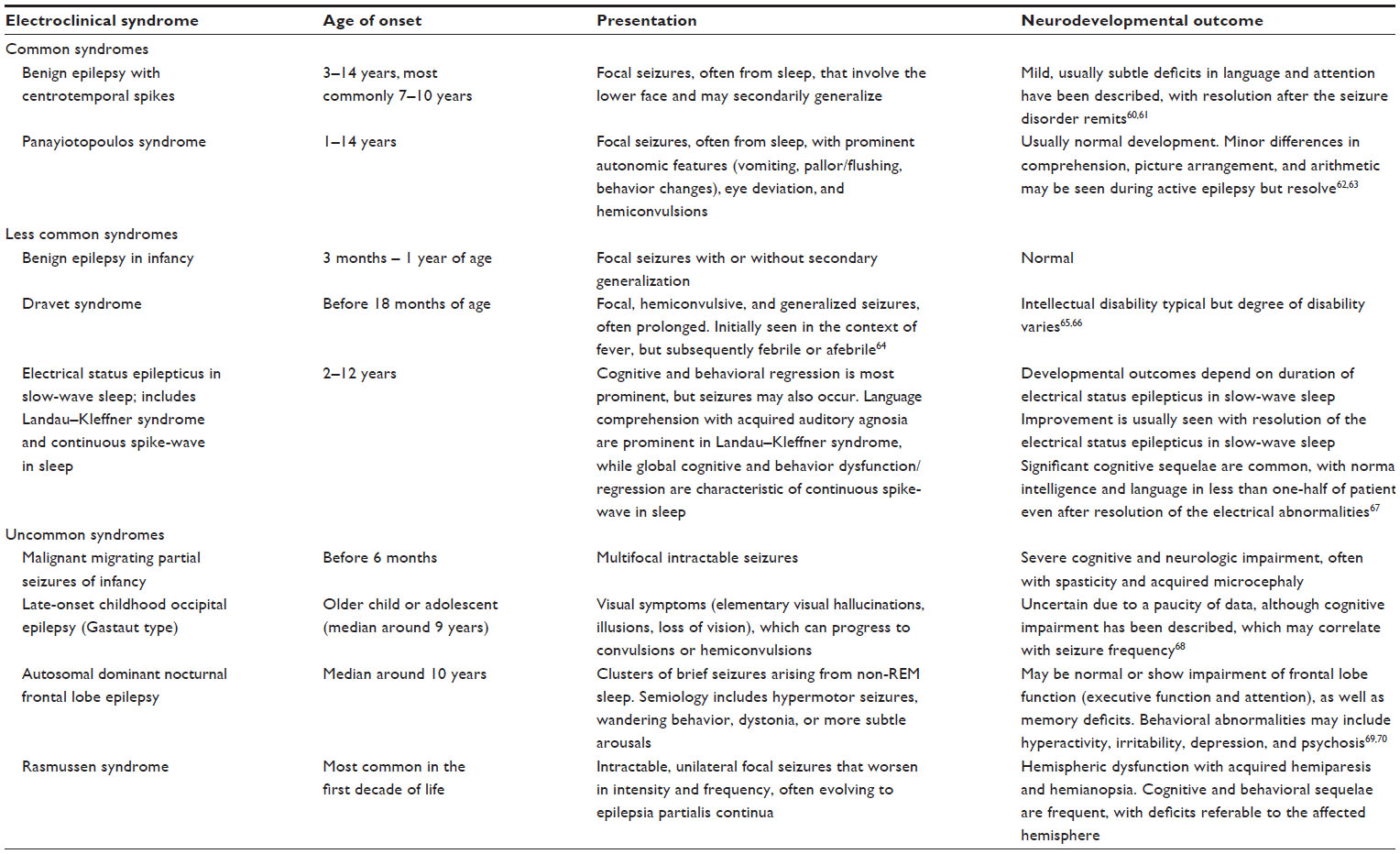 | Table 1 Cognitive and behavioral comorbidities in selected focal electroclinical syndromes |
Symptomatic focal epilepsies
The cognitive and behavioral outcomes of children with focal epilepsies due to known metabolic, genetic, immunologic, or structural causes are often very dependent on the specific etiology. For example, children with tuberous sclerosis are at a high risk for pervasive developmental disorder, while children with multifocal seizures secondary to Angelman syndrome are at great risk of profound developmental delay. Treatment of the underlying cause of the disorder is critical for managing the neurodevelopmental sequelae of the disease.
Suggested investigations for focal epilepsy
Investigations for new-onset focal epilepsy
EEG
In children with focal epilepsy, the first routine EEG will show epileptiform discharge in approximately 30%–50% of cases, but, not uncommonly, can be entirely normal. Furthermore, abnormal EEGs can be seen in well children without epilepsy, so careful clinical correlation is needed. The patient ideally should be sleep deprived, as yield is higher if sleep is recorded. Furthermore, the yield may be greater if the EEG is done in the first 24 hours after a seizure. Findings that may be helpful for localization and identification of pathology include asymmetries in amplitude, focal slowing, and epileptiform discharges.
MRI
With the exception of straightforward cases of benign epilepsy with centrotemporal spikes, an MRI using seizure protocol should be strongly considered in all children with new-onset focal seizures. In our population-based study of children with new-onset epilepsy,5 of whom 247 underwent MRI, this examination was abnormal in 76 patients (31%), showing generalized or multifocal changes in 40 and focal or hemispheric changes in 36.
Other investigations that may be considered in medically-intractable focal epilepsy
Long-term video-EEG monitoring
Long-term video-EEG monitoring is strongly recommended for children and teenagers with focal epilepsy with uncontrolled seizures despite trials of two or more antiepileptic medications at adequate therapeutic doses. The goals of such monitoring are to confirm the diagnosis of focal epilepsy (and, if events are non-epileptic, to determine the type of event) and ascertain whether seizures may arise from a single epileptogenic focus that may be amenable to surgical resection. Long-term video-EEG monitoring is most informative if events are occurring with sufficient frequency (generally weekly or more) or if they can be triggered (ie, by medication reduction).
