")
Back to Journals » Infection and Drug Resistance » Volume 16
Pathogen Diagnosis Value of Nanopore Sequencing in Severe Hospital-Acquired Pneumonia Patients
Authors Zhao X, Ge Y, Zhang Y, Zhang W , Hu H, Li L, Sha T, Zeng Z, Wu F, Chen Z
Received 16 March 2023
Accepted for publication 9 May 2023
Published 26 May 2023 Volume 2023:16 Pages 3293—3303
DOI https://doi.org/10.2147/IDR.S410593
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Xin Zhao,1,2,* Yue Ge,1,3,* Yuan Zhang,1 WenJie Zhang,1 HongBin Hu,1 LuLan Li,1 Tong Sha,1 ZhenHua Zeng,1 Feng Wu,1 ZhongQing Chen1
1Department of Critical Care Medicine, Nanfang Hospital, Southern Medical University, Guangzhou, People’s Republic of China; 2The First Clinical Medical School, Southern Medical University, Guangzhou, People’s Republic of China; 3Department of School of Nursing, Southern Medical University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: ZhongQing Chen; Feng Wu, Department of Critical Care Medicine, Nanfang Hospital, Southern Medical University, Guangzhou, People’s Republic of China, Tel +86 13503049103 ; +86 15915751705, Email [email protected]; [email protected]
Background: Next-generation sequencing of the metagenome (mNGS) is increasingly used in pathogen diagnosis for infectious diseases due to its short detection time. The time for Oxford Nanopore Technologies (ONT) sequencing-based etiology detection is further shortened compared with that of mNGS, but only a few studies have verified the time advantage and accuracy of ONT sequencing for etiology diagnosis. In 2022, a study confirmed that there was no significant difference in sensitivity and specificity between ONT and mNGS in suspected community-acquired pneumonia patients, which there was no clinical study verified in patients with SHAP.
Methods: From October 24 to November 20, 2022, 10 patients with severe hospital-acquired pneumonia (SHAP) in the Nanfang Hospital intensive care unit (ICU) were prospectively enrolled. Bronchoalveolar lavage fluid (BALF) was collected for ONT sequencing, mNGS, and traditional culture. The differences in pathogen detection time and diagnostic agreement among ONT sequencing, mNGS, traditional culture method, and clinical composite diagnosis were compared.
Results: Compared with mNGS and the traditional culture method, ONT sequencing had a significant advantage in pathogen detection time (9.6± 0.7 h versus 24.7± 2.7 h versus 132± 58 h, P < 0.05). The agreement rate between ONT sequencing and the clinical composite diagnosis was 73.3% (kappa value=0.737, P < 0.05).
Conclusion: ONT sequencing has a potential advantage for rapidly identifying pathogens.
Keywords: Oxford Nanopore Technologies, Nanopore sequencing, metagenomic next-generation sequencing, severe hospital-acquired pneumonia, pathogen
Introduction
Severe hospital-acquired pneumonia (SHAP) is the most common acquired infection in the intensive care unit (ICU) and one of the main causes of increased mortality in ICU patients.1,2 Potential multidrug-resistant pathogens are very common in ICU-acquired pneumonia, including Pseudomonas aeruginosa, Acinetobacter spp., methicillin-resistant Staphylococcus aureus (MRSA), and ultrabroad spectrum β-lactamase (ESBL)-producing and carbapenem-resistant Enterobacteriaceae (CRE).3 Timely and appropriate antibiotic treatment is effective in reducing patient mortality, but it often takes up to 48 hours from respiratory sampling to the acquisition of definitive microbiological test results, which leads to untimely antibiotic therapy and increased mortality.3 Therefore, early identification of the pathogen and timely use of targeted antibiotics is particularly important to improve prognosis.
The traditional culture of bronchoscopy specimens remains the gold standard of pathogenic detection. However, the turnaround time of bacterial/fungal cultures is long (3–7 days) in conventional microbiological testing.4 The low positive rates cannot meet the need for early pathogen diagnosis.5 mNGS, known as a powerful, low-cost, shorter turn round-time method, has been used in pathogen diagnosis in recent years. The turnaround time of mNGS is usually 24–36 hours, and the positive rate is 15% higher than that of culture.6 Several studies have shown that the diagnostic accuracy of mNGS was not inferior to traditional culture (61.7% vs 76.7%, P=0.11), and the 28-day mortality of the mNGS group was significantly lower than culture group (47.32% vs 62.2%, P=0.043) due to its shorter pathogen detection time.7,8 Nevertheless, each step of mNGS requires sequence restriction to short 200–500 bp read lengths, and these shorter read lengths make genome, transcriptome, and metagenome assembly more challenging.9
Third-generation sequencing, also known as Nanopore sequencing, uses a long-read length assay to compensate for the shortcomings of mNGS.10 The turnaround time of Nanopore sequencing is only 6–9 hours, which is much shorter than that of mNGS. However, it is not widely used in the clinic because of its high error rate, which is close to 30%.11 In recent years, research has shown that removing human background gene interference significantly improves the pathogenic detection accuracy of Nanopore sequencing, which exhibits striking agreement with culture and mNGS.12 The study has shown that there was no significant difference in the consistency between ONT sequencing and mNGS with clinical diagnosis (59.38% vs 57.81%, P>0.05) in suspected community-acquired pneumonia patients, and the detection time of ONT sequencing was significantly shorter than mNGS.13 However, fewer clinical studies have yet validated the consistency between clinical composite diagnosis with Nanopore sequencing with human background genes removed for pathogen detection in bronchoalveolar lavage fluid (BALF) obtained from patients with SHAP. In this study, we proposed to evaluate the value of pathogen diagnosis based on BALF Nanopore sequencing application in adults with SHAP.
