")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Patient considerations and drug selection in the treatment of idiopathic pulmonary fibrosis
Authors Trawinska M, Rupesinghe R, Hart S
Received 18 September 2015
Accepted for publication 14 December 2015
Published 8 April 2016 Volume 2016:12 Pages 563—574
DOI https://doi.org/10.2147/TCRM.S81144
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Maria A Trawinska,1 Ruwani D Rupesinghe,1 Simon P Hart1,2
1Hull and East Yorkshire Hospitals NHS Trust, 2Hull York Medical School, Academic Respiratory Medicine, Castle Hill Hospital, Cottingham, East Yorkshire, UK
Abstract: Idiopathic pulmonary fibrosis (IPF) is a progressive interstitial lung disease of unknown cause. Approximately 5,000 people are diagnosed with IPF in the UK every year. People with IPF suffer significant morbidity and, without any curative treatment at present, survival rates remain poor with a median survival of 3 years. While treatment remains largely supportive, many drug therapies have been trialed in IPF over the years. Pirfenidone and nintedanib are newly licensed treatments for IPF and the first drugs to have shown convincing evidence of slowing disease progression. In addition to evaluating clinical evidence, we also discuss elements affecting drug choice from the viewpoint of patients and health care professionals. We discuss pharmacological and nonpharmacological aspects of providing best supportive care for patients with IPF. However, few good quality studies exist focusing on controlling symptoms specifically in patients with IPF, and recommendations are often extrapolated from evidence in other chronic diseases. In covering these topics, we hope to provide readers with a comprehensive review of the available evidence pertaining to all aspects of care for patients suffering with IPF.
Keywords: interstitial lung disease, high-resolution computed tomography, forced vital capacity, usual interstitial pneumonia, clinical trials, decision making, idiopathic pulmonary fibrosis
Introduction
Idiopathic pulmonary fibrosis (IPF) is characterized by chronic progressive scarring of the gas-exchanging interstitial tissue of the lung. Despite extensive research into the pathogenesis and treatment of IPF, 5-year survival is less than 45%.1 In this review, we describe the drugs used in the treatment of IPF and how patients and clinicians make decisions about commencing therapy.
Incidence and pathophysiology
IPF is a specific form of chronic and progressive interstitial lung disease (ILD) that primarily affects older adults. Its incidence increases with age, with presentation typically occurring in the sixth and seventh decades. Patients with IPF aged younger than 50 years are rare.2
Over the years, the terminology used to describe the various types of ILD has caused confusion. Nomenclature has altered several times, making reliable epidemiology of IPF difficult. The incidence of IPF in the UK has been investigated using death certificates3 and general practice data.1 However, these data may also include patients with similar diseases such as nonspecific interstitial pneumonia, since the particular data sets used did not include such specific subclassifications. Data from the Office for National Statistics included “post inflammatory fibrosis,” while data from The Health Improvement Network database included cryptogenic fibrosing alveolitis and diffuse pulmonary fibrosis.1,3 With these limitations in mind, it has been suggested that there are more than 5,000 new patients diagnosed with IPF each year in the UK.3 Overall, the incidence of IPF is estimated to be between 4.6 per 100,000 person-years1 and 7.44 per 100,000 person-years.3 The rate is higher in men than in women. IPF is a fatal disease, though its progression is highly variable. Three- and five-year survival rates are 57% and 43%, respectively, and the median survival time is less than 4 years.1
The pathophysiology of IPF is thought to begin with an inciting risk factor, which subsequently triggers a proinflammatory and profibrotic response involving the alveolar epithelial cells.4 Various cell types have been implicated, including epithelial cells, fibroblasts, myofibroblasts, alveolar macrophages, and endothelial cells. It is unclear why dysregulation of normal tissue repair occurs, but there is also evidence to suggest that there may be failed cellular regeneration, mediated by shortened telomere length.5 Abnormal mucin production may be involved in the development of fibrosis, either through impairing host defense systems or disrupting normal alveolar repair mechanisms.6 Eventually, excessive extracellular matrix is deposited in the lungs, which distorts the normal delicate architecture and disrupts function (Figure 1). This lung injury and scarring lead to a decline in the lung function and culminates in respiratory failure and death.7
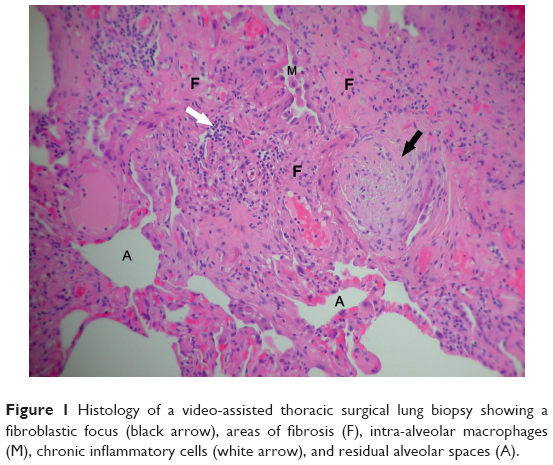 | Figure 1 Histology of a video-assisted thoracic surgical lung biopsy showing a fibroblastic focus (black arrow), areas of fibrosis (F), intra-alveolar macrophages (M), chronic inflammatory cells (white arrow), and residual alveolar spaces (A). |
Diagnosis
The diagnosis of IPF is typically based on the combination of clinical features and typical high-resolution computed tomography (HRCT) appearances,8 following the exclusion of known causes of ILD. These causes include connective tissue diseases, drug toxicity, and environmental exposures.2 Patients classically present with worsening chronic exertional dyspnea and cough. On chest auscultation, they have bibasal end-inspiratory crackles and they may be hypoxemic. The presence of these clinical features and a pattern of usual interstitial pneumonia (UIP) on HRCT (Figure 2) is now considered sufficiently characteristic to confidently diagnose IPF, and therefore obviate the need for surgical lung biopsy.9 A UIP pattern, as seen on HRCT, consists of the following four features: honeycombing, reticular abnormality, subpleural and basal predominance, and an absence of features inconsistent with a UIP pattern. Features inconsistent with UIP include upper- or mid-lung predominance, peribronchovascular predominance, extensive ground-glass abnormality, profuse micronodules, discrete cysts, diffuse mosaic attenuation or air trapping, and consolidation in bronchopulmonary segments or lobes.2 The radiological pattern of UIP is also seen in rheumatoid arthritis-related ILD, chronic hypersensitivity pneumonitis, asbestosis, and amiodarone toxicity;10 hence, there is the need to exclude these conditions before proceeding further.