Functional neuroimaging
Functional neuroimaging is most useful in medically intractable focal epilepsy with either nonlesional or multifocal MRI abnormalities, or if the EEG or semiology is discordant with the MRI focus, with the goal of localizing the epileptogenic zone. Techniques include subtraction ictal single-photon emission computed tomography (SPECT), SPECT coregistered to MRI, or positron emission tomography (PET).
Functional MRI is a noninvasive way to map out motor and language function and ascertain the likelihood that the epileptogenic zone contains eloquent cortex, which may result in deficits if it is resected. Additionally, mapping of eloquent cortex is often done at the time of invasive EEG monitoring.
Magnetoencephalography
Magnetoencephalography measures the associated magnetic fields of the electrical current, and thus provides complementary information to EEG. It is better for tangential currents that arise from neurons situated along the lateral bank of a fissure.
Treatment of focal epilepsy
Pharmacologic treatment with AEDs, dietary therapies, and surgical treatment are all recognized modalities of therapy for focal seizure disorders. The efficacy of pharmacologic therapy is often poorer in focal structural epilepsies, and early consideration of surgery is often warranted.
Pharmacologic treatment
Initiation of prophylactic AEDs is the usual first step in the treatment of focal epilepsy. Given the large variety of medications with efficacy against focal seizures, AED properties as well as patient-specific and epilepsy-specific factors must be considered in determining the best option.
Choosing an AED
Important questions to consider when initiating an anticonvulsant in a patient include:
- Will the common adverse effects likely to be experienced by the patient limit the drug’s tolerance?
- How is the drug administered? Young children, for example, may be limited in their ability to swallow tablets.
- How often must the drug be administered? More frequent dosing schedules often result in decreased compliance.
- What sort of monitoring is required? Generally speaking, if two therapeutic options seem equally good in all other respects, it is better to go with the option that requires less monitoring, both from the perspective of patient satisfaction (fewer blood draws) and resource utilization.
Patient-specific issues
Serious idiosyncratic adverse effects: Severe adverse effects are often idiosyncratic and not usually dose dependent.23 While such adverse effects are not entirely preventable, care is needed to screen patients for factors that may place them at higher risk for these reactions. Certain patient populations are more vulnerable to serious adverse effects of specific AEDs by virtue of their clinical presentation (higher risk of hepatotoxicity with valproate in developmentally delayed children under age 2 years or those with signs of possible mitochondrial disease) or ethnic background (high risk of Stevens–Johnson syndrome to carbamazepine in Han Chinese in association with the HLA-B*1502 allele).24,25 Further advances in pharmacogenetics will likely aid in identification of genetic variants associated with serious adverse drug effects.
“Minor” adverse effects: “Minor” side effects are not life-threatening and frequently demonstrate dose dependence.23 Common dose-dependent, adverse effects associated with many classes of AEDs include sedation, imbalance, nausea, and anorexia. Often, minor reduction in the dose or slowing of the up-titration alleviates symptoms and discontinuation of the AED is not needed. However, other adverse effects can be more problematic. For example, valproic acid can be associated with significant weight gain, so the habitus of the patient prior to therapy must be considered.26 The possibility of hirsutism with phenytoin may limit its use in young women, who may find this adverse effect particularly distasteful.26,27 Levetiracetam and phenobarbital are associated with increased behavioral disturbances.28 Topiramate or zonisamide may lead to academic difficulties due to slowing of verbal processing and fluency, which may be particularly problematic in academically inclined, highly achieving patients.29,30 In rare patients with histories of substance abuse or trafficking, the use of benzodiazepines must be approached with caution. The patient’s entire picture, including social factors, must be considered in the use of AED therapies.
Other medical conditions or drugs: Drug–drug interactions among AEDs and between AEDs and other drugs can be problematic. Prominent enzyme inducers (such as carbamazepine) can lower blood levels of other drugs, necessitating a dose increase or a change in dosing schedule. In other instances, toxicity can occur, either due to the inhibition of an enzyme that metabolizes a drug or active metabolite, induction of an enzyme resulting in higher levels of a toxic metabolite, or because of altered protein-binding kinetics. Several commercial online tools (eg, micromedex.com) are available to check drug–drug interactions and should be consulted when a new medication is being added to a patient on polytherapy.
Gastrointestinal disease may impact on drug absorption, and hepatic and/or renal impairment on the metabolism and excretion of medication. Many medical conditions or use of co-medications can impact protein-binding of drugs, which must be considered when using AEDs with prominent protein binding, such as phenytoin or valproate.
Birth control and pregnancy: Given the risk of contraceptive failure and the concerns for potential teratogenicity with many AEDs, it is essential to discuss contraception in adolescent women with epilepsy. It is reasonable to recommend additional contraceptive measures beyond the oral contraceptive pill, including the use of barrier contraception, progestin implants, or an intrauterine device for women on enzyme inducing agents, as these are known to reduce the efficacy of oral contraceptives.31 Long-acting reversible contraception, such as the progestin implant, is a very reasonable option, and the concentration of progestin is high enough that efficacy is maintained even with enzyme inducing drugs. In addition, lamotrigine levels can be lowered by the use of combination (estrogen and progestin) hormonal birth control, so levels need to be monitored and the dose must often be increased. The teratogenicity of certain AEDs (in particular, valproic acid and phenytoin) must also be reviewed before starting therapy for any female patient with the potential for childbearing.
Psychiatric comorbidities and anticonvulsant therapy: Special consideration must be given to the use of AEDs in patients with psychiatric comorbidities. Some AEDs (eg, levetiracetam, benzodiazepines) have a greater association with symptoms of depression, inattention, oppositional behavior, and other mood or behavioral symptoms. On the other hand, other AEDs (eg, valproate, carbamazepine) are sometimes used as mood stabilizers, and bipolar patients in particular may show some improvement on these drugs. Benzodiazepines have a prominent anxiolytic effect and may be useful in patients with anxiety and seizures. On the whole, it is often very helpful to coordinate care with a patient’s psychiatry team if possible, to avoid drug interactions and to help monitor for adverse mood-related effects.
