")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Pathophysiology of hypophosphatasia and the potential role of asfotase alfa
Authors Orimo H
Received 23 February 2016
Accepted for publication 18 April 2016
Published 17 May 2016 Volume 2016:12 Pages 777—786
DOI https://doi.org/10.2147/TCRM.S87956
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Garry Walsh
Hideo Orimo
Division of Metabolism and Nutrition, Department of Biochemistry and Molecular Biology, Nippon Medical School, Tokyo, Japan
Abstract: Hypophosphatasia (HPP) is an inherited systemic bone disease that is characterized by bone hypomineralization. HPP is classified into six forms according to the age of onset and severity as perinatal (lethal), perinatal benign, infantile, childhood, adult, and odontohypophosphatasia. The causative gene of the disease is the ALPL gene that encodes tissue-nonspecific alkaline phosphatase (TNAP). TNAP is expressed ubiquitously, and its physiological role is apparent in bone mineralization. A defect in bone mineralization can manifest in several ways, including rickets or osteomalacia in HPP patients. Patients with severe forms suffer from respiratory failure because of hypoplastic chest, which is the main cause of death. They sometimes present with seizures due to a defect in vitamin B6 metabolism resulting from the lack of alkaline phosphatase activity in neuronal cells, which is also lethal. Patients with a mild form of the disease exhibit rickets or osteomalacia and a functional defect of exercise. Odontohypophosphatasia shows only dental manifestations. To date, 302 mutations in the ALPL gene have been reported, mainly single-nucleotide substitutions, and the relationships between phenotype and genotype have been partially elucidated. An established treatment for HPP was not available until the recent development of enzyme replacement therapy. The first successful enzyme replacement therapy in model mice using a modified human TNAP protein (asfotase alfa) was reported in 2008, and subsequently success in patients with severe form of the disease was reported in 2012. In 2015, asfotase alfa was approved in Japan in July, followed by in the EU and Canada in August, and then by the US Food and Drug Administration in the USA in October. It is expected that therapy with asfotase alfa will drastically change treatments and prognosis of HPP.
Keywords: hypophosphatasia, alkaline phosphatase, mutation, asfotase alfa, rickets, respiratory failure
Introduction
Hypophosphatasia (HPP; OMIM #241500, 241510, 146300), which was first reported by the Canadian pediatrician John Campbell Rathbun in 1948, is a systemic bone disease caused by the deficiency of tissue-nonspecific alkaline phosphatase (TNAP).1,2 HPP is classified into six forms: perinatal (lethal), perinatal benign, infantile, childhood, adult, and odontohypophosphatasia, according to the age of onset and severity.2,3 The perinatal form occurs in utero and has the most severe manifestations; it is lethal for patients, and these patients are usually stillborn or die during the early postnatal period. Recently, however, a group of patients with such early manifestation of HPP that have good prognosis was reported, and this group is now classified as a perinatal benign form.4 The infantile form occurs before 6 months of age, and the phenotypes are generally less severe than the perinatal form. The childhood form occurs after 6 months of age and shows milder manifestations. Their prognosis for life is good, although the patients usually exhibit functional musculoskeletal disorder. The adult form occurs at middle age. Odontohypophosphatasia is only a dental disorder without skeletal manifestations. The symptoms of HPP vary depending on the form involved. Except for odontohypophosphatasia, a common feature is rickets or osteomalacia leading to bone deformity and disorder of musculoskeletal function.2 These symptoms are due to systemic hypomineralization of bone. Severe forms (perinatal and infantile forms) also show respiratory failure as a result of hypoplasia of the chest. Patients with severe forms sometimes suffer from seizures because of a defect of γ-aminobutyric acid (GABA) in neuronal cells in the brain. Respiratory failure and seizures are the main causes of death. No established therapy has been available until the recent success of enzyme replacement therapy (ERT) using asfotase alfa.
Alkaline phosphatase
Tissue-nonspecific alkaline phosphatase
Alkaline phosphatases (orthophosphoric-monoester phosphohydrolase [alkaline optimum], EC 3.1.3.1) are membrane-bound ectoenzymes that hydrolyze monophosphate esters between pH 8 and 10.5 Alkaline phosphatases exist in organisms ranging from bacteria to mammals, and DNA and amino acid sequences of these enzymes are fairly conserved:5 ~57.8% of the amino acids that make up the human TNAP sequence are conserved among mammalian alkaline phosphatases.6 Human TNAP is one of four isoenzymes of human alkaline phosphatase. The other three isozymes, namely, intestinal, placental, and germ cell (placental-like) alkaline phosphatases, are tissue-specific, whereas TNAP is expressed ubiquitously, with an especially marked expression reported in the liver, bone, kidney, neuronal cells, and neutrophils.5,7 TNAP is, therefore, also known as the liver/bone/kidney (LBK) alkaline phosphatase, although no precise physiological roles in the liver and kidney are known. TNAP is encoded by a gene on the short arm of chromosome 1 (1p36.1–34) and is spread over 50 kb, whereas the genes for the tissue-specific alkaline phosphatases are clustered on the long arm of chromosome 2 (2q34–37).8–13 The TNAP gene (ALPL) is ~1.5 kb in length and consists of 12 exons, of which exons 2–12 are coding exons, and there are also two alternative noncoding exons 1 (1B and 1L).9,14 Functional TNAP enzyme is considered to be existed as a homodimer.5 The molecular weight of each monomer is ~80 kDa and is linked to the outer membrane of the cells via a glycosylphosphatidylinositol (GPI) anchor.5 After the TNAP peptide is synthesized as a native protein with a molecular weight of 66 kDa, carbohydrate chains are added as O- and N-linked sugar chains in the endoplasmic reticulum, and the modified protein is then processed in the Golgi apparatus and is eventually localized on the outer membrane via a GPI anchor.5,15 There are also isoforms of TNAP itself that are tissue-specific (liver-specific, bone-specific, etc). These isoforms have different sugar chains, mainly because of different O-linked glycosylation, although they all have the same peptide sequence.5,16 TNAP isoforms and isoenzymes can be distinguished by through electrophoresis and antibody, and this can be used for diagnosis to determine the tissue origin of the enzyme, which can suggest defective organs.