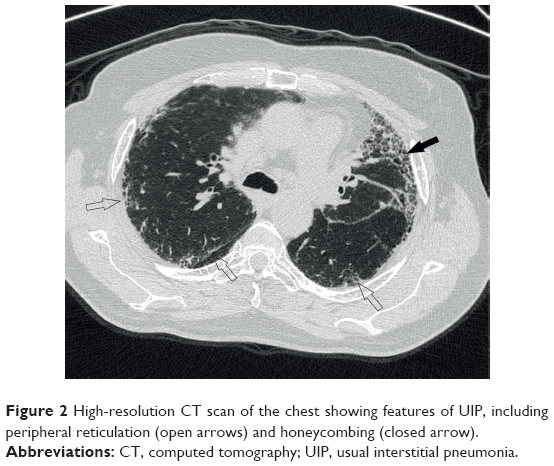 | Figure 2 High-resolution CT scan of the chest showing features of UIP, including peripheral reticulation (open arrows) and honeycombing (closed arrow). |
If HRCT scanning is not characteristic of UIP, it may be necessary to proceed to biopsy and histological evaluation. However, it is important to remember that the majority of patients diagnosed with IPF are elderly and may not be fit enough for such an invasive investigation. This diagnostic difficulty has implications for organizing clinical trials into IPF and its potential treatments.
Monitoring
Patients diagnosed with IPF should have pulmonary function testing and a six-minute walk (6MWD) test performed at baseline. It has been shown that a decline in forced vital capacity (FVC) of >5%–10% over 6 months or 12 months is a poor prognostic factor,11 so this should be measured in order to identify patients at increased risk of mortality. Similarly, the 6MWD can also be used for prognostication, as a shorter walk distance and a fall in oxygen saturation with exercise have been associated with an increased risk of subsequent mortality.12
Management
The overall management of IPF is complex, and the practice has varied greatly over the years. We will discuss the evidence for drug therapies associated with IPF, including the current guidelines and best practice, those treatments that have been used in the past but are no longer recommended, as well as aspects of palliative care and what the future may hold.
Drug treatment
Many drugs have been studied for the treatment of IPF, and evidence both for and against their use has altered over the years. There are currently no curative treatments for patients with IPF, but there have been some recent advances in terms of disease-modifying drugs.
Here, we will refer to the current recommendations from the National Institute for Health and Care Excellence (NICE),13 the joint American Thoracic Society (ATS)/European Respiratory Society (ERS)/Japanese Respiratory Society (JRS)/Latin American Thoracic Association (ALAT) guidelines,2,14 and the US Food and Drug Administration.15
Pirfenidone
Pirfenidone is a pyridone derivative and is licensed for the treatment of IPF in Europe and North America. The mechanism of action of pirfenidone is not fully understood, but it is thought to suppress fibroblast proliferation, reducing the production and activity of fibrosis-associated proteins and cytokines, including the key profibrotic cytokine transforming growth factor-β.16
Two clinical trials, CAPACITY 004 and CAPACITY 006, were carried out to investigate the benefits of oral pirfenidone in IPF17 across 110 centers in Australia, Europe, and North America. Patients were aged between 40 and 80 years, with measured FVC ≥50% predicted and diffusion capacity for carbon monoxide [DLco] ≥35%. Patients were randomized to receive pirfenidone or placebo and were followed up for a minimum of 72 weeks. The primary endpoint was change in percent predicted FVC at week 72. However, the primary outcomes were different between the individual studies, with CAPACITY 004 favoring the treatment arm (FVC -8.0% vs -12.4%), while CAPACITY 006 showed no statistically significant change in percent predicted FVC. The trial group went on to do a preplanned pooled analysis of the patients in both trials that suggested an overall benefit from pirfenidone treatment.
There was no statistically significant difference in the risk of death between the treatment and placebo groups. Patients in the treatment arm had a higher incidence of nausea, dyspepsia, vomiting, rashes, and dizziness than the placebo group.17 A common side effect was photosensitivity (Figure 3), which was a reason for discontinuation in some patients. It is now recommended that patients taking pirfenidone should avoid sun exposure, wear protective clothing, and use sunblock with high UVA and UVB protection.18
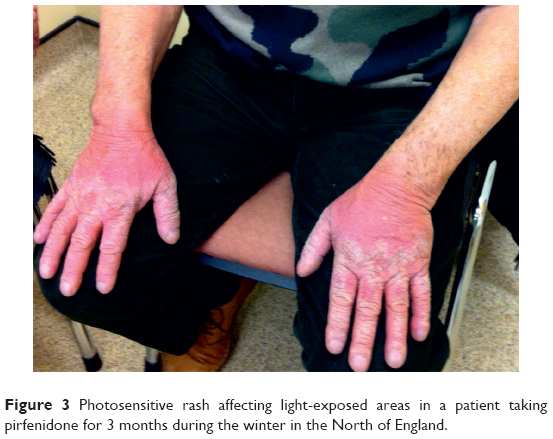 | Figure 3 Photosensitive rash affecting light-exposed areas in a patient taking pirfenidone for 3 months during the winter in the North of England. |
In the CAPACITY studies, 15% of patients discontinued treatment due to side effects.17 Although this may seem high, it has been shown that between 4% and 26% of patients discontinue placebos due to perceived adverse events.19 The level of adverse effects reported for pirfenidone is therefore not unusual in the context of a trial. Most of the patients who completed the CAPACITY trials were enrolled into an open-label extension study (RECAP) to evaluate the long-term safety of pirfenidone.20 An interim analysis was published, reviewing data from enrollment in September 2008 until August 2013. The median duration of treatment was 163 weeks, with 99% of patients reporting at least one treatment-emergent adverse event. In more than one-third of patients, these included dyspnea, cough, or bronchitis, as well as upper respiratory symptoms of infection or nasopharyngitis. In total, 33% of patients also experienced nausea, with just less than one-third complaining of fatigue and dizziness. Rash and photosensitivity were less of a problem in RECAP20 (16% and 9%) than in the CAPACITY trials17 (32% and 12%), although 45% of patients went on to discontinue treatment due to treatment-emergent adverse events. The most common cause for this was progression of IPF. Published reports of “real-life” experience with pirfenidone suggest that it is well tolerated.21,22
Because of the discrepancy between the primary outcomes in the two CAPACITY trials, a further randomized controlled trial (ASCEND) was designed.23 ASCEND was performed in a similar manner to the CAPACITY trials, but diagnosis, spirometry, and decisions regarding deaths were centralized. There were also differences in eligibility criteria aimed at recruiting patients who were at higher risk of disease progression.