Materials and Methods
Study Participants and Study Design
A prospective cohort study was conducted on patients with severe hospital-acquired pneumonia admitted to the intensive care unit of Nanfang Hospital from October 24, 2022, to November 20, 2022. Both Oxford Nanopore Technologies (ONT) sequencing and mNGS were used to detect pathogens in BALF. Hospital-acquired pneumonia (HAP) was defined as pneumonia that manifested 48 h after hospital admission and encompasses two entities: ventilator-associated pneumonia (VAP) and severe pneumonia that developed in the hospital.14 SHAP was defined for patients who met one major or at least three minor Infectious Diseases Society of America (IDSA)/American Thoracic Society (ATS) criteria based on HAP.15 Patients were excluded if they left the ICU within 48 hours or if bronchoalveolar lavage was contraindicated. This study was approved by the Medical Ethics Committee of Nanfang Hospital (NFEC-2022-399), and informed consent was obtained from each patient or guardian. The study was performed in line with the Declaration Helsinki.
In our study, we compared the mNGS, ONT sequencing results for BALF from patients who had a matched BALF culture result for bacteria and fungi. Due to the absence of an available conventional test, we did not compare the numbers of sequences in the mNGS and ONT sequencing results for BALF with respiratory viral tests for viruses.
DNA Extraction
Five milliliters of BALF were collected, 600 µL was added to 250 µL of 0.5 mm glass beads for physical degradation, followed by 7.2 µL of lyticase lysis enzyme (RT410-TA, Tiangen Biotech, Beijing, China) for the enzyme degradation reaction, mixed and shaken, and centrifuge at 8000 g for 5 min. For mNGS, 300 μL of the sample was extracted according to the TIANamp Micro DNA Kit (DP316, Tiangen Biotech, Beijing, China) to extract DNA. As for ONT sequencing, we applied a human DNA removal step in BALF samples to improve the correct rate, as recently described.16 And we extracted the genomic DNA with MagNA Pure compact (Roche Diagnostics GmbH, Germany).
Library Preparation and Sequencing
Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific Inc.) was used to construct mNGS libraries and the MGISEQ-2000 platform (BGI-Tianjin, Tianjin, China) was used for sequencing. The Rapid Barcoding kit SQK-RBK004 (Oxford Nanopore) was used to construct ONT sequencing libraries and MinION flowcells (Oxford Nanopore) were used for sequencing.
Bioinformatics Analyses
High-quality data were obtained by removing low-quality data and short (length < 35bp) reads. The high-quality data were filtered by BWA (BWA: http://bio-bwa.sourceforge.net/) to match the human reference genome to exclude human sequences data.17 The remaining data were compared with the BGI-PMDB pathogen database (including 6350 bacteria, 1064 fungi, 4945 viruses, and 234 parasites) after removing low-complexity reads to obtain the number of sequences that could match a certain pathogen.
Identification of Pathogens
Culture-Identified Pathogens
(i) Bacteria: A bacterium concentration more e than 104 CFU/mL was regarded as a positive criterion. (ii) Fungi and parasites: Positive BALF culture and smear of fungi and parasites were used to determine positivity. (iii) Tuberculosis: Tuberculosis (TB) pneumonia was diagnosed by sputum smear exams for Mycobacterium tuberculosis and/or TB culture.
Positive mNGS/ONT Criteria
Due to the lack of criteria for the interpretation of mNGS results, we used the developed criteria from previous study.18 The infectious bacteria (excluding mycobacteria), fungi, and parasites were considered positive if they met any of the following standards of the mNGS/ONT sequencing: (i) if mNGS/ONT detected the same pathogen as culture with the number of unique reads from a single species exceeded 50; (ii) if the unique reads of pathogen less than 50, the diagnosis of infection can still be made based on the clinical manifestations; (iii) mycobacterium tuberculosis: at least one read was identified to a species, and TB was considered positive; (iv) if mNGS/ONT detected the pathogen with the number of unique reads exceeded 50 while culture missed this pathogen, it was considered potential pathogenic microbe.
Furthermore, mNGS/ONT sequencing results could not be used to determine if microorganisms were infected, colonized, or contaminated. After receiving the mNGS/ONT sequencing results, two physicians analyzed the clinical features to reach a consensus.
Clinical Data Collection
Data were obtained from hospital records, including demographic, laboratory data, APACHE II scores, and SOFA scores. Data on pathogen species and unique reads were collected from reports from the sequencing company.
Statistical Analysis
The one-way analysis of variance (ANOVA) was used to compare the turnaround time of three pathogen-testing methods. The kappa value measured the agreement between the results of mNGS and ONT tests. According to the research of Landis and Koch19 the importance of consistency was considered as follows: a kappa value of 0.8–1 denoted close to a perfect consistency; 0.6–0.8 denoted significant consistency; 0.4–0.6 denoted moderate consistency, and less than 0.4 denoted low consistency. SPSS 26.0 software was utilized for data analysis. P values less than 0.05 were deemed significant.