Structure of alkaline phosphatase
To express enzymatic activity, TNAP needs two Zn2+, one Mg2+, and one Ca2+ ion as cofactors.5,17 The amino acid sequence of the human TNAP molecule is 57% identical and 74% homologous with the human placental alkaline phosphatase (PLAP) molecule. The three-dimensional (3D) structure of TNAP based on the crystallography of the human TNAP protein has not been elucidated. However, the 3D structure of human TNAP can be obtained using a simulation model18 (Figure 1) based on human PLAP19 and rat-intestinal alkaline phosphatase,20 whose 3D structures have been solved. The core structure of the PLAP protein consists of an extended β-sheet and flanking α-helices. PLAP also has an N-terminal α-helix. The active site comprises a catalytic serine residue (S92) and metal ion-binding sites (two Zn2+-binding sites and a Mg2+-binding site).19 Mammalian alkaline phosphatases have a characteristic crown domain of an interfacial flexible loop consisting of 60 residues, which may interact with extracellular proteins, as well as a long N-terminal α-helix and a Ca2+-binding domain.18–21 Although basic structures of the alkaline phosphatase proteins are well conserved, the crown domain is lacking in bacterial (Escherichia coli) alkaline phosphatase.22 In the C-terminus of the PLAP protein, 29 hydrophobic amino acid residues are removed after translation in the endoplasmic reticulum and the GPI anchor is then added to an aspartate residue (D484).23 The GPI anchor consists of an ethanolamine phosphate, three residues of mannose, a glucosamine, and a phosphatidylinositol.
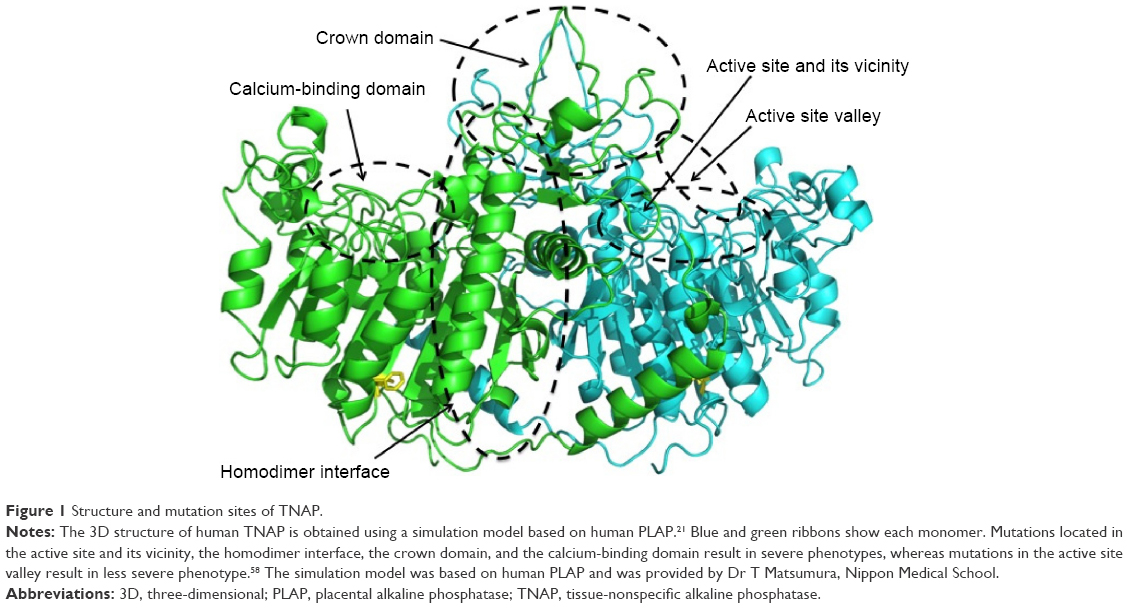 | Figure 1 Structure and mutation sites of TNAP. |
Nomenclature of the ALPL gene and protein
Nucleotides are numbered by reference to the first nucleotide (+1), which corresponds to the A of the ATG initiation codon,9,24 and are represented by a c.number. The standardized nomenclature of amino acid number follows the HGVS recommendations, in which the first codon is the ATG initiation codon, and amino acid numbers are represented by a p.number.25,26
Alkaline phosphatase and mineralization
TNAP is essential for tissue biomineralization (Figure 2).27 Mineralization takes place in two distinct processes.28 Hypertrophic chondrocytes, osteoblasts, and odontoblasts bud matrix vesicles when mineralization begins. Matrix vesicles are membrane-invested vesicles of 50–200 nm in diameter and are rich in annexins A2, A5, and A6, and in Ca2+-ATPase, TNAP, nucleotide pyrophosphate phosphodiesterase 1 (NPP1, formerly known as PC-1), Pit1 (a sodium–phosphate [Na/Pi] cotransporter), and PHOSPHO1.28 The first step of the mineralization process occurs within the matrix vesicles, in which hydroxyapatite (Ca10(PO4)6(OH)2) crystals are formed.28 Phosphate is derived from membrane phospholipids, which are hydrolyzed by phospholipase C to produce phosphocholine and phosphoethanolamine.29 These phosphocompounds are hydrolyzed by PHOSPHO1, a cytosolic phosphatase that is abundant in the matrix vesicles, to yield inorganic phosphate (Pi).30,31 Another source of Pi in the matrix vesicles is Pi that is transported through the Na/Pi cotransporter Pit1 that is also abundant on the matrix vesicle membrane.28 Calcium is incorporated into the matrix vesicles through annexin Ca2+ channels that consist of annexins A2, A5, and A6.28 Developing hydroxyapatite crystals then penetrate the matrix vesicle membrane, are elongated in the extracellular space, and eventually deposit in the spaces between collagen fibrils to complete extracellular matrix mineralization.28 The concentration ratio of Pi to inorganic pyrophosphate (PPi) in the extracellular matrix is crucial in the second step of mineralization because PPi is an inhibitor of hydroxyapatite formation.32 Two mechanisms are used for PPi formation.27,33 PPi is formed in the extracellular matrix from ATP by the matrix vesicle membrane enzyme NPP1.34 PPi is also provided through the PPi transporter ANKH (a homologue of the mouse progressive ankylosis gene product) from the cytoplasm, in which PPi is routinely formed by cellular metabolism.35 ANKH is distributed on the plasma membrane of hypertrophic chondrocytes and osteoblasts.35 TNAP on the membrane of the matrix vesicles hydrolyzes PPi and yields Pi, thereby reducing the levels of the PPi inhibitor and promoting hydroxyapatite formation. The importance of the Pi/PPi ratio has been proved by knockout (KO) mice of the respective genes as described in the “KO mice as models of HPP” section. TNAP KO mice (Akp2−/−) show hypomineralization (as will be described). NPP1 KO mice (Enpp1−/−) and naturally generated mutant mice (tiptoe walking mice; ttw/ttw) show calcification of articular cartilage and aorta.36,37 The human ENPP1 gene mutation causes idiopathic infantile arterial calcification.38 Naturally occurring progressive ankylosis (ank/ank) mice present hypercalcification of vertebrae and joints, leading to progressive ankylosis.39 Double KO animals of TNAP (Akp2−/−) and NPP1 (Enpp1−/−) or ANK (ank/ank) show a seemingly normal skeleton, indicating that TNAP, NPP1, and ANK(H) regulate mineralization in concert.40,41
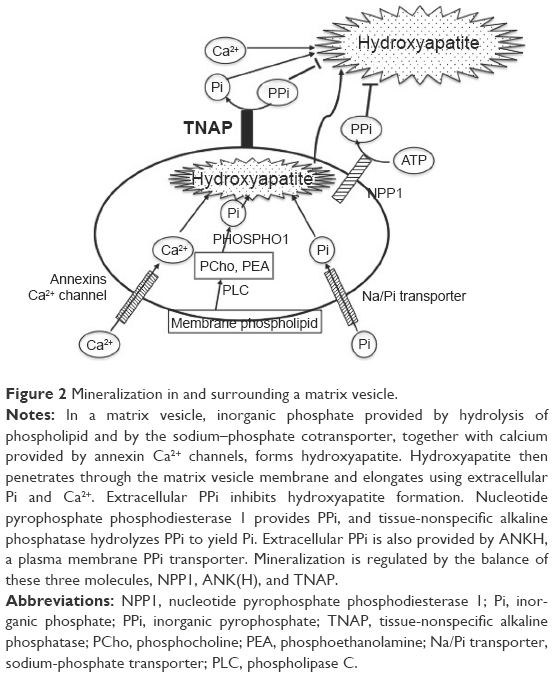 | Figure 2 Mineralization in and surrounding a matrix vesicle. |
Pathophysiology of HPP
Bone mineralization