Epilepsy-specific issues: From the perspective of efficacy, superiority of any specific drug for the treatment of focal seizures in children, in general, has not been established. However, specific electroclinical syndromes require specific considerations. For example, in Dravet syndrome, the prophylactic use of sodium channel blockers (carbamazepine, oxcarbazepine, phenytoin, and lamotrigine) is contraindicated as they result in an exacerbation of seizures. On the other hand, valproate, stiripentol, clobazam, or topiramate can be particularly effective in this syndrome. Vigabatrin often has marked efficacy in focal epilepsy or infantile spasms associated with tuberous sclerosis complex.
Optimizing anticonvulsant therapy
Starting a drug
Table 2 lists medications that are used to treat focal epilepsy. In order to minimize adverse effects, most AEDs are started at low doses and incrementally increased over a period of time. The initial target dose of an AED is typically at the lower end of the range, and further up-titration is based on clinical response. Some drugs (particularly those with more favorable side effect profiles) can be increased fairly rapidly, while others must be introduced much more slowly. In particular, lamotrigine is associated with Stevens–Johnson syndrome, and this risk is greater with rapid dose escalation. Specific guidelines for up-titration of lamotrigine must be followed to mitigate this risk. Starting slowly with a medication does not, however, guarantee a lower risk of serious idiosyncratic drug reactions.
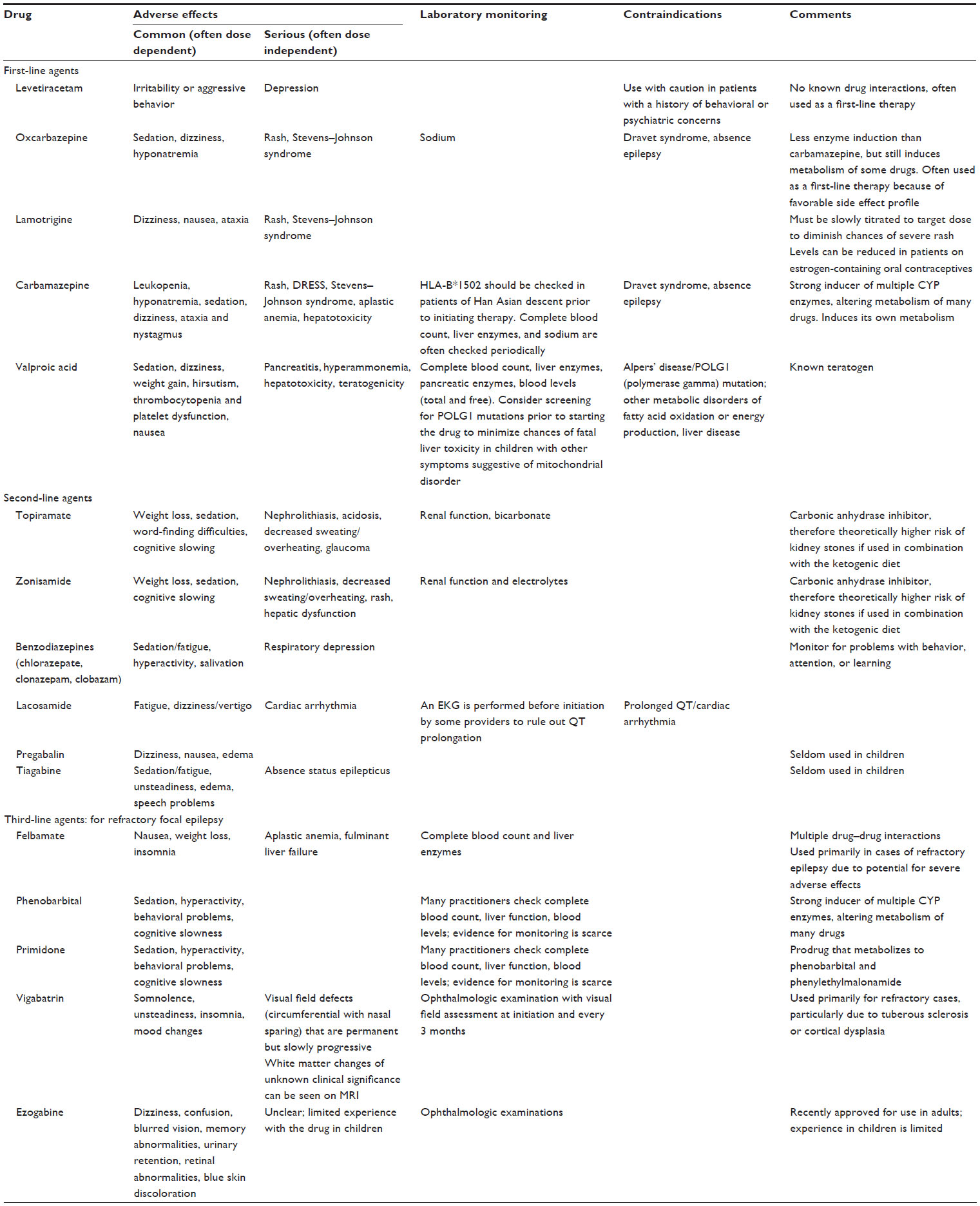 | Table 2 List of drugs that are useful in controlling focal seizures, with partial listing of adverse effects, monitoring parameters, and contraindications |
In cases of status epilepticus, a maintenance AED is commonly started after the seizure is stopped with IV medication, particularly if the underlying etiology suggests a high risk of seizure recurrence. In patients presenting with clusters of seizures, many clinicians will administer an IV load of AED to more rapidly stop seizures, followed by commencement of a maintenance AED, although no clinical trial has evaluated this practice.