Results
Sample and Patient Characteristics
10 patients diagnosed with critical hospital-acquired pneumonia with a median age of 63 years were included in this study with the majority of patients being male (n=7, 70%). Laboratory findings and disease severity are shown in Table 1. Among the underlying diseases, hypertension (n=3, 30%), cerebral infarction (n=3, 30%), and cardiovascular disease (n=2, 20%) were the most prevalent (Table 2).
 |
Table 1 Clinical and Laboratory Characteristics of 10 Patients with SHAP |
 |
Table 2 Characteristics and Detection Results of Patients |
Identification of Pathogens by Traditional Culture Methods, mNGS, and ONT Sequencing
In terms of the type of pathogens detected, the ONT sequencing and clinical composite diagnosis displayed consistency in 9 (90%) cases, including 7 diagnostically positive cases versus 2 diagnostically negative cases. The ONT sequencing and mNGS results were consistent in 8 (80%) cases, including 6 mNGS-positive and 2 mNGS-negative cases. And a total of 50 bacteria, 16 fungi, and 2 parasites were identified in 30 samples that 36 bacteria, 5 fungi, and 2 parasites were the true-positive pathogenic microbes (Figure 1A). 11 pathogens were identified by culture method, and 8 pathogens were considered the true positive. The most frequently detected bacteria were Pseudomonas aeruginosa (two cases), Acinetobacter baumannii (two cases) (Figure 1B). A total of 18 pathogens were identified by mNGS in 10 samples, while 12 pathogens were true-positive microbes. The most commonly detected bacteria were Acinetobacter baumannii (two cases), Pseudomonas aeruginosa (two cases), and Enterococcus faecalium (two cases) (Figure 1C). The samples were also sequenced by ONT. A total of 39 pathogens were identified while 16 strains (41%) were false-positive pathogenic microbes. And the most frequently bacteria were Acinetobacter baumannii (three cases), Pseudomonas aeruginosa (three cases), and Klebsiella pneumonia (three cases) (Figure 1D). Notably, Strongyloides steroralis was detected in 1 case by mNGS and traditional culture, but it was not discovered by ONT sequencing. Overall, ONT sequencing identified more potential kinds of pathogens than mNGS. The detailed pathogen results of ONT sequencing, mNGS, and culture for each patient are shown in Supplementary Materials.
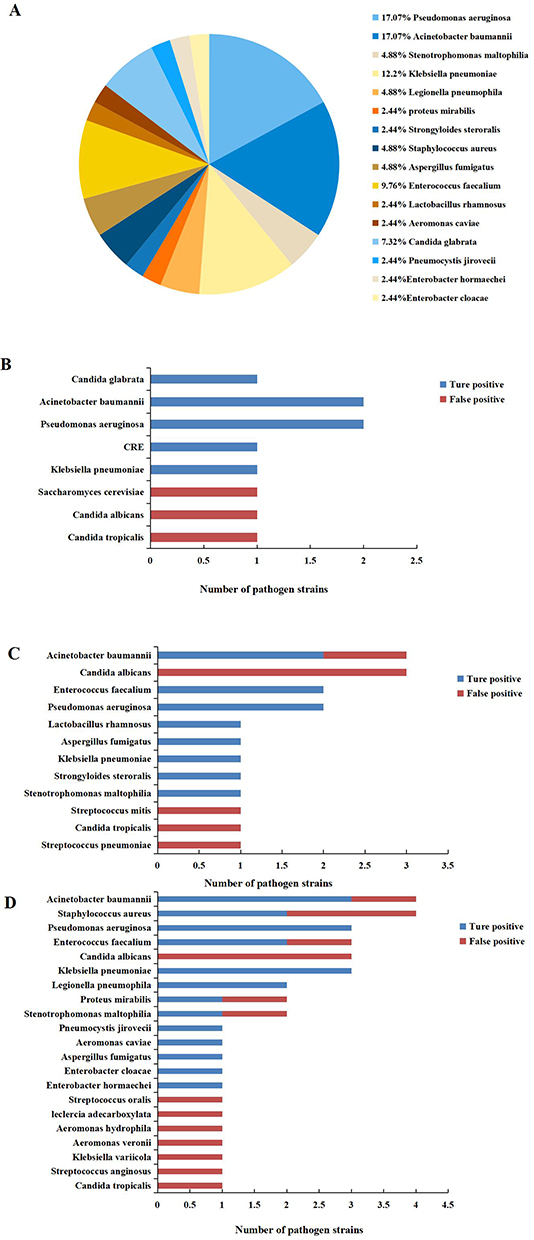 |
Figure 1 Identification of pathogenic microbes by traditional culture, mNGS, and ONT sequencing. (A) The pathogen distribution of 43 true-positive microbes. (B) Histogram of traditional culture method to detect pathogenic microbes. (C) Histogram of mNGS to detect pathogenic microbes. (D) Histogram of ONT sequencing detection for pathogenic microbes. |
Detection Time
Among the 10 cases in this study, the overall average time for the ONT sequencing assay was 9.6±0.7 h, which was significantly shorter than that for mNGS (24.7±2.7 h, P<0.05) and traditional culture (132±58 h, P <0.05).