The pathophysiology of HPP is derived from the defective function of TNAP. TNAP deficiency causes not only defective enzymatic activity but also defective mineralizing activity. The common feature of HPP is hypomineralization of bones and teeth. In HPP patients, extracellular hydroxyapatite crystals are reduced and mineralization is impaired.42 However, mineralization does occur within the matrix vesicles formed in these patients42 because PHOSPHO1 is expressed normally in the patients’ matrix vesicles. This has been proven using double KO mice of TNAP and PHOSPHO1, which show little formation of hydroxyapatite either in the matrix vesicles or in the extracellular space.43 HPP patients exhibit an increase in osteoid tissue that contains abundant nonmineralized bone extracellular matrix without hydroxyapatite crystals, leading to rickets and osteomalacia.2 Mineralization can be assessed by relatively easy procedures. An expression plasmid is introduced into U2OS osteoblastic cells that express trace levels of TNAP enzymatic activity, and then the cells are cultured with a mineralization medium that contains β-glycerophosphate as an artificial TNAP substrate with or without ascorbic acid. The cells in which the wild-type TNAP cDNA is introduced mineralize around the cells within 5 days, whereas cells in which mutant cDNAs are introduced show less mineralization.44 Mineralizing ability parallels TNAP enzymatic activity in this assay, and therefore the level of TNAP activity can reflect phenotype, especially with regard to bone mineralization.44
Central nervous system and seizures
Another physiological role of TNAP is associated with neurotransmitter synthesis in the central nervous system.45 One of the physiological substrates of TNAP is pyridoxal 5′-phosphate, a derivative of vitamin B6, which is necessary for the biosynthesis of GABA, a cofactor of glutamic acid carboxylase in neuronal cells.45,46 GABA is known to act as a repressive neurotransmitter.46 For pyridoxal 5′-phosphate entry into cells, the phosphate moiety has to be first released, and then pyridoxal is incorporated into the cells, followed by rephosphorylation within the cells.47 It has been confirmed in fibroblasts from normal and HPP patients that TNAP hydrolyzes pyridoxal 5′-phosphate to release phosphate outside the cell membrane,47 and therefore a similar mechanism is presumed to operate in the neuronal cell membrane.45 Failure of this role of TNAP leads to epileptic seizures in HPP patients because reduced GABA in the neuronal cells results in hyperactivity of the central nervous system.48,49 However, although almost all patients with seizures are pyridoxine responsive, the etiology of seizures other than deteriorated vitamin B6 metabolism may be considered, such as high intracranial pressure because of craniosynostosis, hypercalcinemia, and hypoxia.2
Inheritance of HPP
Although HPP is inherited as an autosomal recessive trait, autosomal dominant cases have been reported in patients with milder HPP.50,51 The dominant negative effect of severe alleles accounts for almost all autosomal dominant cases.52 Severe forms are mostly compound heterozygotes of severe alleles, and residual activity is important to decide clinical forms.53 Infrequently, however, siblings with different phenotypes have been reported;54,55 this phenomena may raise the suspicion of epigenetic factors being involved in the expression of phenotypes.
Mutations in alkaline phosphatase
The first patient reported by Rathbun was suffering from an infantile form of HPP. Fifty years later, his mutations were identified using the surviving parents’ DNA as a compound heterozygote of p.A114T (c.340G > A) and p.D294A (c.881A > C).56 Mutations found in HPP patients are scattered throughout the molecule, and 302 mutations, mainly missense mutations, have been described to date in the ALPL gene.57 In addition, although some deletions, insertions, and splice mutations have been reported, no large deletion or insertion of the ALPL gene area was reported.57 Regarding the relationships between genotype and phenotype, mutation sites are classified by the protein regions in which they occur.58 Mutations located in the active site and in its vicinity, the homodimer interface, the crown domain, and the calcium-binding domain are mainly associated with a severe phenotype, whereas mutations in the active site valley show less severe phenotypes (Figure 1). The phenotypic severity of mutations of specific residues depends on the type of mutation at each residue. For example, although p.F327 is a frequently mutated site, it is a moderate allele,59 but p.F327del is a severe allele.60 The difference in the severity of the phenotype probably depends on the stability of the molecule. As the phenylalanine residue at 327 is in a β-sheet that consists of 10 core β-sheets near the Mg2+-binding site, its deletion may result in deterioration of the structure, resulting in a severe phenotype. Mutations in the C-terminus also result in a severe phenotype. Deletion of nucleotide 1559 (c.1559del) causes a frameshift mutation, which adds 80 amino acids to the C-terminus, resulting in the failure of GPI-anchor formation.61 The mutant protein cannot be processed properly in the cells, aggregates in the Golgi apparatus, and is then treated in the proteasome; therefore, this deletion results in almost no enzymatic activity and is a severe allele.62 Nevertheless, most HPP patients are compound heterozygotes, indicating that interaction of the two mutated alleles may determine the phenotype.63,64 The relationships between genotype and phenotype have been previously discussed.65 Generally, phenotypes are related to residual enzymatic activity and the mineralizing ability of the cells that express the mutant enzymes.44 However, the relationship between genotype and phenotype is not so simple, and more research is needed to clarify this relationship.