Treatment failure and starting a new medication
After the failure of the first appropriately chosen, therapeutically dosed AED, the odds of success with subsequent AEDs are reduced. In adults, approximately one-half of patients achieve seizure control with their first AED, but only 13% with the second monotherapy trial.32,33 In children, around 60% achieve seizure freedom with their first anticonvulsant, with around 40% of the remainder experiencing seizure freedom with the second drug. The odds of further success with subsequent drug attempts are far lower.34,35
Typically, with ongoing seizures, AED dose is increased until either adverse effects become limiting or the maximal dose is reached. Trough serum levels can be useful in the evaluation of treatment failure. If serum trough levels are low despite a reasonable prescribed dose, patient compliance must be addressed. In the event of low levels with documented compliance, increases in either medication dose and/or dosing frequency may be needed. Younger children often metabolize drugs more quickly and may require more frequent doses. In the event of ongoing seizures despite maximal therapeutic trough drug levels or intolerable adverse effects, a second AED, ideally with a different mechanism of action to the first, is typically started. In most cases, the second AED is brought to therapeutic dose prior to weaning off the first medication, to reduce the risk of worsening breakthrough seizures. If, however, a drug needs to be stopped suddenly due to a severe adverse reaction, there may be a period of less anticonvulsant coverage while the new drug is being started. In situations in which serious breakthrough seizures may occur, consideration can be given to bridging with a long-acting benzodiazepine until the new drug has reached a therapeutic level.
Monotherapy is typically preferable to polytherapy whenever possible. Polytherapy increases the chance of drug interactions and adverse effects and makes patient adherence to a regimen more difficult. If polytherapy is used, drug interactions and mechanisms must be considered to minimize adverse effects.36 Some adverse effects, such as sedation and cognitive slowing, may be additive across multiple medications. It makes sense to use drugs that have different mechanisms of action. For example, if a patient is already taking a drug with sodium channel-blocking properties, addition of a drug with a GABAergic mechanism would be reasonable. Among AEDs, lamotrigine and valproate are thought to have a synergistic (not merely additive) anticonvulsant effect. While this can be a very effective combination for seizure control, their complex pharmacokinetics must be considered, as valproate increases serum levels of lamotrigine, with a concomitant increase in the risk of adverse effects (including the serious Stevens–Johnson syndrome).
It is not uncommon, particularly in those with refractory epilepsy, to experience an excellent but transient response to a new AED (the “honeymoon period”). The underlying physiologic basis for this is not understood, but it can result in the appearance of a drug working well for a variable period of time, followed by an apparent loss of efficacy with no other reason uncovered.
Monitoring drug levels
Assessment of the efficacy and tolerability of an AED is best done clinically, based on reported seizure control and adverse effects. In a patient who is seizure free with no AED adverse effects, there is little indication to monitor drug levels. However, AED levels can be very useful to distinguish whether apparent treatment failure is due to low drug levels or inefficacy. Levels may also help determine whether a new symptom is due to drug toxicity. For drugs with a narrow therapeutic window, like phenytoin, many clinicians will routinely monitor levels. However, there is little evidence to guide practice in this regard.
If drug levels are to be taken, it is most useful to measure the trough concentration (30–60 minutes prior to taking the next dose), as this is the time on which the normative range of AED levels are based. The utility of peak or random drug levels is very limited, and changing doses based on these results is likely to result in poorer seizure control. For drugs with substantial protein binding (like valproic acid or phenytoin), it can be useful to measure both the protein-bound and the physiologically active free drug concentrations, particularly in those on multiple other medications or with underlying medical conditions that can impact serum protein levels.
Monitoring for adverse effects
When starting a new medication, it is always important to counsel family and other caregivers about both the common, dose-dependent adverse effects and those that are rare, idiosyncratic, and life-threatening. In addition, for certain medications, some screening may be performed before initiating treatment (for example, checking for elevated transaminases and/or POLG1 [polymerase gamma] mutations in children with concerns in their medical history before starting valproic acid). There is not good evidence for routine laboratory monitoring of children for adverse effects of anticonvulsants. In addition to causing problems with patient tolerance of a medication regimen, routine monitoring can lead to a sense of false security if labwork normal (since the time course of most serious adverse effects is usually much too acute). Also, incidentally discovered laboratory abnormalities (eg, transient leukopenia in the context of a viral infection) could lead to discontinuation of an effective drug. While parents and caregivers must be vigilant of the symptoms of adverse medication effects, the need for laboratory monitoring in a patient must be approached based on that specific patient’s clinical context.37
Surgery
In appropriately selected children and teenagers with medically intractable seizures, epilepsy surgery can offer the opportunity for cure or substantial palliation. It remains underused, with patients often going years before referral to a center with expertise in epilepsy surgery.38 Far from being a last resort, early consideration and screening for surgery in medically refractory patients with focal epilepsy is the standard of care.39
Drug resistance (ie, refractoriness) is defined by the ILAE as “failure of adequate trials of two tolerated, appropriately chosen and used antiepileptic drug schedules (whether as monotherapies or in combination) to achieve sustained seizure freedom”.30 In addition to this, some practitioners will attempt one trial of an appropriate combination therapy. After this, surgical evaluation should be strongly considered.
Appropriate patients for consideration will be those who suffer disabling seizures of focal onset. Generally, patients who have seizures arising from a single focus, in a non-eloquent region of the brain, are the best surgical candidates. Long-term video-EEG monitoring is usually obtained both to confirm the diagnosis of focal epilepsy and ascertain whether seizures may arise from a single epileptogenic focus that may be amenable to surgical resection. Various imaging modalities are available to localize the seizure focus; however, a high-quality MRI scan, using an epilepsy-specific protocol, is the usual first step. Further functional imaging studies, such as SPECT or PET, as well as magnetoencephalography, are often considered in MRI-negative cases, or where there is lack of concordance between ictal EEG, semiology, and MRI. Functional MRI may also assist in defining the location of eloquent cortex.