Diagnostic Performance
The diagnostic positivity rates for ONT sequencing, mNGS, and traditional culture were 80%, 60%, and 60%, respectively. Take clinical composite diagnosis as the reference standard, consistent with previous studies, mNGS had good agreement with the final diagnosis (Kappa value=0.783, p<0.05). ONT sequencing also had good agreement with the final diagnosis (Kappa value=0.737, p<0.05). However, the agreement rate between traditional culture and the final diagnosis was notably only 34.8% in the 10 samples, which was not statistically significant (Kappa value=0.348, P>0.05) (Table 3).
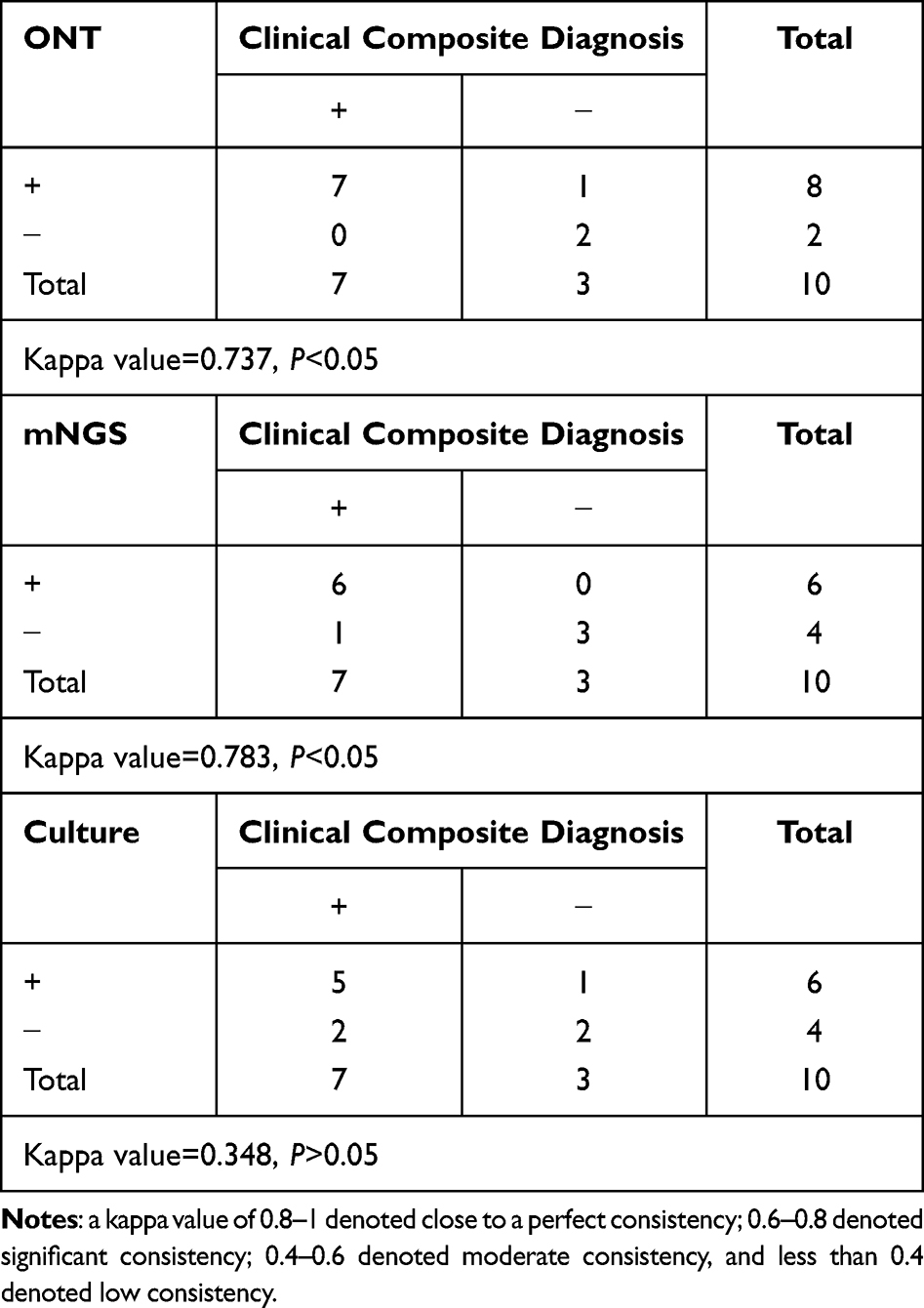 |
Table 3 Diagnostic Performance of ONT Sequencing, mNGS, and Traditional Culture |
“False Positive” and “False Negative” of ONT Sequencing and mNGS
“False Positive” of ONT Sequencing
In the 7 clinical composite diagnosed positive cases, there were no “False negative” cases for ONT sequencing which identified all pathogenic microbes and were consistent with the final diagnosis. For the “ONT false positive” cases, possible reasons included colonization (2/4), and potential causes of infection (2/4). In 12 pathogens that did not meet positive criteria, only 2 microbes were considered potential causes of infection, which was shown in Table 4.