Patients were recruited from 127 centers across nine countries if they met the following criteria: age between 40 and 80 years, diagnosis of UIP on HRCT or surgical lung biopsy, FVC 50%-90%, DLco 30%-90% predicted, FEV1/FVC >80%, and 6MWD of >150 m. The primary endpoint of the study was a change in the percent predicted FVC at the end of 52 weeks. Those who were eligible were randomized to receive oral pirfenidone or placebo for 1 year. A total of 555 people were enrolled and assigned to placebo or pirfenidone in a 1:1 ratio. The groups were well matched, and 94.1% went on to complete the study. In total, 46 patients in the pirfenidone arm and 88 in the placebo arm had a decline in FVC of 10% or more, equating to a reduction of 47.9% with treatment. A total of 63 patients taking pirfenidone showed no decline in the percent predicted FVC compared to 27 receiving placebo. The mean decline in FVC was 235 mL in the pirfenidone group compared to 428 mL in the placebo group. These results also translated into fewer patients showing deterioration in their 6MWD of 50 m or more at the end of the 52-week period. All of these results had statistical significance.
All-cause mortality was lower in the pirfenidone group, but not by a statistically significant amount. None of the secondary outcomes appeared to translate into a symptomatic benefit for patients, with no statistically significant difference in their Shortness of Breath Questionnaire scores.
NICE has released guidelines for the use of pirfenidone based on its evaluation of the trial data and costing information.24 It recommends that, as pirfenidone has been trialed only in patients with IPF, it is important to have an accurate diagnosis prior to commencing treatment. NICE has recommended that only patients with IPF and FVC of 50%-80% predicted should be eligible to receive pirfenidone. Lung function should be monitored and treatment discontinued if the FVC continues to fall by >10% in 12 months. It is important to counsel patients that pirfenidone is not a cure for their condition and is instead aimed at slowing the decline in lung function, although even this result cannot be guaranteed. As such, the provision of symptomatic and supportive care is essential in this group of patients.
Nintedanib
Nintedanib is an intracellular inhibitor of multiple receptor-associated tyrosine kinases, including receptors for vascular endothelial growth factor and platelet-derived growth factor (PDGF).25 It is believed that these pathways play roles in the development of pulmonary fibrosis, as high plasma concentrations of vascular endothelial growth factor have been found in patients with the disease.26 Fibrotic lung tissue from patients with IPF has also been found to express PDGF, and this is thought to be an important contributing factor in the pathophysiology of pulmonary fibrosis through abnormal fibroblast proliferation and collagen production.27
INPULSIS-1 and 2 were two Phase III clinical trials of oral nintedanib treatment for IPF. The study design for both was the same, and these were international, multicenter, randomized, double-blind, placebo-controlled trials.25 Patients were monitored over a 52-week period, and the primary endpoint for both trials was the annual rate of decline in FVC. Inclusion criteria included FVC ≥50% predicted and DLco 30%-79% predicted. Patients included in the INPULSIS studies had a mean FVC of ~80% predicted, indicating rather milder disease compared with subjects receiving pirfenidone in the CAPACITY and ASCEND trials. In all, >1,000 patients underwent randomization, with 515 in INPULSIS-1 and 551 in INPULSIS-2. This was well over the sample size calculated to be able to detect a difference of 100 mL in the annual decline in FVC. Patients were randomized on a 1:2 ratio of placebo to nintedanib. Discontinuation rates were slightly higher in the treatment arm in INPULSIS-1 (25.2% vs 17.6%), but INPULSIS-2 had similar discontinuation rates in each arm (23.7% nintedanib vs 20.1% placebo). The most common reason for discontinuation was an adverse event. Patients who had received at least one dose of trial drug were reviewed over the full 52 weeks, though some were lost to follow-up. Both trials demonstrated a smaller change in FVC in the treatment group compared to placebo (INPULSIS-1, -114.7 mL/year for nintedanib vs -239.9 mL/year for placebo and INPULSIS-2, -113.6 mL/year for nintedanib vs -207.3 mL/year for placebo). These were statistically significant differences in both studies. A preplanned pooled analysis revealed a between-group difference of -109.9 mL/year change in FVC.
In INPULSIS-1, there was no difference in time to first acute exacerbation and St George’s Respiratory Questionnaire score. INPULSIS-2 showed slight improvements, which were statistically significant, in both of these secondary endpoints.
The most frequent adverse event in the nintedanib group within both trials was diarrhea. However, it only led to discontinuation in 14 patients receiving the drug (4.5%). In both trials, the patients within the nintedanib arm had a higher incidence of derangement of hepatic enzymes, more specifically a rise in aspartate aminotransferase, alanine aminotransferase, or both. It is unclear if this led to a discontinuation in treatment and whether that then led to a subsequent improvement in enzyme levels. There was a <2% incidence of cardiac events, but five patients in each nintedanib group had a myocardial infarction compared to one in each placebo group. Deaths were reported from the pooled analysis rather than the individual trials, with no statistically significant between-group differences for all-cause mortality or death from a respiratory cause.
As with pirfenidone, it is essential patients understand that treatment is not curative but aimed at slowing disease progression, and therefore, supportive measures should not be neglected. There are no trials directly comparing pirfenidone with nintedanib. While both had similar recruitment criteria with regard to disease severity, progression was measured by slightly different methods, but it is likely that they are similarly efficacious.
NICE is currently in the process of evaluating nintedanib for use in IPF. The drug will be compared against both pirfenidone and best supportive care in terms of the following outcome measures: function parameters, physical function, exacerbation rate, progression-free survival, mortality, adverse effects of treatment, and health-related quality of life. In a consultation document, NICE suggested imposing a similar restriction to pirfenidone in terms of lung function, namely, FVC 50%–80% predicted.
After initially declining an application for the use of pirfenidone in IPF in 2010, in October 2014 the US Food and Drug Administration approved both pirfenidone (trade name Esbriet) and nintedanib (Ofev) for the treatment of IPF.15 The agency found that, after three clinical trials of each drug, both were safe and significantly slowed the decline in FVC. Both nintedanib and pirfenidone have received conditional recommendations for their use in a recent update to the IPF international clinical practice guideline.14
Decision making: pirfenidone or nintedanib or supportive care alone?