Clinical features of HPP
Clinical features of HPP vary depending on the form.3 The perinatal (lethal) form is the most severe form, which shows shortened and deformed limbs during gestation and at birth.2 The cranium presents as membranous and the ribs are hypomineralized and deformed, resulting in respiratory failure after birth. Epileptic seizures sometimes occur. Respiratory failure and/or seizures precipitate a lethal course of the disease. The infantile form also displays rickets and deformity of limbs and ribs because of hypomineralization of bones. In addition, craniosynostosis often occurs. Patients fail to thrive and often need respiratory aid, and the main cause of death is respiratory failure; the elongation of lifespan recently observed can be accounted for by developments in respiratory management. Hypercalcinemia and hypercalciuria are often seen, and the latter causes nephrocalcinosis. Patients with childhood form of the disease exhibit a wide range of clinical findings,66 including deformity of limbs, delayed walking, and waddling gait. Radiography shows characteristic focal bone defect at the ends of long bones as a tongue-like radiolucent projection from the rachitic growth plate into the metaphysis.2 These patients also show premature loss of deciduous teeth because of disturbed cementum formation.67 Craniosynostosis occurs in some cases, leading to raised intracranial pressure.68 Patients also exhibit problems in doing exercise because of not only rickets but also muscle weakness and functional muscular failure. The adult form occurs during middle age. Although the natural history of the adult form has not been well characterized,69 patients sometimes have a history of rickets and/or premature exfoliation of deciduous teeth. In the adult form, osteomalacia develops with pain associated with often recurrent metatarsal stress fractures.2 In some patients, calcium pyrophosphate dehydrate crystals are deposited on articular cartilage due to an increase in endogenous levels of PPi.2 Odontohypophosphatasia affects only dental tissue without any manifestations of rickets or osteomalacia, but should present with a low serum alkaline phosphatase level. The perinatal benign form is a newly recognized form in which patients manifest symptoms in utero, but this form is nonlethal.4
HPP can be diagnosed using ultrasonography and radiography in utero for the perinatal form.2,3 In any case, serum alkaline phosphatase activity is strong evidence of the disease. In some cases, carriers show subnormal enzymatic activity. In addition, some natural TNAP substrates have diagnostic value. Alkaline phosphatase activity is measured in the clinical laboratory using the artificial substrate p-nitrophenylphosphate. There is some controversy regarding natural substrates of TNAP;2 PPi and pyridoxal 5′-phosphate are natural substrates (as already mentioned). The status of phosphoethanolamine is controversial; it is probably not a natural TNAP substrate, but it does have diagnostic value because the measurement of urine phosphoethanolamine is easier, and so is more widely used than that of serum PPi.2 HPP patients show higher serum PPi and urine phosphoethanolamine; low alkaline phosphatase activity with high PPi or phosphoethanolamine indicates strong evidence of HPP. In some milder cases, however, an increase in phosphoethanolamine is not shown, and, in some cases, phosphoethanolamine is slightly elevated in carriers.70
Prevalence of HPP
The prevalence of HPP was estimated to be 1 in 100,000 live births in the Toronto area in Canada.71 It is known to be as high as 1 in 2,500 for Mennonite families in Manitoba, Canada.72 For severe forms, prevalence has been estimated at 1 in 300,000 in Europe.73 A recent report from Japan estimated a prevalence of 1 in 450,000 for patients who have the particular mutant allele c.1559delT,70 which is possessed by 46.8% of Japanese patients with HPP.74
A possible role of asfotase alfa
KO mice as models of HPP
KO mice of the murine TNAP gene (Akp2) were created45,75 and were confirmed as a suitable model of HPP.76 The KO mice show hypomineralization of bone, rickets and osteomalacia, failure to thrive, abnormal dental manifestations,77 and seizures.45 The latter finding is essential, and they die within 20 days after birth. Although these mice mimic the manifestations of severe forms of HPP patients, one difference between them is the presence of epileptic seizures, which HPP patients rarely exhibit but almost all KO mice show.
History of treatment of HPP
Trials of treatment of HPP have been performed and have failed.5 Artificial respiration is used for respiratory failure. Other symptomatic treatments including dietary therapy (calcium restriction, vitamin D supplementation, etc) are also used.2,3 As a fundamental therapy, ERT has been attempted. Symptoms of female patients with mild form of the disease are known to improve when they are pregnant because of the activity of PLAP.5 Therefore, infusion of PLAP into an infant with severe form of HPP was attempted, but no improvement was observed.5 TNAP infusion was also attempted using the sera of patients with Paget disease who exhibit hyperphosphatemia, but almost no improvement was observed.78 On the other hand, mesenchymal stem-cell transplantation has shown slight improvement.79 Those results suggested that cells expressing TNAP on the cell surface is necessary for successful treatment. In 2008, successful treatment of TNAP null mice (Akp2−/−) with human TNAP bioengineered with the C-terminus extended by the Fc region of human IgG and a deca-aspartate sequence was reported.80 In this experiment, TNAP was cut at the C-terminal membrane-bound region to yield the soluble type but without a loss of enzymatic activity. The Fc region was added in order to purify the enzyme by a one-step procedure. Given that deca-aspartate was proven to bind to hydroxyapatite with high affinity,81 it was used for bone targeting (Figure 3). The modified enzyme also prevents dental defects in Akp2−/− mice.82 As Akp2−/− is a good model for perinatal form HPP,76 this report gave hope of ERT to the patients. A clinical trial was conducted, and the first results of the trials in the USA were reported in 2012.83 The recruited eleven patients (five perinatal and six infantile) were treated with 2 mg/kg of ENB-0040 (asfotase alfa; 40 mg/mL) provided by Enobia Pharma, Inc. (San Carlos, CA, USA) as a single intravenous infusion followed by subcutaneous injections three times per week at a dose of 1 mg/kg for 48 weeks. With the exception of one case who died of respiratory failure that was unrelated to asfotase alfa, the recruited patients showed great improvement in rickets, developmental milestones, and pulmonary function.83 Asfotase alfa is the same bioengineered enzyme that was used in the animal experiment described earlier.80 A successful case of ERT of the perinatal lethal form with asfotase alfa was also reported from Japan.84 In July 2015, asfotase alfa (Strensiq™; Alexion Pharmaceuticals, Inc., New Haven, CT, USA) was approved in Japan, followed by approval in the EU and Canada in August and then by the US Food and Drug Administration in the USA in October 2015.