Curative surgery
If the ictal onset zone is clear and does not involve potentially eloquent cortex, single-step resective surgery may be performed. Single-step resective surgery most commonly involves temporal lobectomy in cases of documented temporal lobe pathology, or lesionectomy in cases in which the epileptogenic lesion is completely apparent (ie, clear MRI findings and EEG monitoring indicating all epileptogenic activity and seizures coming only from the area of the lesion) and is remote from eloquent cortex. However, intracranial monitoring is often required to more clearly define the extent of the epileptogenic zone and demarcate it from eloquent cortex. Prior to intracranial monitoring, a clear hypothesis on the location of the likely epileptogenic zone should exist, and a plan for limited placement of electrodes in this suspicious region be devised. In this situation, subdural or depth electrodes are placed in the suspicious brain region, seizures are captured, and the epileptogenic zone is defined. Following that, cortical mapping can be performed by stimulation of the intracranial electrodes to determine if the epileptogenic zone contains eloquent cortex. If all of the above indicates a focal epileptogenic lesion that can be safely resected without endangering eloquent cortex or critical neurovascular structures, then resection (or, in some cases, disconnection) can proceed.
Temporal lobe resection is an effective therapy in well-selected patients.40 Mesial temporal sclerosis is less commonly found in children than adults, and, if present, is more commonly associated with other pathologies such as cortical dysplasia or low-grade tumors. The procedure is generally well tolerated, although language dysfunction with dominant temporal resection and visual field cuts (most commonly a small loss of the upper temporal field contralateral to the side of resection [“pie-in-the-sky” defect]) can occur. Additionally, the usual complications of neurosurgery (hydrocephalus, bleeding, stroke, or infection) are known possible complications.
Extratemporal resection is more common than temporal resection in children, with frontal lobe foci being the most common location. Cortical dysplasia is the most common etiology, although neurocutaneous disorders, prior brain injury, low-grade tumors, and vascular abnormalities are also frequent causes. Given that the extent of cortical dysplasia can be difficult to visualize on MRI, and that extratemporal foci more commonly abut or involve eloquent cortex, children undergoing extratemporal resection are more likely to require invasive monitoring and cortical mapping than those with temporal resection. The chances of seizure freedom after surgery seem greater with wider resection, unilobar lesion, lesional MRI, and older age at onset of seizures.41–43 Seizure freedom in selected patients undergoing extratemporal resection ranges from 50%–75%, with poorer outcomes among patients with negative MRIs.42 Generally speaking, extratemporal lesionectomy for epilepsy does not seem to have a deleterious effect on cognition.44
Hemispherectomy, or hemispheric disconnection, is indicated for diseases that affect one hemisphere (large anatomical abnormalities, Rasmussen encephalitis, etc). Rather than anatomical resection of the hemisphere, the typical technique has now evolved to performing a disconnection so as to minimize later bleeding and hemosiderosis. Post- and perioperative complications include excessive blood loss, infarct, intracranial hematoma, infections, and hydrocephalus, with one series reporting a complication rate of 27%, including minor complications.45 Approximately 60% of patients have seizure freedom at 5 years, with bilateral PET abnormalities and acute postoperative seizures identified as risk factors for recurrence.45,46 Because of the preexisting pathology, many of the children undergoing hemispherectomy had hemispheric dysfunction prior to surgery, and linguistic and developmental outcome is often stable or shows a slight improvement correlating with seizure frequency.45 Because of brain plasticity, earlier resection is correlated with better neurodevelopmental outcome.
After resective surgery, anticonvulsants are usually continued in the short term; however, there is not good evidence to guide duration of therapy. A tapered discontinuation is often considered after 1 year of seizure freedom in patients. The likelihood of seizure freedom after surgery differs depending on the completeness of resection of the epileptogenic zone, the specific pathology, and the location of the lesion. Long-term seizure freedom is achieved in 70%–80% of children after temporal lobectomy for mesial temporal sclerosis but in only 50%–60% with nonlesional extratemporal resections.47,48
Palliative surgical options
Neurostimulation
For patients with drug-resistant epilepsy who are not candidates for resective surgery, neurostimulation can provide another option for seizure control. Vagal nerve stimulation has been used in the US for nearly 20 years. A number of other modalities are under investigation, including deep brain stimulation of the anterior thalamus (already approved for use in Europe), responsive neurostimulation, and trigeminal nerve stimulation.
Vagal nerve stimulation is the only neurostimulatory modality that currently has US Food and Drug Administration approval for children. Generally speaking, around one-half of patients with refractory epilepsy will see a reduction in their seizure frequency of 50%.49 The therapy is generally well tolerated, with the most common side effects being chronic cough and hoarseness. Vocal cord paralysis and dysphagia are recognized adverse effects that may manifest in the postoperative period.
Corpus callosotomy
Corpus callosotomy is a palliative surgical therapy that involves transecting either the anterior two-thirds or complete corpus callosum. It is considered most effective in generalized epilepsies, particularly atonic seizures (“drop attacks”) such as those encountered in Lennox–Gastaut syndrome. It has demonstrated little effectiveness for focal epilepsy with exacerbation of seizures in 25%.50
Dietary therapy
The ketogenic diet is the most well-known and studied dietary therapy, but the modified Atkins (MAD) and low glycemic index (LGID) diets are gaining traction as better-tolerated alternatives that may be appropriate for selected, medically refractory patients with focal epilepsy who are not ideal candidates for resective surgery.