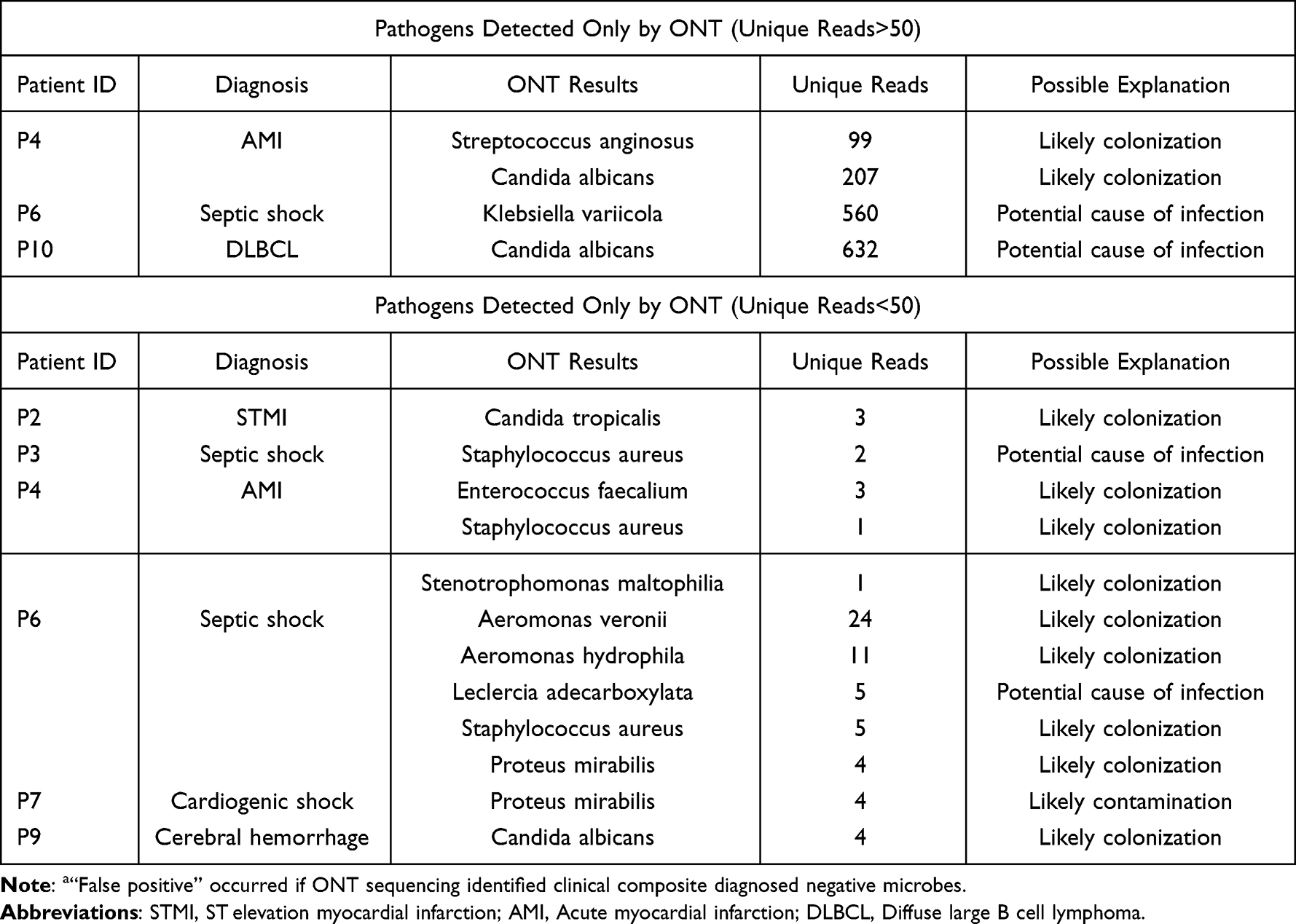 |
Table 4 Analysis of “False Positive”a Results of ONT Sequencing |
“False Positive” and “False Negative” of mNGS
In the clinical composite diagnosed positive cases, up to 11 pathogens were missed by mNGS, and were unidentifiable by mNGS. As for the “false positive” cases of mNGS, the most commonly reasons was potential causes of infection (2/3) (Table 5).