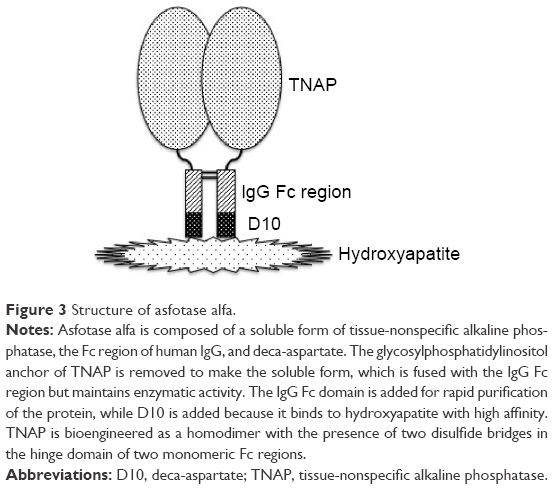 | Figure 3 Structure of asfotase alfa. |
Usage and evaluation of asfotase alfa
Symptomatic and radiographic evaluation, motor and cognitive development, and laboratory findings should confirm the outcome of the treatment. The recommended administration method of asfotase alfa is subcutaneous injection six times a week at a dose of 1 mg/kg or three times a week at 2 mg/kg, and the maximal volume of injection is 1 mL.85 In the report of the first trial, the authors employed laboratory data (serum alkaline phosphatase activity, plasma pyridoxal phosphate level, and serum PTH level), radiography (to check the severity of rickets, craniosynostosis, and nephrocalcinosis), respiratory state, feeding state, and gross motor function to evaluate the outcome of the treatment.83 Gross motor function was assessed with the use of the Bayley Scales of Infant and Toddler Development, third edition (Bayley-III) instrument. The main adverse event of the trial directly associated with the administration was transient erythema at the injection site, although some patients showed serious respiratory distress, craniosynostosis, and conductive hearing loss, which were considered possibly related to the study treatment.83 Regarding immunogenicity, about half of the patients showed the presence of neutralizing antibody, but as yet no clinical adverse effects have been observed.83 Common adverse reactions were hypersensitivity reactions, localized lipodystrophy at the injection site, ectopic calcifications of the eye, including the cornea and conjunctiva, and nephrocalcinosis.85 Although calcification of blood vessels was not reported,83 since medial artery calcifications are the most important pathological state associated with elevated alkaline phosphatase expression,86 cautious follow-up is necessary.
Future perspective
The targets of clinical trials of asfotase alfa were perinatal and infantile forms of HPP.83,84 For milder forms, namely, the childhood form, adult form, and odontohypophosphatasia, feasibility and safety have not been established. The current indication of asfotase alfa is therefore for perinatal/infantile- and juvenile-onset HPP.85 Furthermore, the natural course of the adult form of the disease is not well understood68 and should be elucidated, and more evidence regarding the feasibility of asfotase alfa for the milder form is needed. In addition, there are some problems with administration methods of asfotase alfa. Asfotase alfa must be administered at least three times per week, which is a big burden for the growing patients. The end point of the drug is also not known. It is reasonable that TNAP is necessary for growing children, but the enzyme is also required for bone remodeling in adulthood. It is still unclear whether asfotase alfa should be administered to help bone remodeling in patients who reach middle age. Other problems with administration are the frequency and dosage. The half-life of asfotase alfa following subcutaneous administration is 5 days.85 Modification of the drug can be considered to increase the interval of the injection. Otherwise, combination treatment can also be considered; asfotase alfa could be combined with stem-cell transplantation. Bone marrow stem-cell transplantation has been attempted, but the outcome was not very effective.87,88 Currently, the combination treatment is being attempted. In addition, a recent successful attempt of ERT in Akp2−/− mice using a soluble chimeric alkaline phosphatase, which is a chimeric molecule of human intestinal alkaline phosphatase with the crown domain of human PLAP,89 showed therapeutic benefit of the nonmineral-targeting recombinant enzyme.90 Another method to reduce the injection burden is gene therapy, especially ex vivo gene therapy into bone marrow stem cells. Gene therapy was successfully attempted in KO mice.91,92 Although administration of a viral vector containing the TNAP gene into blood is effective, the effect is transient. Administration into muscular tissue or combination with stem-cell transplantation (ex vivo gene therapy)93 may be effective with a one-shot administration. Nevertheless, those experiments were performed in the KO mouse model, and it is necessary to prove the safety of vector administration for application to human patients. Furthermore, it is necessary to investigate the optimal vector that has the highest transduction efficacy.
Conclusion
HPP is an inherited systemic bone disease characterized by bone hypomineralization. The responsible gene ALPL encodes TNAP, which is a GPI-anchored membrane-bound ectoenzyme expressed ubiquitously and plays an essential role in bone mineralization. Since the 3D structure of human TNAP has not been solved, a simulation model based on human PLAP is used to discuss the relationship between TNAP structure and mutations seen in the disease. The symptoms of HPP vary depending on the clinical form. Patients with severe forms suffer from respiratory failure, which is the main cause of death. An established treatment for HPP was not available until the recent development of ERT with a modified human TNAP (asfotase alfa). Treatment for HPP has changed drastically since the advent of asfotase alfa. However, there is still not enough data regarding the outcome of its administration, especially regarding the end points. In addition, daily or thrice-weekly injection of asfotase alfa may be a burden for patients. Further improvement of the therapy for HPP patients as well as more knowledge of the pathophysiology is needed.