The ketogenic diet
The ketogenic diet is a high-fat, low-carbohydrate diet that results in the patient maintaining a state of ketosis. The mechanism of action is as yet not well understood. The 50% responder rate for this diet is approximately 50%; however, seizure freedom is generally not achieved for patients with intractable focal epilepsy.51,52 Although highly efficacious in selected patients, the ketogenic diet has the potential for serious adverse effects and requires input from an experienced dietician and careful monitoring. The ketogenic ratio (ratio of grams of fats to grams of protein and carbohydrates combined) is adjusted based on degree of seizure control and level of ketosis. In many centers, the diet is often started during an inpatient admission, so that adverse medical effects (hypoglycemia, excessive ketosis and acidosis, metabolic decompensation) and overall tolerability (food refusal) can be monitored. Possible longer-term adverse effects include cardiomyopathy, dyslipidemia, hyperketosis, hypoglycemia, renal stones, pancreatitis, and perhaps, in some cases, immune dysfunction (Table 3). In addition, unless the patient is on a ketogenic diet formula, the diet is not nutritionally complete, and supplementation and monitoring of micronutrients is necessary. Additional supplements that are often required include a carbohydrate-free multivitamin, calcium supplementation, and citrate preparations in children with increased excretion of calcium and risk of renal stones. Patients should be seen by a dietician every 3 months. Discontinuation of the diet may be considered after 2 years by slowly weaning the ketogenic ratio by about 0.25 every 2–3 months.
 | Table 3 Possible side effects of the ketogenic diet |
Other dietary therapies
Other dietary therapies include the MAD and LGID. The MAD is based on limiting carbohydrate intake to between 10 and 20 g of carbohydrate per day and encouraging fat. Unlike the ketogenic diet, there are no caloric or fluid restrictions. Efficacy is similar to the ketogenic diet, with increased efficacy for children started at 10 g of carbohydrate per day.53,54 The LGID is a less-restrictive option that involves mild carbohydrate restriction (40–60 g per day) and elimination of carbohydrates with a glycemic index higher than 50.55,56 These diets have the advantage of greater flexibility in food selection than the ketogenic diet and therefore increased tolerability, especially for adolescent patients.
Long-term seizure outcomes in focal epilepsy in children and adolescents
The long-term seizure outcome of focal epilepsy in children is highly dependent on the underlying etiology and/or electroclinical syndrome. The long-term prognosis of benign epilepsy with centrotemporal spikes is excellent, even if seizures are initially frequent and troublesome. In a meta-analysis, 50% of patients were in remission at age 6 years, 92% at age 12 years, and 99.8% at age 18 years.57 Similarly, in Panayiotopoulos syndrome, remission of active epilepsy occurs 1–2 years from onset. The prognosis is less clear in late-onset benign occipital epilepsy, although Gastaut reported that 5% of his 63 cases had recurrent seizures into adulthood.58
Focal epilepsy due to either malformations of cortical development or mesial temporal sclerosis carries a much lower likelihood of favorable long-term seizure outcome without surgery.5,6 In a population-based study of new-onset epilepsy in children, 83% with malformations of cortical development and 67% of those with mesial temporal sclerosis had either undergone surgery for medically intractable epilepsy or had persistent, drug-resistant seizures at final follow-up.5 Encephalomalacia had a somewhat better outcome, with intractable epilepsy seen in only one-third of cases and seizure freedom in over one-half.
Nonlesional focal epilepsy that does not fit an electroclinical syndrome accounts for approximately one-quarter to one-third of all cases of new-onset focal seizures in children. The outcome in this group appears much more favorable than with lesional causes, with 81% achieving seizure freedom after long-term follow-up, and 55% achieving terminal remission (seizure and medication freedom).59
Conclusion
Focal epilepsy accounts for over half of new-onset epilepsy in children and teens, and can arise from a diverse group of etiologies with varying long-term prognoses. In addition to seizure control, care must be given to identify and manage the cognitive, behavioral, and psychiatric comorbidities that frequently co-exist. Investigations and treatment must be individualized; however, intractrable cases should be referred to an epilepsy center for consideration of surgical or dietary therapies.
Disclosure
The authors report no conflicts of interest in this work.
References
Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–685. | |
Blume WT, Lüders HO, Mizrahi E, Tassinari C, van Emde Boas W, Engel J Jr. Glossary of descriptive terminology for ictal semiology: report of the ILAE task force on classification and terminology. Epilepsia. 2001;42:1212–1218. | |
Camfield CS, Camfield PR, Gordon K, Wirrell E, Dooley JM. Incidence of epilepsy in childhood and adolescence: a population-based study in Nova Scotia from 1977 to 1985. Epilepsia. 1996;37:19–23. | |
Wirrell EC, Grossardt BR, Wong-Kisiel LC, Nickels KC. Incidence and classification of new-onset epilepsy and epilepsy syndromes in children in Olmsted County, Minnesota from 1980 to 2004: a population-based study. Epilepsy Res. 2011;95:110–118. | |