In the UK, it is likely that both drugs will be offered to patients with IPF and that the final decision will be based on contraindications and patient choice when faced with the different side effect profiles. Another factor that may be important for some patients to consider is dosing – pirfenidone is titrated to a dose of three capsules taken three times a day, whereas nintedanib is taken as one capsule twice a day. It is important to note that patients taking full-dose anticoagulant therapy or high-dose antiplatelet therapy were excluded from the INPULSIS trials.25 The recommendation is that nintedanib should be used cautiously in patients taking anticoagulant therapy, and this may therefore restrict its use.28 As the mechanisms of action for these drugs are thought to be different, there may be an option to use both pirfenidone and nintedanib together in patients with IPF in the future, in the hope that they will have an additive effect or work synergistically. This is an area for further research. It is also important to consider the high costs of these drugs. According to the Scottish Medicines Consortium,29 even through a scheme that improves cost-effectiveness, treatment with either nintedanib or pirfenidone will cost £26,100 per patient per year.
There have been no studies looking specifically at patient decision making in the context of treatment options in IPF. The majority of research looking at patient choice with regard to picking between therapies is based on patients with malignant disease. Thompson et al suggest that patients are more likely to take part in choosing between two equally effective drugs, which “differ only in lifestyle or side effect implications,” if they think they have the relevant expertise.30 However, according to Haynes et al, patients may have either no views or unshakable views on their treatment options, depending on factors such as “personal values and experiences, degree of aversion to risk (…), family, willingness to take medicines” and the accuracy of the information they have been given.31 Chewning and Sleath wrote that a patient’s condition can affect how involved they are in making decisions about their health, with some suggesting that they “prefer their health care provider to take a stronger role in decision-making as their condition worsens.”32 A study based in primary care found that the main barrier to patients being involved in decisions about their medications was that their beliefs were not taken seriously, which made it difficult for general practitioners and patients to reach an agreement on which treatment to implement.33 As this study was carried out in 2000 and there has been a greater focus on communication skills both in medical schools and during postgraduate medical training in recent years, this may be a lesser issue now.
From another perspective, Schwartz et al looked at what motivates clinicians when they prescribe, other than cost, efficacy and alternative drugs.34 The authors found that clinicians most frequently explained their suboptimal prescribing by attributing it to patient demand, utilizing the placebo effect, or relying on their own clinical experience. However, since evidence-based medicine emerged in the early 1990s, there has been much more of an emphasis on informing clinical decisions with research and guidelines.
There is an increasing move toward focusing on quality of life outcomes for patients with chronic illness, and the same should be true when considering treatment approaches and goals. Chewning and Sleath suggest that asking for a patient’s “priorities and treatment goals could alter both the regimen selection and evaluation of subsequent regimen outcomes,”32 and we feel this is particularly pertinent to therapy selection in IPF.
It has been suggested that evidence-based tools, such as patient decision aids, could facilitate shared decision making between doctors and patients. According to National Health Service England, patient decision aids are:
Designed to help patients understand and consider the pros and cons of possible treatment options and to encourage communication between them and their health care professionals.35
They should feature information that is evidence based and may contain images or diagrams. A systematic review looking at helping patients with treatment choices by using decision aids found that, although they improved knowledge and encouraged patients to be more involved in decision making, they had little effect on satisfaction and a variable effect on decisions. Effects on quality of life were uncertain.36 Unfortunately, none of the studies in this review involved decisions regarding IPF treatment.
A review of discussion forums about IPF at Patient UK,37 one of the most popular online medical information and support web sites, suggests that patients who are taking pirfenidone are experiencing either no or minimal side effects and they are encouraging other users to try the drug as they have noticed a benefit. Clearly, this does not account for the placebo effect. The specific benefits that forum users discussed were that they felt their exercise tolerance had stopped declining and their cough had either improved or resolved. The side effects that were mentioned were tiredness and feeling anywhere from slightly to moderately hungover. Forum users also discussed the need for wearing high-factor sunblock and either staying inside when the sun is brightest or covering up when going outdoors to minimize the risk of photosensitivity, though none of them had experienced this side effect. One of the commenters discussed that, at the age of 70, they do not feel ready to start pirfenidone yet, as “this will mean for me I have to give up all alcohol and wear sun block 50 every day.” Some patients also mentioned that they would be commencing treatment with nintedanib, but none had actually started the therapy yet.
Drugs that should not be used for treatment of IPF
The following drugs have a strong recommendation against their use in IPF according to the most recent ATS/ERS/JRS/ALAT guidelines.14
Anticoagulation
There is evidence that in IPF, there is a procoagulant state both systemically and in the lung tissue, and this led to investigations into the potential benefits of anticoagulation.38 Studies have focused on anticoagulation with vitamin K antagonists and low molecular-weight heparins.
Kubo et al reported an unblinded, randomized trial comparing warfarin plus prednisolone vs prednisolone alone.39 A total of 56 patients were prospectively recruited from five different Japanese hospitals over a 3-year period. In all, 60% of patients were readmitted to hospital during their follow-up period. Those in the anticoagulation arm were switched to low molecular-weight heparins while in hospital. Steroids were discontinued in those admitted with heart failure or bacterial infection, while those admitted with an acute exacerbation of IPF were given high-dose methylprednisolone. The results seemed to support the use of dual therapy with patients in the anticoagulation arm having fewer exacerbations (48% vs 64%) and better survival rates at both 1 year (58% vs 87%) and 3 years (35% vs 63%). However, the exacerbation and mortality rates in the patients treated with prednisolone were very high and are difficult to explain. A larger randomized, controlled trial was subsequently carried out comparing warfarin against placebo.38 The trial was discontinued before the planned 48-week study period was complete due to safety concerns. The mean follow-up at this stage was 28 weeks with the results showing an increased mortality within the warfarin arm (relative risk 4.73; 95% confidence interval 1.42–15.77), with no statistically significant difference in FVC or its rate of decline. For this reason, treatment with warfarin for disease-modifying purposes is not recommended in IPF. Anticoagulation for alternative indications such as atrial fibrillation or venous thrombotic events should follow treatment guidelines independent of underlying IPF,14 especially as the option exists for using the newer direct oral anticoagulants. None of these agents has so far specifically been studied for use in IPF, but the role of coagulation, as well as the profibrotic effect of platelets, is being actively investigated.40
Imatinib
Imatinib is an inhibitor of PDGF, which has been implicated in the pathogenesis of IPF.27 It was initially studied for use in chronic myeloid leukemia and was approved as a treatment for this in the UK in 2001. As such, safety data were already available, and a randomized, double-blind, placebo-controlled trial was carried out to know its potential benefits in IPF.41 The primary outcome was a combined measure of disease progression or death. There was no benefit in either of these at completion of the trial after 96 weeks, hence the recommendation against its use.