Acknowledgment
The author thanks Dr Tomohiro Matsumura, Division of Metabolism and Nutrition, Department of Biochemistry and Molecular Biology, Nippon Medical School, for arranging the 3D structure model of TNAP.
Disclosure
The author received honoraria from Alexion Pharmaceuticals, Inc. The author reports no other conflicts of interest in this work.
References
Rathbun JC. “Hypophosphatasia”: a new developmental anomaly. Am J Dis Child. 1948;75:822–831. | ||
Whyte MP. Hypophosphatasia: nature’s window on alkaline phosphatase function in man. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles of Bone Biology. 2nd ed. San Diego, CA: Academic Press; 2002:1229–1248. | ||
Bianchi ML. Hypophosphatasia: an overview of the disease and its treatment. Osteoporos Int. 2015;26:2742–2757. | ||
Wenkert D, McAlister WH, Coburn SP, et al. Hypophosphatasia: nonlethal disease despite skeletal presentation in utero (17 new cases and literature review). J Bone Miner Res. 2011;26:2389–2398. | ||
Millán JL. Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co; 2006. | ||
Silvent J, Gasse B, Mornet E, Sire JY. Molecular evolution of the tissue-nonspecific alkaline phosphatase allows prediction and validation of missense mutations responsible for hypophosphatasia. J Biol Chem. 2014;289:24168–24179. | ||
Harris H. The human alkaline phosphatases: what we know and what we don’t know. Clin Chim Acta. 1989;186:133–150. | ||
Smith M, Weiss MJ, Griffin CA, et al. Regional assignment of the gene for human liver/bone/kidney alkaline phosphatase to chromosome 1p36.1-34. Genomics. 1988;2:139–143. | ||
Weiss MJ, Ray K, Henthorn PS, Lamb B, Kadesch T, Harris H. Structure of the human liver/bone/kidney alkaline phosphatase gene. J Biol Chem. 1988;263:12002–12010. | ||
Henthorn PS, Raducha M, Kadesch T, Weiss MJ, Harris H. Sequence and characterization of the human intestinal alkaline phosphatase gene. J Biol Chem. 1988;263:12011–12019. | ||
Knoll BJ, Rothblum KN, Longley M. Nucleotide sequence of the human placental alkaline phosphatase gene: evolution of the 5′ flanking region by deletion/substitution. J Biol Chem. 1988;263:12020–12027. | ||
Millán JL, Manes T. Seminoma-derived Nagao isozyme is encoded by a germ-cell alkaline phosphatase gene. Proc Natl Acad Sci U S A. 1988;85:3124–3028. | ||
Griffin CA, Smith M, Henthorn PS, et al. Human placental and intestinal alkaline phosphatase genes map to 2q34-q37. Am J Hum Genet. 1987;41:1025–1034. | ||
Matsuura S, Kishi F, Kajii T. Characterization of a 5′-flanking region of the human liver/bone/kidney alkaline phosphatase gene: two kinds of mRNA from a single gene. Biochem Biophys Res Commun. 1990;168:993–1000. | ||
Shibata H, Fukushi M, Igarashi A, et al. Defective intracellular transport of tissue-nonspecific alkaline phosphatase with an Ala162 → Thr mutation associated with lethal hypophosphatasia. J Biochem. 1998;123:968–977. | ||
Nosjean O, Koyama I, Goseki M, Roux B, Komoda T. Human tissue-nonspecific alkaline phosphatases: sugar-moiety-induced enzymic and antigenic modulation and genetic aspects. Biochem J. 1997;321:297–303. | ||
Hall SL, Dimai HP, Farley JR. Effects of zinc on human skeletal alkaline phosphatase activity in vitro. Calcif Tissue Int. 1999;64:163–172. | ||
Le Du MH, Millán JL. Structural evidence of functional divergence in human alkaline phosphatases. J Biol Chem. 2002;277:49808–49814. | ||
Le Du MH, Stigbrand T, Taussig MJ, Ménez A, Stura EA. Crystal structure of alkaline phosphatase from human placenta at 1.8 Å resolution. J Biol Chem. 2001;276:9158–9165. | ||
Ghosh K, Tagore DM, Anumula R, et al. Crystal structure of rat intestinal alkaline phosphatase – role of crown domain in mammalian alkaline phosphatases. J Struct Biol. 2013;184:182–192. | ||
Llinas P, Stura EA, Ménez A, et al. Structural studies of human placental alkaline phosphatase in complex with functional ligands. J Mol Biol. 2005;350:441–451. | ||
Sowadski JM, Handschumacher MD, Murthy HM, Foster BA, Wyckoff HW. Refined structure of alkaline phosphatase from Escherichia coli at 2.8 Å resolution. J Mol Biol. 1985;186:417–433. | ||
Micanovic R, Gerber LD, Berger J, Kodukula K, Udenfriend S. Selectivity of the cleavage/attachment site of phosphatidylinositol-glycan-anchored membrane proteins determined by site-specific mutagenesis at Asp-484 of placental alkaline phosphatase. Proc Natl Acad Sci U S A. 1990;87:157–161. | ||
Antonarakis SE, the Nomenclature Working Group. Recommendation for a nomenclature system for human gene mutations. Hum Mutat. 1998;11:1–3. | ||
den Dunnen J, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. | ||
den Dunnen J. Nomenclature for the description of sequence variants. Melbourne, Australia: HGVS. Available from: http://www.hgvs.org/mutnomen/. Accessed April 18, 2016. | ||
Orimo H. The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J Nippon Med Sch. 2010;77:4–12. | ||
Anderson HC. The role of matrix vesicles in physiological and pathological calcification. Curr Opin Orthop. 2007;18:428–433. | ||
Mebarek S, Abousalham A, Magne D, et al. Phospholipases of mineralization competent cells and matrix vesicles: roles in physiological and pathological mineralizations. Int J Mol Sci. 2013;14:5036–5129. | ||
Roberts SJ, Stewart AJ, Sadler PJ, Farquharson C. Human PHOSPHO1 exhibits high specific phosphoethanolamine and phosphocholine phosphatase activities. Biochem J. 2004;382:59–65. | ||
Roberts S, Narisawa S, Harmey D, Millán JL, Farquharson C. Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization. J Bone Miner Res. 2007;22:617–627. | ||