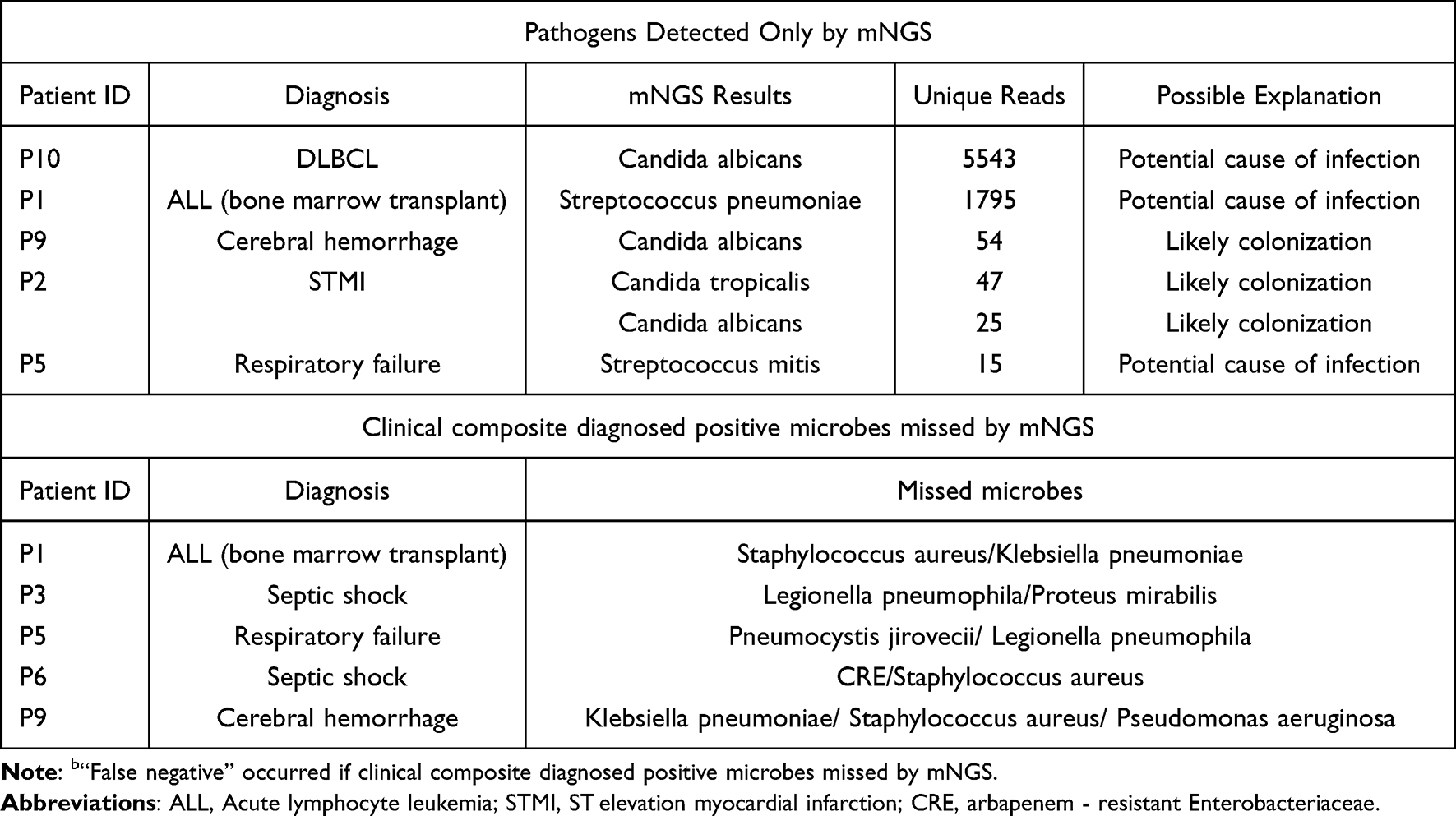 |
Table 5 Analysis of “False Negative”b Results of mNGS |
Discussion
Oxford Nanopore Technologies (ONT) was prospective to be a viable method out of its broad breadth, long reading sequencing, and real-time production.20 However, the interference of human DNA limited the application of ONT sequencing in clinical practice.16 This prospective self-controlled study explored the consistency of pathogen diagnosis in SHAP patients by ONT sequencing which depleted human DNA before sequencing. In this study, we showed that ONT sequencing may be a promising method to detect pathogens in SHAP patients. Future large-sample clinical studies can further verify the sensitivity and specificity of this method and explore whether it can guide personalized antibiotic therapy to improve prognosis.
Previous studies had already demonstrated that the mNGS can improve the prognosis by guiding targeted antibiotic treatment with high sensitivity and specificity.7,8 However, fewer clinical studies have verified the diagnostic accuracy and detection time of ONT sequencing.13 And there was no clinical study exploring the consistency between ONT sequencing with the clinical diagnosis in patients with SHAP.
In this study, we confirmed the superior time requirements and result agreement of ONT sequencing compared with mNGS and traditional microbiological methods in pathogen diagnosis in patients with SHAP. First, the mNGS and ONT sequencing tests had a higher positive rate than traditional methods, which was consistent with earlier researches.21 Charalampous, Kay, Richardson, Aydin, Baldan, Jeanes, Rae, Grundy, Turner, Wain, Leggett, Livermore and O’Grady16 found that the sensitivity of ONT sequencing with human DNA depletion is as high as 96.6% in lower respiratory tract infections which was consistent with our study. The kappa value between ONT sequencing and clinical composite diagnosis was high in SHAP patients (kappa value=0.737, indicating substantial agreement). However, the kappa value between ONT sequencing and mNGS was low (kappa value=0.545, P >0.05), which was not statistically significant. Possible reasons for the lack of concordance are as follows. First, since the study’s limited sample size, inconsistent results in individual cases may cause significant bias. In 2 cases where the results of ONT sequencing were different with mNGS, one case was the false positive of ONT which was considered a colonization pathogen. Another case was the false negative of mNGS, Pneumocystis jirovecii, and Legionella pneumophila were not detected; the second possibility is that more potential pathogens are detected by ONT sequencing.22
Second, according to the HAP/VAP treatment guidelines, the patients strongly suspected of VAP who accepted the early antibiotic treatment had lower mortality, compared with inadequate therapy (38% versus 91%).1 A prospective cohort study also indicated that the patients who accepted inappropriate antibiotic treatment had a higher mortality rate than those who accepted correct therapy.23 Therefore, early adequate and appropriate antibiotic therapy is extremely important in the treatment of HAP, which means that rapid pathogen diagnosis tests, such as mNGS and ONT sequencing, are helpful to guide targeted antibiotic treatment due to their high sensitivity.