Ambrisentan
Increased activity of endothelin A and B receptors has been found in fibrotic lungs.42 Ambrisentan is a selective type A endothelin receptor blocker. The ARTEMIS-IPF trial was a randomized, controlled study comparing ambrisentan with placebo.43 The primary endpoint was time-to-disease progression, defined as death from any cause, hospitalization due to respiratory illness, or deterioration in the lung function as per prespecified criteria. The study was terminated early, as analysis indicated that the probability of meeting the primary endpoint was <5%. Analysis of the acquired data revealed that more patients in the treatment arm showed disease progression as well as a higher number of serious adverse events. As such, its use is not recommended.
Prednisolone/azathioprine/N-acetylcysteine
The 2005 IFIGENIA trial proposed that N-acetylcysteine (NAC), in combination with prednisolone and azathioprine, was beneficial in IPF compared with prednisolone and azathioprine alone.44 The results of the trial suggested that this triple therapy preserved FVC and DLco and was superior to standard therapy. However, there had been a substantial number of treatment changes, withdrawals and deaths during the trial, and there was no placebo group.
PANTHER-IPF was a landmark study evaluating this previously popular combination of drugs (“triple therapy”).45 It was a multicenter, randomized trial evaluating the three-drug regimen against NAC alone, as compared with matched placebos for each of the active therapies. The triple therapy arm was discontinued early due to an excess number of deaths, hospitalizations, and serious adverse events in the combination group. For this reason, clinicians should avoid this combination of drugs in IPF.
The other arms of the trial were allowed to continue, and the results were published in 2014.46 Approximately one-quarter of patients in the NAC vs placebo arms discontinued the study medication. The primary endpoint was change in FVC at 60 weeks, and there was no statistically significant difference between the two groups. There was also no significant difference between the rates of acute exacerbations, hospitalization, or death. Adverse events were similar in both groups, with the only major differences occurring in cardiac disorders (6.8% NAC vs 1.8% placebo) and gastrointestinal disorders (0% NAC vs 4.6% placebo).
A randomized, controlled trial was carried out in Japan with a smaller number of patients comparing inhaled NAC with a control. There was no statistical difference in the primary outcome of change in FVC between the two groups.47
The most recent ATS/ERS/JRS/ALAT guidelines do not recommend the use of NAC, but as there are no data suggesting harm, they make no strong recommendations to discontinue treatment in those who are already taking it.14
Similarly, despite the findings of the PANTHER-IPF trial,45 there is no strong recommendation to stop this combination in patients who have been safely taking it already.14 However, it is important that patients are made aware of this evidence and given the option of whether to continue the treatment or not.
Drugs with a conditional recommendation against use
The following treatments have a conditional recommendation against their use in the treatment of IPF according to the most recent ATS/ERS/JRS/ALAT guidelines.14
Sildenafil
Sildenafil is a phosphodiesterase-5 inhibitor, commonly used in the treatment of pulmonary hypertension. It has been studied for use in the treatment of IPF in two randomized, controlled trials.48,49 The larger of the two trials included 180 patients, but only randomized them to sildenafil or placebo for a period of 12 weeks, at which point all patients continued on sildenafil for a further 12 weeks.48 The primary endpoint was >20% improvement in 6MWD after the first 12 weeks. There was no statistically significant difference between the two groups. There was also no difference in serious adverse events, but those who received sildenafil reported improvement in shortness of breath and quality of life. There were also benefits in DLco and arterial oxygen saturations initially, but ultimately, this was not statistically significant. A subgroup analysis of 119 patients who had documented echocardiogram data found that those with right ventricular systolic dysfunction showed a statistically significant improvement in their 6MWD, but not in other secondary outcomes.
The smaller study also compared sildenafil to placebo, but only included 29 patients who had more mild disease.49 They were followed up for a longer 6-month period. Those with known pulmonary hypertension or right ventricular systolic dysfunction were excluded. There was no statistically significant difference in 6MWD, breathlessness, pulmonary function testing, or arterial oxygen saturation. There were more adverse events in the treatment group, but none of them were considered serious. The NICE guidelines consider the data in these trials to be of low quality.13
Bosentan and macitentan
Both drugs are dual endothelin receptor antagonists acting on type A and B receptors. Two randomized, controlled trials have been conducted comparing bosentan to placebo.50,51 BUILD-1 was a multicenter trial that recruited 158 patients and followed them up for a period of 12 months.50 Of note is the fact that patients with pulmonary hypertension were excluded, which is important because of the well-documented benefits of bosentan in treating idiopathic pulmonary arterial hypertension. The primary endpoint was a change in 6MWD, which revealed that there was no statistically significant difference between the groups. The authors felt that there was a tendency to improvement in disease progression in the treatment arm, despite the lack of any significant difference, and that further investigation into the use of bosentan was warranted. This was particularly true in the subgroup of patients in whom the diagnosis had been confirmed on surgical lung biopsy. BUILD-3 was a larger study with >600 participants,51 all of whom had biopsy-proven IPF in view of the subgroup analysis of BUILD-1. Patients were randomized on a 2:1 ratio, and therefore, ~400 received bosentan and the rest placebo. The primary endpoint was time to IPF worsening, and there was no statistically significant difference in this outcome between the two groups nor were there any statistically significant differences in the secondary outcomes of dyspnea and quality of life.
MUSIC was a randomized trial comparing macitentan with placebo.52 In total, 178 patients were recruited, assigned on a 2:1 ratio, and followed-up for 12 months. The primary endpoint was change in FVC, and again no statistically significant difference was observed between the groups.
Best supportive care
The control of symptoms is an important part of IPF management and includes both pharmacological and nonpharmacological methods. The NICE IPF guideline13 includes a review of the evidence for various methods used to palliate symptoms. The authors identified studies about palliation of breathlessness, using oxygen therapy and managing cough. It is important to consider referral to multidisciplinary palliative care services when advancing IPF is identified, as disease progression is unpredictable. An assessment tool is being developed to aid clinicians in identifying the palliative care needs of patients with ILD.53
Breathlessness
There are no pharmacological studies aimed specifically at treating breathlessness in IPF. Opioids are the most common drug class used palliatively. The majority of studies have been aimed at either chronic obstructive pulmonary disease (COPD) or lung cancer, with positive results. Currow et al studied the benefit of low-dose morphine in patients with chronic lung disease, of which ten had ILD.54 They found that most patients benefited from treatment with 10 mg of sustained-release oral morphine once daily, without experiencing any serious adverse events. There are limitations in that this was only a pharmacovigilance trial, with small sample size and no placebo control or other comparator.