Dhamija R, Moseley BD, Cascino GD, Wirrell EC. A population-based study of long-term outcome of epilepsy in childhood with a focal or hemispheric lesion on neuroimaging. Epilepsia. 2011;52: 1522–1526. | |
Semah F, Picot MC, Adam C, et al. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology. 1998;51: 1256–1262. | |
Blümcke I, Spreafico R. An international consensus classification for focal cortical dysplasias. Lancet Neurology. 2011;10:26–27. | |
Shinnar S, Bello JA, Chan S, et al. MRI abnormalities following febrile status epilepticus in children: the FEBSTAT study. Neurology. 2012;79:871–877. | |
Tsai MH, Pardoe HR, Perchyonok Y, et al. Etiology of hippocampal sclerosis: evidence for a predisposing familial morphologic anomaly. Neurology. 2013;81:144–149. | |
Dlugos DJ, Sammel MD, Strom BL, Farrar JT. Response to first drug trial predicts outcome in childhood temporal lobe epilepsy. Neurology. 2001;57:2259–2264. | |
Guilioni M, Rubboli G, Marucci G, et al. Focal epilepsies associated with glioneuronal tumors: review article. Panminerva Med. 2013;55(22):225–238. | |
Odding E, Roebroeck ME, Stam HJ. The epidemiology of cerebral palsy: incidence, impairments and risk factors. Disabil Rehabil. 2006;28:183–191. | |
Rosenfeld MR, Dalmau JO. Paraneoplastic disorders of the CNS and autoimmune synaptic encephalitis. Continuum (Minneap Minn). 2012;18(2):366–383. | |
Hernandez MT, Sauerwein HC, Jambaqué I, et al. Attention, memory, and behavioral adjustment in children with frontal lobe epilepsy. Epilepsy Behav. 2003;4:522–536. | |
Exner C, Boucsein K, Lange C, et al. Neuropsychological performance in frontal lobe epilepsy. Seizure. 2002;11:20–32. | |
McDonald CR, Delis DC, Kramer JH, Tecoma ES, Iragui VJ. A componential analysis of proverb interpretation in patients with frontal lobe epilepsy and temporal lobe epilepsy: relationships with disease-related factors. Clin Neuropsychol. 2008;22:480–496. | |
Upton D, Thompson PJ. Epilepsy in the frontal lobes: neuropsychological characteristics. J Epilepsy. 1996;9:215–222. | |
Helmstaedter C, Kemper B, Elger CE. Neuropsychological aspects of frontal lobe epilepsy. Neuropsychologia. 1996;34:399–406. | |
Glosser G, Zwil AS, Glosser DS, O’Connor MJ, Sperling MR. Psychiatric aspects of temporal lobe epilepsy before and after anterior temporal lobectomy. J Neurol Neurosurg Psychiatry. 2000;68:53–58. | |
Salpekar JA, Berl MM, Havens K, et al. Psychiatric symptoms in children prior to epilepsy surgery differ according to suspected seizure focus. Epilepsia. 2013;54:1074–1082. | |
Hermann B, Seidenberg M, Jones J. The neurobehavioural comorbidities of epilepsy: can a natural history be developed? Lancet Neurol. 2008;7:151–160. | |
Battaglia D, Chieffo D, Tamburrini G, et al. Posterior resection for childhood lesional epilepsy: neuropsychological evolution. Epilepsy Behav. 2012;23:131–137. | |
St Louis EK. Minimizing AED adverse effects: improving quality of life in the interictal state in epilepsy care. Curr Neuropharmacol. 2009;7:106–114. | |
Mindikoglu AL, King D, Magder LS, Ozolek JA, Mazariegos GV, Shneider BL. Valproic acid-associated acute liver failure in children: case report and analysis of liver transplantation outcomes in the United States. J Pediatr. 2011;158:802–807. | |
Shi YW, Min FL, Qin B, et al. Association between HLA and Stevens-Johnson syndrome induced by carbamazepine in Southern Han Chinese: genetic markers besides B*1502? Basic Clin Pharmacol Toxicol. 2012;111:58–64. | |
Sahota P, Prabhakar S, Kharbanda PS, et al. Seizure type, antiepileptic drugs, and reproductive endocrine dysfunction in Indian women with epilepsy: a cross-sectional study. Epilepsia. 2008;49:2069–2077. | |
Morrell MJ. Reproductive and metabolic disorders in women with epilepsy. Epilepsia. 2003;44 Suppl 4:11–20. | |
Helmstaedter C, Fritz NE, Kockelmann E, Kosanetzky N, Elger CE. Positive and negative psychotropic effects of levetiracetam. Epilepsy Behav. 2008;13:535–541. | |
Salinsky M, Storzbach D, Oken B, Spencer D. Topiramate effects on the EEG and alertness in healthy volunteers: a different profile of antiepileptic drug neurotoxicity. Epilepsy Behav 2007;10:463–469. | |
Park SP, Hwang YH, Lee HW, Suh CK, Kwon SH, Bee BI. Long-term cognitive and mood effects of zonisamide monotherapy in epilepsy patients. Epilepsy Behav. 2008;12:102–108. | |
Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697–714. | |
Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51: 1069–1077. | |
Zhang Y, Yu N, Su L, Di Q. A prospective cohort study of prognosis for newly diagnosed epilepsy in east China. BMC Neurol. 2013;13:116. | |
Camfield PR, Camfield CS, Gordon K, Dooley JM. If a first antiepileptic drug fails to control a child’s epilepsy, what are the chances of success with the next drug? J Pediatr. 1997;131:821–824. | |
Wirrell E, Camfield C, Camfield P, Dooley J. Prognostic significance of failure of the initial antiepileptic drug in children with absence epilepsy. Epilepsia. 2001;42:760–763. | |
Kwan P, Sills GJ, Brodie MJ. The mechanisms of action of commonly used antiepileptic drugs. Pharmacol Ther. 2001;90:21–34. | |
Crepeau AZ, Moseley BD, Wirrell EC. Specific safety and tolerability considerations in the use of anticonvulsant medications in children. Drug Healthc Patient Saf. 2012;4:39–54. | |
Engel J Jr, McDermott MP, Wiebe S, et al; Randomized Surgical Epilepsy Trial (ERSET) Study Group. Early surgical therapy for drug-resistant temporal lobe epilepsy: a randomized trial. JAMA. 2012;307: 922–930. | |
Follett PL, Vora N, Cross JH. Paediatric intractable epilepsy syndromes: changing concepts in diagnosis and management. Adv Tech Stand Neurosurg. 2012;39:45–60. | |