In this study, consistent with a previous study,13 ONT sequencing had a shorter turnaround time than mNGS (9.6±0.7 h versus 24.7±2.7 h, P<0.05) and traditional culture methods (9.6±0.7 h versus 132±58 h, P <0.05), which means that pathogens can be identified as early as possible to guide personalized antibiotic therapy.24
In addition, more types of species were detected by ONT sequencing than mNGS, especially among bacteria. ONT sequencing applied the enrichment method to improve the detection rate of pathogens, while mNGS was performed directly on extracted nucleic acids. This may be a reason for the differences in microbial detection between the two methods. In this study, the most frequent pathogens were P. aeruginosa, A. baumannii, K. pneumonia, and fungi, which is in agreement with former reports.25 The most frequently detected viruses included Epstein-Barr virus (EBV), cytomegalovirus (CMV), and human herpesvirus 1, 4, 5, and 7, which were considered to have no pathogenic significance when detected in BALF in most studies.26,27
According to our data, ONT sequencing with the removal of human DNA is a powerful method for microbiological diagnosis. As such, ONT sequencing has potential clinical application value. Large-sample clinical research is required to validate the sensitivity and specificity of ONT sequencing in microbiological detection and determine whether it can guide targeted therapy and reduce clinical antibiotic overuse.
However, there are still some disadvantages of ONT sequencing. The fecal round nematode was identified by mNGS which was considered a pathogenic microbe, while ONT sequencing did not detect this pathogen. There have been no clinical studies investigating the accuracy of ONT sequencing for parasites. The reasons we considered are the following. (i) During the process of removing human DNA, most nucleic acids of the fecal round nematode were removed, which led to failure in the amplification and library preparation process. (ii) Alternatively, the database for analysis was not comprehensive and did not include the sequence of the fecal round nematode. On the other hand, one case showed a positive ONT sequencing result but negative culture and mNGS results (Streptococcus pyogenes and C. albicans), which may be a false-positive ONT sequencing result due to the high content of oropharyngeal pathogens.28
Our research also has some shortcomings. First, our study was conducted in a single location with a limited sample size, there existed selection bias. Second, as a self-controlled study, we divided the bronchoalveolar lavage fluid of one patient into three-part to send for ONT sequencing, mNGS, and culture at the same time. So we were unable to compare the difference in change of antibiotic treatment between ONT sequencing and mNGS, which was to be carried out by a large sample cohort/case-control study. Third, we did not conduct the real-time ONT sequencing analysis because our objective was to verify its turnaround time and accuracy. In addition, the antibiotic resistance gene sequencing outcomes were not analyzed due to the small sample size of this study. To investigate further, we are conducting a prospective cohort study.
Conclusion
Our results demonstrated that ONT sequencing had a substantial agreement with clinical composite diagnosis in pathogen diagnosis. With shorter turn-round times of 8–10 h, ONT sequencing may play an important role in the creation of quick and accurate SHAP pathogen diagnostic tools. Further investigations are required to determine the direct influence on antibiotic options and prognosis.
Acknowledgments
We would like to thank Dr. Qitao Yan from JingXun Medicals (Guangzhou, China) for his helpful technical support. The authors also thank all the patients for participating in this study. Xin Zhao and Yue Ge are co-first authors for this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Natural Science Foundation of China (Grant Number 81871604) and the Guangdong Basic and Applied Basic Research Foundation (Grant Number 2021A1515111028).
Disclosure
All authors declare that they have no potential conflicts of interest in this work.