Concerns into respiratory depression secondary to opioid use have led to further evaluations of safety. A 2014 review of the evidence found three randomized, placebo-controlled trials and five prospective studies.55 In total, six trials showed opioids significantly benefited breathlessness. There was no evidence to suggest that they affected oxygen saturations or caused carbon dioxide retention at the low doses which were being used for symptomatic relief.
More recently, a team in the Netherlands investigated the potential benefits of physical training in patients with IPF.56 They also included patients with sarcoidosis-related fibrosis who were reaching the end stages of the disease. It was hypothesized that the two conditions might follow the same natural history. The study had a very small sample size of 24 patients. All partook in a 12-week physical training program with promising results: 13 participants had an improvement in 6MWD of >10%. NICE has included pulmonary rehabilitation as one of its quality standards for improving the care of patients with IPF.57 The authors suggest that this should include “education, exercise training, psychosocial support, and advice on nutrition,” with the aim that these would “contribute to improved health-related quality of life and exercise capacity.” The evidence for pulmonary rehabilitation in IPF is predominantly extrapolated from research into its use in the context of COPD, where it has been found to be of benefit.58
Oxygen
There is very limited evidence looking into the use of oxygen in IPF for either palliative or prognostic benefits. In 2001, Crockett et al carried out a retrospective search for evidence on the use of oxygen in ILD, though not specifically IPF, and found only one randomized, controlled trial that was unpublished.59 A total of 62 patients were followed up, and there was no statistically significant difference in mortality between the group that received oxygen and the control group, with both having mortality rates of ~91% at 3 years. Obi et al looked into the effects of oxygen on patients with various advanced lung diseases.60 Within this relatively small study, 22 patients had IPF. The measured outcome was 6MWD, which showed a mean change of +19.17 m.
The British Thoracic Society guidelines for home oxygen61 suggest the use of long-term oxygen therapy (LTOT) in patients with ILD. Although the quality of supporting evidence is poor, the authors recommend LTOT for patients with ILD who have a resting PaO2 ≤7.3 kPa, or ≤8 kPa and peripheral edema, polycythemia, or features of pulmonary hypertension. The guidelines also cover nocturnal oxygen therapy, and the authors advise that this should not be given to patients with ILD with only nocturnal hypoxemia, who do not fulfill the LTOT criteria. This is based on evidence showing that, although nocturnal oxygen therapy may improve nocturnal hypoxemia, tachycardia, and tachypnea, it does not have a positive impact on sleep quality. If patients are on LTOT and mobile outdoors, the guidelines suggest that they could be offered ambulatory oxygen therapy. Similarly, patients should be offered ambulatory oxygen therapy for use during exercise in a pulmonary rehabilitation program only if they have demonstrated an improvement in exercise endurance. However, a study by Nishiyama et al showed that there was no additional benefit from oxygen over air in terms of exertional dyspnea for patients with IPF who did not have resting hypoxemia.62 Similarly, there have been several studies, which have failed to demonstrate a symptomatic benefit from palliative oxygen over air in patients with breathlessness,63 even in those with hypoxemia,64 which corrected with administration of oxygen. However, these studies may be less applicable in the IPF population, as the patients involved were suffering from malignant disease. The British Thoracic Society guidelines61 suggest that patients with end-stage ILD and severe breathlessness could be considered for palliative oxygen therapy, though the authors would not recommend this in those who are nonhypoxemic or have mild levels of hypoxemia. The guidelines also recommend a handheld fan, which has been shown to result in a significant reduction in breathlessness after only 5 minutes of use when directed across the nose and mouth.65
Cough
Many different theories on the pathogenesis of cough in IPF have been pursued, and its treatment remains problematic. In addition, patients with IPF tend to be in the age group where comorbidities will add to diagnostic and therapeutic difficulties. Harrison reviewed the data available and found limited evidence regarding the successful treatment of cough in IPF.66 This is consistent with the literature review carried out by the authors of the NICE guidelines.13
There are three studies investigating the use of thalidomide: two observational and one randomized, controlled. All three of these had low patient numbers. The randomized trial ran over a 12-week period and was placebo controlled, with a short washout period before treatment cross-over.67 The primary endpoint was quality of life, measured using a questionnaire. There was a statistically significant improvement in scores with thalidomide. NICE guidelines recommend that, if thalidomide is being considered in the management of cough, this decision should only be made by respiratory physicians who specialize in ILD, as its provision would be unlicensed.13 A small study in 2003 suggested that high-dose prednisolone might cause a reduction in cough sensitivity in patients with IPF,68 though this would require further research prior to being recommended as a treatment. Cough in patients with IPF, as in other respiratory diseases, could well be the result of cough hypersensitivity syndrome69 or gastroesophageal reflux disease (GORD).70
Gastroesophageal reflux disease
Studies have shown that patients with IPF have a very high incidence of acid and nonacid reflux when compared to controls.71 Despite 24-hour pH monitoring and esophageal manometry revealing that 87% of a cohort of patients in an extended prospective study had abnormal acid reflux, only half of the subjects reported symptoms typically associated with GORD.72 There is also a suggestion that GORD may play a role in the pathogenesis and progression of IPF.73 This requires further research, but in the meantime, it is recommended that all patients with symptoms of GORD should be treated with proton-pump inhibitor therapy.74 It is our practice to look for clinical evidence of reflux as a cause of cough in patients with IPF and to offer trials of antireflux treatment if appropriate.
Conclusion
We have discussed the pathophysiology and diagnosis of IPF, the various drug treatments that are currently used to treat the disease as well as those which were used previously but are no longer recommended, the importance of concurrent best supportive care, and some of the aspects of how patients and doctors make treatment decisions. It is important for clinicians to be aware of the different treatments available for the management of IPF and the factors that may influence how patients choose between them.
Acknowledgment
The authors are grateful to Dr Michael Crooks for his helpful comments.
Disclosure
SPH has received financial and non-financial support for research and to attend conferences from Boehringer Ingelheim and Roche. MAT and RDR report no conflicts of interest.