Addison WN, Azari F, Sørensen ES, Kaartinen MT, McKee MD. Pyrophosphate inhibits mineralization of osteoblast cultures by binding to mineral, upregulating osteopontin, and inhibiting alkaline phosphatase activity. J Biol Chem. 2007;282:15872–15883. | ||
Millán JL. The role of phosphatases in the initiation of skeletal mineralization. Calcif Tissue Int. 2013;93:299–306. | ||
Johnson K, Moffa A, Chen Y, Pritzker K, Goding J, Terkeltaub R. Matrix vesicle plasma cell membrane glycoprotein-1 regulates mineralization by murine osteoblastic MC3T3 cells. J Bone Miner Res. 1999;14:883–892. | ||
Wang W, Xu J, Du B, Kirsch T. Role of the progressive ankylosis gene (ank) in cartilage mineralization. Mol Cell Biol. 2005;25:312–323. | ||
Johnson K, Goding J, Van Etten D, et al. Linked deficiencies in extracellular PPi and osteopontin mediate pathologic calcification associated with defective PC-1 and ANK expression. J Bone Miner Res. 2003;18:994–1004. | ||
Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, Ikegawa S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat Genet. 1998;19:271–273. | ||
Rutsch F, Ruf N, Vaingankar S, et al. Mutations in ENPP1 are associated with “idiopathic” infantile arterial calcification. Nat Genet. 2003;34:379–381. | ||
Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289:265–270. | ||
Hessle L, Johnson KA, Anderson HC, et al. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci U S A. 2002;99:9445–9449. | ||
Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R, Millán JL. Concerted regulation of inorganic pyrophosphate and osteopontin by Akp2, Enpp1, and Ank. Am J Pathol. 2004;164:1199–1209. | ||
Anderson HC, Hsu HH, Morris DC, Fedde KN, Whyte MP. Matrix vesicles in osteomalacic hypophosphatasia bone contain apatite-like mineral crystals. Am J Pathol. 1997;151:1555–1561. | ||
Yadav M, Simão AM, Narisawa S, et al. Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: a unified model of the mechanisms of initiation of skeletal calcification. J Bone Miner Res. 2011;26:286–297. | ||
Orimo H, Goseki-Sone M, Hosoi T, Shimada T. Functional assay of the mutant tissue-nonspecific alkaline phosphatase gene using U2OS osteoblast-like cells. Mol Genet Metab. 2008;94:375–381. | ||
Waymire KG, Mahunen JD, Jaje JM, Guilarte TR, Coburn SP, MacGregor GR. Mice lacking tissue nonspecific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat Genet. 1995;11:45–51. | ||
Guilarte TR. Regional changes in the concentrations of glutamate, glycine, taurine, and GABA in the vitamin B-6 deficient developing rat brain: association with neonatal seizures. Neurochem Res. 1989;14:889–897. | ||
Fedde KN, Whyte MP. Alkaline phosphatase (tissue-nonspecific isoenzyme) is a phosphoethanolamine and pyridoxal-5′-phosphate ectophosphatase: normal and hypophosphatasia fibroblast study. Am J Hum Genet. 1990;47:767–775. | ||
Whyte MP, Mahunen JD, Fedde KN, Cole S, McCabe ER, Coburn SP. Perinatal hypophosphataia: tissue levels of vitamin B6 are unremarkable despite markedly increased circulating concentrations of pyridoxal-5′-phosphate. J Clin Invest. 1988;81:1234–1239. | ||
Baumgartner-Sigl S, Haberlandt E, Mumm S, et al. Pyridoxine-responsive seizures as the first symptom of infantile hypophosphatasia caused by two novel missense mutations (c.677T > C, p.M226T; c.1112C > T, p.T371I) of the tissue-nonspecific alkaline phosphatase gene. Bone. 2007;40:1655–1661. | ||
Silverman JL. Apparent dominant inheritance of hypophosphatasia. Arch Int Med. 1962;110:191–198. | ||
Moore CA, Curry CJ, Henthorn P, et al. Mild autosomal dominant hypophosphatasia: in utero presentation in two families. Am J Med Genet. 1999;86:410–415. | ||
Fauvert D, Brun-Heath I, Lia-Baldini AS, et al. Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles. BMC Med Genet. 2009;10:51. | ||
Mornet E. Hypophosphatasia. Orphanet J Rare Dis. 2007;2:40. | ||
Macfarlane JD, Kroon HM, van den Harten JJ. Phenotypically dissimilar hypophosphatasia in two sibships. Am J Med Genet. 1992;42:117–121. | ||
Peach CA, Zhang Y, Wordsworth BP. Mutations of the tissue-nonspecific alkaline phosphatase gene (TNAP) causing a non-lethal case of perinatal hypophosphatasia. Rheumatology. 2007;46:1037–1040. | ||
Mumm S, Jones J, Finnegan P, Whyte MP. Hypophosphatasia: molecular diagnosis of Rathbun’s original case. J Bone Miner Res. 2001;16:1724–1727. | ||
Mornet E. The tissue nonspecific alkaline phosphatase gene mutations database. Available from: http://www.sesep.uvsq.fr/03_hypo_mutations.php. Accessed April 18, 2016. | ||
Mornet E, Stura E, Lia-Baldini AS, Stigbrand T, Ménez A, Le Du MH. Structural evidence for a functional role of human tissue-nonspecific alkaline phosphatase in bone mineralization. J Biol Chem. 2001;276:31171–31178. | ||
Cai G, Michigami T, Yamamoto T, et al. Analysis of localization of mutated tissue-nonspecific alkaline phosphatase proteins associated with neonatal hypophosphatasia using green fluorescent protein chimeras. J Clin Endocrinol Metab. 1998;83:3936–3942. | ||
Orimo H, Goseki-Sone M, Sato S, Shimada T. Detection of deletion 1154–1156 hypophosphatasia mutation using TNSALP exon amplification. Genomics. 1997;42:364–366. | ||
Goseki-Sone M, Orimo H, Iimura T, et al. Expression of the mutant (1735T-DEL) tissue-nonspecific alkaline phosphatase gene from hypophosphatasia patients. J Bone Miner Res. 1998;13:1827–1834. | ||
Komaru K, Ishida Y, Amaya Y, Goseki-Sone M, Orimo H, Oda K. Novel aggregate formation of a frame-shift mutant protein of tissue-nonspecific alkaline phosphatase is ascribed to three cysteine residues in the C-terminal extension: retarded secretion and proteasomal degradation. FEBS J. 2005;272:1704–1717. | ||