Maton B, Jayakar P, Resnick T, Morrison G, Ragheb J, Duchowny M. Surgery for medically intractable temporal lobe epilepsy during early life. Epilepsia. 2008;49:80–87. | |
Wyllie E, Comair YG, Kotagal P, Bulacio J, Bingaman W, Ruggieri P. Seizure outcome after epilepsy surgery in children and adolescents. Ann Neurol. 1998;44:740–748. | |
Liava A, Francione S, Tassi L, et al. Individually tailored extratemporal epilepsy surgery in children: anatomo-electro-clinical features and outcome predictors in a population of 53 cases. Epilepsy Behav. 2012;25:68–80. | |
Babini M, Giulioni M, Galassi E, et al. Seizure outcome of surgical treatment of focal epilepsy associated with low-grade tumors in children. J Neurosurg Pediatr. 2013;11:214–223. | |
D’Argenzio L, Colonnelli MC, Harrison S, et al. Cognitive outcome after extratemporal epilepsy surgery in childhood. Epilepsia. 2011;52:1966–1972. | |
Jonas R, Nguyen S, Hu B, et al. Cerebral hemispherectomy: hospital course, seizure, developmental, language, and motor outcomes. Neurology. 2004;62:1712–1721. | |
Moosa AN, Jehi L, Marashly A, et al. Long-term functional outcomes and their predictors after hemispherectomy in 115 children. Epilepsia. 2013;54:1771–1779. | |
Englot DJ, Rolston JD, Wang DD, Sun PP, Chang EF, Auguste KI. Seizure outcomes after temporal lobectomy in pediatric patients. J Neurosurg Pediatr. 2013;12:134–141. | |
Englot DJ, Breshears JD, Sun PP, Chang EF, Auguste KI. Seizure outcomes after resective surgery for extra-temporal lobe epilepsy in pediatric patients. J Neurosurg Pediatr. 2013;12:126–133. | |
Morris GL 3rd, Gloss D, Buchhalter J, Mack KJ, Nickels K, Harden C. Evidence-based guideline update: vagus nerve stimulation for the treatment of epilepsy: report of the guideline development subcommittee of the american academy of neurology. Epilepsy Curr. 2013;13: 297–303. | |
Spencer SS. Corpus callosum section and other disconnection procedures for medically intractable epilepsy. Epilepsia. 1988;29 Suppl 2:S85–S99. | |
Payne NE, Cross JH, Sander JW, Sisodiya SM. The ketogenic and related diets in adolescents and adults – a review. Epilepsia. 2011;52: 1941–1948. | |
Cervenka MC, Kossoff EH. Dietary treatment of intractable epilepsy. Continuum (Minneap Minn). 2013;19(3 Epilepsy):756–766. | |
Kossoff EH, Dorward JL. The modified Atkins diet. Epilepsia. 2008; 49 Suppl 8:37–41. | |
Kossoff EH, Turner Z, Bluml RM, Pyzik PL, Vining EP. A randomized, crossover comparison of daily carbohydrate limits using the modified Atkins diet. Epilepsy Behav. 2007;10:432–436. | |
Pfeifer HH, Lyczkowski DA, Thiele EA. Low glycemic index treatment: implementation and new insights into efficacy. Epilepsia. 2008; 49 Suppl 8:42–45. | |
Muzykewicz DA, Lyczkowski DA, Memon N, Conant KD, Pfeifer HH, Thiele EA. Efficacy, safety, and tolerability of the low glycemic index treatment in pediatric epilepsy. Epilepsia. 2009;50:1118–1126. | |
Bouma PA, Bovenkerk AC, Westendorp RG, Brouwer OF. The course of benign partial epilepsy of childhood with centrotemporal spikes: a meta-analysis. Neurology. 1997;48:430–437. | |
Gastaut H. [The benigne epilepsy in childhood with occipital spike wave complexes (author’s transl)]. EEG EMG Z Elektroenzephalogr Elektromyogr Verwandte Geb. 1982;13:3–8. German. | |
Wirrell EC, Grossardt BR, So EL, Nickels KC. A population-based study of long-term outcomes of cryptogenic focal epilepsy in childhood: cryptogenic epilepsy is probably not symptomatic epilepsy. Epilepsia. 2011;52:738–745. | |
Giordani B, Caveney AF, Laughrin D, et al. Cognition and behavior in children with benign epilepsy with centrotemporal spikes (BECTS). Epilepsy Res. 2006;70:89–94. | |
Goldberg-Stern H, Gonen OM, Sadeh M, Kivity S, Shuper A, Inbar D. Neuropsychological aspects of benign childhood epilepsy with centrotemporal spikes. Seizure. 2010;19:12–16. | |
Lada C, Skiadas K, Theodorou V, Loli N, Covanis A. A study of 43 patients with panayiotopoulos syndrome, a common and benign childhood seizure susceptibility. Epilepsia. 2003;44:81–88. | |
Specchio N, Trivisano M, Di Ciommo V, et al. Panayiotopoulos syndrome: a clinical, EEG, and neuropsychological study of 93 consecutive patients. Epilepsia. 2010;51:2098–2107. | |
Morse RP. Dravet syndrome: inroads into understanding epileptic encephalopathies. J Pediatr. 2011;158:354–359. | |
Wolff M, Casse-Perrot C, Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia. 2006;47 Suppl 2:45–48. | |
Caraballo RH, Fejerman N. Dravet syndrome: a study of 53 patients. Epilepsy Res. 2006;70 Suppl 1:S231–S238. | |
Nickels K, Wirrell E. Electrical status epilepticus in sleep. Sem Pediatr Neurol. 2008;15:50–60. | |
Gagliano A, Ferlazzo E, Germanò E, et al. Neuropsychological deficits in monozygotic twins with childhood epilepsy with occipital paroxysms. J Clin Exp Neuropsychol. 2007;29:488–495. | |
Wood AG, Saling MM, Fedi M, et al. Neuropsychological function in patients with a single gene mutation associated with autosomal dominant nocturnal frontal lobe epilepsy. Epilepsy Behav. 2010;17: 531–535. | |
Derry CP, Heron SE, Phillips F, et al. Severe autosomal dominant nocturnal frontal lobe epilepsy associated with psychiatric disorders and intellectual disability. Epilepsia. 2008;49:2125–2129. | |
Kossoff EH, Zupec-Kania BA, Amark PE, et al; International Ketogenic Diet Study Group. Optimal clinical management of children receiving the ketogenic diet: recommendations of the International Ketogenic Diet Study Group. Epilepsia. 2009;50(2):304–317. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.