References
Gribbin J, Hubbard RB, Jeune IL, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61(11):980–985. | ||
Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011; 183(6):788–824. | ||
Navaratnam V, Fleming KM, West J, et al. The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax. 2011;66(6):462–467. | ||
Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134(2):136–151. | ||
Alder JK, Chen JJ-L, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008; 105(35):13051–13056. | ||
Stock CJ, Sato H, Fonseca C, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax. 2013;68(5):436–441. | ||
Loveman E, Copley V, Colquitt J, et al. The effectiveness and cost-effectiveness of treatments for idiopathic pulmonary fibrosis: systematic review, network meta-analysis and health economic evaluation. BMC Pharmacol Toxicol. 2014;15(1):63. | ||
Wells AU, Hirani N. Interstitial lung disease guideline. Thorax. 2008;63(suppl 5):v1–v58. | ||
American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med. 2002;165(2):277–304. | ||
Wuyts WA, Cavazza A, Rossi G, Bonella F, Sverzellati N, Spagnolo P. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur Respir Rev. 2014;23(133):308–319. | ||
Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med. 2005;171(6):639–644. | ||
Hallstrand TS, Boitano LJ, Johnson WC, Spada CA, Hayes JG, Raghu G. The timed walk test as a measure of severity and survival in idiopathic pulmonary fibrosis. Eur Respir J. 2005;25(1):96–103. | ||
National Clinical Guideline C. National Institute for Health and Clinical Excellence: guidance. Diagnosis and Management of Suspected Idiopathic Pulmonary Fibrosis: Idiopathic Pulmonary Fibrosis. London: Royal College of Physicians (UK) National Clinical Guideline Centre; 2013:1–30. | ||
Raghu G, Rochwerg B, Zhang Y, et al; American Thoracic Society; European Respiratory society; Japanese Respiratory Society; Latin American Thoracic Association. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192(2):e3–e19. | ||
FDA U.S. Food and Drug Administration. [webpage on the Internet]. FDA Approves Esbriet to Treat Idiopathic Pulmonary Fibrosis. 2014. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm418991.htm. Accessed December 16, 2015. | ||
Oku H, Shimizu T, Kawabata T, et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol. 2008;590(1–3):400–408. | ||
Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779): 1760–1769. | ||
Kreuter M. Pirfenidone: an update on clinical trial data and insights from everyday practice. Eur Respir Rev. 2014;23(131):111–117. | ||
Rief W, Avorn J, Barsky AJ. Medication-attributed adverse effects in placebo groups: implications for assessment of adverse effects. Arch Intern Med. 2006;166(2):155–160. | ||
Costabel U, Albera C, Fagan E, et al. Long-term safety of pirfenidone in RECAP, an open-label extension study in patients with idiopathic pulmonary fibrosis, interim results. Eur Respir J. 2014;44(suppl):58. | ||
Gan Y, Herzog EL, Gomer RH. Pirfenidone treatment of idiopathic pulmonary fibrosis. Ther Clin Risk Manag. 2011;7:39–47. | ||
Chaudhuri N, Duck A, Frank R, Holme J, Leonard C. Real world experiences: pirfenidone is well tolerated in patients with idiopathic pulmonary fibrosis. Respir Med. 2014;108(1):224–226. | ||
King TE, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–2092. | ||
National Institute for Health and Care Excellence. [webpage on the Internet]. Pirfenidone for Treating Idiopathic Pulmonary Fibrosis Technology Appraisal Guidance. 2013. Available from: https://www.nice.org.uk/guidance/ta282. Accessed December 16, 2015. | ||
Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. | ||
Simler NR, Brenchley PE, Horrocks AW, Greaves SM, Hasleton PS, Egan JJ. Angiogenic cytokines in patients with idiopathic interstitial pneumonia. Thorax. 2004;59(7):581–585. | ||
Antoniades HN, Bravo MA, Avila RE, et al. Platelet-derived growth factor in idiopathic pulmonary fibrosis. J Clin Invest. 1990;86(4):1055–1064. | ||
FDA U.S. Food and Drug Administration. [webpage on the Internet]. FDA Approves Ofev to Treat Idiopathic Pulmonary Fibrosis. 2014. Available from: http://www.fda.gov/NewsEvents/Newsroom/Press Announcements/ucm418994.htm. Accessed December 16, 2015. | ||
Scottish Medicines Consortium. [webpage on the Internet]. Nintedanib. 2015. Available from: https://www.scottishmedicines.org.uk/SMC_Advice/Advice/1076_15_nintedanib_Ofev/nintedanib_Ofev. Accessed December 16, 2015. | ||
Thompson SC, Pitts JS, Schwankovsky L. Preferences for involvement in medical decision-making: situational and demographic influences. Patient Educ Couns. 1993;22(3):133–140. | ||
Haynes RB, Devereaux PJ, Guyatt GH. Clinical expertise in the era of evidence-based medicine and patient choice. Evidence Based Med. 2002;7(2):36–38. | ||
Chewning B, Sleath B. Medication decision-making and management: a client-centered model. Soc Sci Med. 1996;42(3):389–398. | ||
Stevenson FA, Barry CA, Britten N, Barber N, Bradley CP. Doctor–patient communication about drugs: the evidence for shared decision making. Soc Sci Med. 2000;50(6):829–840. | ||
Schwartz RK, Soumerai SB, Avorn J. Physician motivations for nonscientific drug prescribing. Soc Sci Med. 1989;28(6):577–582. | ||
NHS England. [webpage on the Internet]. Patient Decision Aids. 2015. Available from: http://www.england.nhs.uk/ourwork/pe/sdm/tools-sdm/pda/. Accessed December 16, 2015. | ||
Connor AM, Rostom A, Fiset V, et al. Decision aids for patients facing health treatment or screening decisions: systematic review. BMJ. 1999;319(7212):731–734. | ||
Patient. [webpage on the Internet]. Pulmonary Fibrosis. 2015. Available from: http://patient.info/forums/discuss/browse/pulmonary-fibrosis-1760. Accessed December 16, 2015. | ||
Noth I, Anstrom KJ, Calvert SB, et al; Idiopathic Pulmonary Fibrosis Clinical Research Network (IPFnet). A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186(1):88–95. | ||
Kubo H, Nakayama K, Yanai M, et al. Anticoagulant therapy for idiopathic pulmonary fibrosis. Chest. 2005;128(3):1475–1482. | ||
Crooks MG, Hart SP. Coagulation and anticoagulation in idiopathic pulmonary fibrosis. Eur Respir Rev. 2015;24(137):392–399. | ||