Orimo H, Goseki-Sone M, Inoue M, Tsubakio Y, Sakiyama T, Shimada T. Importance of deletion of T at nucleotide 1559 in the tissue-nonspecific alkaline phosphatase gene in Japanese patients with hypophosphatasia. J Bone Miner Metab. 2002;20:28–33. | ||
Michigami T, Uchihashi T, Suzuki A, Tachikwa K, Nakajima S, Ozono K. Common mutation F310L and T1559del in the tissue-nonspecific alkaline phosphatase gene are related to distinct phenotypes in Japanese patients with hypophosphatasia. Eur J Pediatr. 2005;164:277–282. | ||
Zurutuza L, Muller F, Gibrat JF, et al. Correlations of genotype and phenotype in hypophosphatasia. Hum Mol Genet. 1999;8:1039–1046. | ||
Whyte MP, Zhang F, Wenkert D, et al. Hypophosphatasia: validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patients. Bone. 2015;75:229–239. | ||
van den Bos T, Handoko G, Niehof A, et al. Cementum and dentin in hypophosphatasia. J Dent Res. 2005;84:1021–1025. | ||
Collmann H, Mornet E, Gattenlöhner S, Beck C, Girschick H. Neurosurgical aspects of childhood hypophosphatasia. Childs Nerv Syst. 2009;25:217–223. | ||
Berkseth KE, Tebben PJ, Drake MT, Hefferran TE, Jewison DE, Wermers RA. Clinical spectrum of hypophosphatasia diagnosed in adults. Bone. 2013;54:21–27. | ||
Watanabe A, Karasugi T, Sawai H, et al. Prevalence of c.1559delT in ALPL, a common mutation resulting in the perinatal (lethal) form of hypophosphatasia in Japanese and effects of the mutation on heterozygous carriers. J Hum Genet. 2011;56:166–168. | ||
Fraser D. Hypophosphatasia. Am J Med. 1957;22:730–746. | ||
Greenberg CR, Taylor CL, Haworth JC, et al. A homoallelic Gly317 → Asp mutation in ALPL causes the perinatal (lethal) form of hypophosphatasia in Canadian mennonites. Genomics. 1990;17:215–217. | ||
Mornet E, Yvard A, Taillandier A, Fauvert D, Simon-Bouy B. A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann Hum Genet. 2011;75:439–445. | ||
Taketani T, Onigata K, Kobayashi H, Mushimoto Y, Fukuda S, Yamaguchi S. Clinical and genetic aspects of hypophosphatasia in Japanese patients. Arch Dis Child. 2014;99:211–215. | ||
Narisawa S, Fröhlander N, Millán JL. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev Dyn. 1997;208:432–446. | ||
Fedde KN, Blair L, Silverstein J, et al. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res. 1999;14:2015–2026. | ||
Beertsen W, VandenBos T, Everts V. Root development on mice lacking functional tissue non-specific alkaline phosphatase gene: inhibition of acellular cementum formation. J Dent Res. 1999;78:1221–1229. | ||
Whyte MP, McAlister WH, Patton LS, et al. Enzyme replacement therapy for infantile hypophosphatasia attempted by intravenous infusions of alkaline phosphatase-rich Paget plasma: results in three additional patients. J Pediatr. 1984;105:926–933. | ||
Tadokoro M, Kanai R, Taketani T, Uchino Y, Yamaguchi S, Ohgushi H. New bone formation by allogenic mesenchymal stem cell transplantation in a patient with perinatal hypophosphatasia. J Pediatr. 2009;154:924–930. | ||
Millán JL, Narisawa S, Lemire I, et al. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res. 2008;23:777–787. | ||
Nishioka T, Tomatsu S, Guitierrez MA, et al. Enhancement of drug delivery to bone: characterization of human tissue-nonspecific alkaline phosphatase tagged with an acidic oligopeptide. Mol Genet Metab. 2006;88:244–255. | ||
McKee MD, Nakano Y, Masica DL, et al. Enzyme replacement therapy prevents dental defects in a model of hypophosphatasia. J Dent Res. 2011;90:470–476. | ||
Whyte MP, Greenberg CR, Salman NJ, et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012;366:904–913. | ||
Okazaki Y, Kitajima H, Mochizuki N, Kitaoka T, Michigami T, Ozono K. Lethal hypophosphatasia successfully treated with enzyme replacement from day 1 after birth. Eur J Pediatr. 2016;175:433–437. | ||
STRENSIQ™ (asfotase alfa) for Strensiq [prescribing information]. Cheshire, CT: Alexion Pharmaceuticals, Inc; 2015. Available from: http://strensiq.com/images/pi.pdf. Accessed April 18, 2016. | ||
Sheen CR, Kuss P, Narisawa S, et al. Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J Bone Miner Res. 2015;30:824–836. | ||
Whyte MP, Kurtzberg J, McAlister WH, et al. Marrow cell transplantation for infantile hypophosphatasia. J Bone Miner Res. 2003;18:624–636. | ||
Taketani T, Oyama C, Mihara A, et al. Ex vivo expanded allogeneic mesenchymal stem cells with bone marrow transplantation improved osteogenesis in infants with severe hypophosphatasia. Cell Transplant. 2015;24:1931–1943. | ||
Sasajima Y, Kohama Y, Kojima-Misaizu M, et al. Simultaneous retension of thermostability and specific activity in chimeric human alkaline phosphatases. Mol Biotechnol. 2014;56:953–961. | ||
Gasque KC, Foster BL, Kuss P, et al. Improvement of the skeletal and dental hypophosphatasia phenotype in Alpl−/− mice by administration of soluble (non-targeted) chimeric alkaline phosphatase. Bone. 2015;72:137–147. | ||
Yamamoto S, Orimo H, Matsumoto T, et al. Prolonged survival and phenotypic correction of Akp2−/− hypophosphatasia mice by lentiviral gene therapy. J Bone Miner Res. 2011;26:135–142. | ||
Matsumoto T, Miyake K, Yamamoto S, et al. Rescue of severe infantile hypophosphatasia mice by AAV-mediated sustained expression of soluble alkaline phosphatase. Hum Gene Ther. 2011;22:1355–1364. | ||
Iijima O, Miyake K, Watanabe A, et al. Prevention of lethal murine hypophosphatasia by neonatal ex vivo gene therapy using lentivirally transduced bone marrow cells. Hum Gene Ther. 2015;26:801–812. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.