Daniels CE, Lasky JA, Limper AH, et al; Imatinib-IPF Study Investigators. Imatinib treatment for idiopathic pulmonary fibrosis: randomized placebo-controlled trial results. Am J Respir Crit Care Med. 2010;181(6):604–610. | ||
Abraham DJ, Vancheeswaran R, Dashwood MR, et al. Increased levels of endothelin-1 and differential endothelin type A and B receptor expression in scleroderma-associated fibrotic lung disease. Am J Pathol. 1997;151(3):831–841. | ||
Raghu G, Behr J, Brown KK, et al; ARTEMIS-IPF Investigators. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158(9):641–649. | ||
Demedts M, Behr J, Buhl R, et al; IFIGENIA Study Group. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005; 353(21):2229–2242. | ||
Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu G, Anstrom KJ, et al. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968–1977. | ||
Idiopathic Pulmonary Fibrosis Clinical Research Network, Martinez FJ, de Andrade JA, et al. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2093–2101. | ||
Homma S, Azuma A, Taniguchi H, et al; Japan NAC Clinical Study Group. Efficacy of inhaled N-acetylcysteine monotherapy in patients with early stage idiopathic pulmonary fibrosis. Respirology. 2012;17(3):467–477. | ||
Idiopathic Pulmonary Fibrosis Clinical Research Network, Zisman DA, Schwarz M, et al. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010;363(7):620–628. | ||
Jackson RM, Glassberg MK, Ramos CF, Bejarano PA, Butrous G, Gómez-Marín O. Sildenafil therapy and exercise tolerance in idiopathic pulmonary fibrosis. Lung. 2010;188(2):115–123. | ||
King TE Jr, Behr J, Brown KK, et al. BUILD-1: a randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177(1):75–81. | ||
King TE Jr, Brown KK, Raghu G, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(1):92–99. | ||
Raghu G, Million-Rousseau R, Morganti A, Perchenet L, Behr J; MUSIC Study Group. Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomised controlled MUSIC trial. Eur Respir J. 2013;42(6):1622–1632. | ||
Reigada C, Bajwah S, Ross J, et al. Adaptation, face and content validation of a palliative care needs assessment tool for people with interstitial lung disease. BMJ Supportive Palliative Care. 2015;5(1): 111–112. | ||
Currow DC, McDonald C, Oaten S, et al. Once-daily opioids for chronic dyspnea: a dose increment and pharmacovigilance study. J Pain Symptom Manage. 2011;42(3):388–399. | ||
Lopez-Saca JM, Centeno C. Opioids prescription for symptoms relief and the impact on respiratory function: updated evidence. Curr Opin Support Palliat Care. 2014;8(4):383–390. | ||
Strookappe B, Elfferich M, Swigris J, et al. Benefits of physical training in patients with idiopathic or end-stage sarcoidosis-related pulmonary fibrosis: a pilot study. Sarcoidosis Vasc Diffuse Lung Dis. 2015;32(1):43–52. | ||
National Institute for Health and Care Excellence. [webpage on the Internet]. Idiopathic Pulmonary Fibrosis NICE Quality Standard. 2015. Available from: http://www.nice.org.uk/guidance/qs79. Accessed December 16, 2015. | ||
Lacasse Y, Goldstein R, Lasserson TJ, Martin S. Pulmonary rehabilitation for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2006;4:Cd003793. | ||
Crockett AJ, Cranston JM, Antic N. Domiciliary oxygen for interstitial lung disease. Cochrane Database Syst Rev. 2001;(3):CD002883. | ||
Obi I, Shlobin OA, Weir N, Ahmad S, Barnett S, Nathan SD. Effects of oxygen therapy in patients with advanced lung diseases. B97. Adjunctive Therapies for Pulmonary Rehabilitation. American Thoracic Society; 2010:A3764–A3764. | ||
Hardinge M, Annandale J, Bourne S, et al; British Thoracic Society Home Oxygen Guideline Development Group; British Thoracic Society Standards of Care Committee. British Thoracic Society guidelines for home oxygen use in adults. Thorax. 2015;70(suppl 1):i1–i43. | ||
Nishiyama O, Miyajima H, Fukai Y, et al. Effect of ambulatory oxygen on exertional dyspnea in IPF patients without resting hypoxemia. Respir Med. 2013;107(8):1241–1246. | ||
Philip J, Gold M, Milner A, Di Iulio J, Miller B, Spruyt O. A randomized, double-blind, crossover trial of the effect of oxygen on dyspnea in patients with advanced cancer. J Pain Symptom Manage. 2006;32(6):541–550. | ||
Uronis HE, Currow DC, McCrory DC, Samsa GP, Abernethy AP. Oxygen for relief of dyspnoea in mildly- or non-hypoxaemic patients with cancer: a systematic review and meta-analysis. Br J Cancer. 2008;98(2):294–299. | ||
Galbraith S, Fagan P, Perkins P, Lynch A, Booth S. Does the use of a handheld fan improve chronic dyspnea? A randomized, controlled, crossover trial. J Pain Symptom Manage. 2010;39(5):831–838. | ||
Harrison NK. Cough, sarcoidosis and idiopathic pulmonary fibrosis: raw nerves and bad vibrations. Cough. 2013;9(1):9. | ||
Horton MR, Santopietro V, Mathew L, et al. Thalidomide for the treatment of cough in idiopathic pulmonary fibrosis: a randomized trial. Ann Intern Med. 2012;157(6):398–406. | ||
Hope-Gill BDM, Hilldrup S, Davies C, Newton RP, Harrison NK. A study of the cough reflex in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003;168(8):995–1002. | ||
Morice AH. Chronic cough hypersensitivity syndrome. Cough. 2013; 9(1):14. | ||
Molyneux ID, Morice AH. Airway reflux, cough and respiratory disease. Ther Adv Chronic Dis. 2011;2(4):237–248. | ||
Tobin RW, Pope CE 2nd, Pellegrini CA, Emond MJ, Sillery J, Raghu G. Increased prevalence of gastroesophageal reflux in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;158(6):1804–1808. | ||
Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J. 2006;27(1):136–142. | ||
Raghu G, Yang ST, Spada C, Hayes J, Pellegrini CA. Sole treatment of acid gastroesophageal reflux in idiopathic pulmonary fibrosis: a case series. Chest. 2006;129(3):794–800. | ||
Bradley B, Branley HM, Egan JJ, et al; British Thoracic Society Interstitial Lung Disease Guideline Group, British Thoracic Society Standards of Care Committee; Thoracic Society of Australia; New Zealand Thoracic Society; Irish Thoracic Society. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax. 2008;63(suppl 5):v1–v58. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.