References
1. Martin-Loeches I, Rodriguez AH, Torres A. New guidelines for hospital-acquired pneumonia/ventilator-associated pneumonia: USA vs. Europe. Curr Opin Crit Care. 2018;24(5):347–352. doi:10.1097/mcc.0000000000000535
2. Peiffer-Smadja N, Bouadma L, Mathy V, et al. Performance and impact of a multiplex PCR in ICU patients with ventilator-associated pneumonia or ventilated hospital-acquired pneumonia. Crit Care. 2020;24(1):366. doi:10.1186/s13054-020-03067-2
3. Bassetti M, Righi E, Vena A, Graziano E, Russo A, Peghin M. Risk stratification and treatment of ICU-acquired pneumonia caused by multidrug- resistant/extensively drug-resistant/pandrug-resistant bacteria. Curr Opin Crit Care. 2018;24(5):385–393. doi:10.1097/mcc.0000000000000534
4. Liu BB, Tian Q, Wang P, et al. Evaluating the diagnostic value of using metagenomic next-generation sequencing on bronchoalveolar lavage fluid and tissue in infectious pathogens located in the peripheral lung field. Ann Palliat Med. 2022;11(5):1725–1735. doi:10.21037/apm-21-3474
5. Zhao D, Guo L, Lian D, et al. Diagnostic value and clinical application of mNGS for post-liver transplantation infection: a cross-sectional study with case reports. Front Microbiol. 2022;13:919363. doi:10.3389/fmicb.2022.919363
6. Jin X, Li J, Shao M, et al. Improving suspected pulmonary infection diagnosis by bronchoalveolar lavage fluid metagenomic next-generation sequencing: a multicenter retrospective study. Microbiol Spectr. 2022;10(4):e0247321. doi:10.1128/spectrum.02473-21
7. Qin CH, Zhang SG, Zhao YY, Ding XF, Yang F, Zhao YC. Diagnostic value of metagenomic next-generation sequencing in sepsis and bloodstream infection. Front Cell Infect Microbiol. 2023;131117987. doi:10.3389/fcimb.2023.1117987
8. Peng JM, Du B, Qin HY, Wang Q, Shi Y. Metagenomic next-generation sequencing for the diagnosis of suspected pneumonia in immunocompromised patients. J Infect. 2021;82(4):22–27. doi:10.1016/j.jinf.2021.01.029
9. Li H, Gao H, Meng H, et al. Detection of pulmonary infectious pathogens from lung biopsy tissues by metagenomic next-generation sequencing. Front Cell Infect Microbiol. 2018;8:205. doi:10.3389/fcimb.2018.00205
10. Jain M, Koren S, Miga KH, et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018;36(4):338–345. doi:10.1038/nbt.4060
11. Madoui MA, Engelen S, Cruaud C, et al. Genome assembly using Nanopore-guided long and error-free DNA reads. BMC Genomics. 2015;16(1):327. doi:10.1186/s12864-015-1519-z
12. Yang L, Haidar G, Zia H, et al. Metagenomic identification of severe pneumonia pathogens in mechanically-ventilated patients: a feasibility and clinical validity study. Respir Res. 2019;20(1):265. doi:10.1186/s12931-019-1218-4
13. Zhang J, Gao L, Zhu C, et al. Clinical value of metagenomic next-generation sequencing by Illumina and Nanopore for the detection of pathogens in bronchoalveolar lavage fluid in suspected community-acquired pneumonia patients. Front Cell Infect Microbiol. 2022;12:1021320. doi:10.3389/fcimb.2022.1021320
14. Saied WI, Mourvillier B, Cohen Y, et al. A comparison of the mortality risk associated with ventilator-acquired bacterial pneumonia and nonventilator ICU-acquired bacterial pneumonia. Crit Care Med. 2019;47(3):345–352. doi:10.1097/ccm.0000000000003553
15. Cillóniz C, Torres A, Niederman MS. Management of pneumonia in critically ill patients. BMJ. 2021;375:e065871. doi:10.1136/bmj-2021-065871
16. Charalampous T, Kay GL, Richardson H, et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat Biotechnol. 2019;37(7):783. doi:10.1038/s41587-019-0156-5
17. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
18. Xu JY, Huang Q, Yu JH, et al. Metagenomic next-generation sequencing for the diagnosis of suspected opportunistic infections in people living with HIV. Infect Drug Resist. 2022;15:1767–1775. doi:10.2147/idr.S350047
19. Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33(1):159–174. doi:10.2307/2529310
20. Dunlap DG, Marshall CW, Fitch A, et al. Improved detection of culprit pathogens by bacterial DNA sequencing affects antibiotic management decisions in severe pneumonia. Am J Case Rep. 2018;19:1405–1409. doi:10.12659/ajcr.912055
21. Wu X, Li Y, Zhang M, et al. Etiology of severe community-acquired pneumonia in adults based on metagenomic next-generation sequencing: a prospective multicenter study. Infect Dis Ther. 2020;9(4):1003–1015. doi:10.1007/s40121-020-00353-y
22. Sun T, Wu X, Cai Y, et al. Metagenomic next-generation sequencing for pathogenic diagnosis and antibiotic management of severe community-acquired pneumonia in immunocompromised adults. Front Cell Infect Microbiol. 2021;11:661589. doi:10.3389/fcimb.2021.661589
23. Leone M, Garcin F, Bouvenot J, et al. Ventilator-associated pneumonia: breaking the vicious circle of antibiotic overuse. Crit Care Med. 2007;35(2):379–85; quizz 386. doi:10.1097/01.ccm.0000253404.69418.aa
24. Liu VX, Fielding-Singh V, Greene JD, et al. The timing of early antibiotics and hospital mortality in sepsis. Am J Respir Crit Care Med. 2017;196(7):856–863. doi:10.1164/rccm.201609-1848OC
25. Luyt CE, Hékimian G, Koulenti D, Chastre J. Microbial cause of ICU-acquired pneumonia: hospital-acquired pneumonia versus ventilator-associated pneumonia. Curr Opin Crit Care. 2018;24(5):332–338. doi:10.1097/mcc.0000000000000526
26. Ho DY, Enriquez K, Multani A. Herpesvirus infections potentiated by biologics. Infect Dis Clin North Am. 2020;34(2):311–339. doi:10.1016/j.idc.2020.02.006
27. Clementi N, Cappelletti F, Criscuolo E, et al. Role and potential therapeutic use of antibodies against herpetic infections. Clin Microbiol Infect. 2017;23(6):381–386. doi:10.1016/j.cmi.2016.12.023
28. Wu N, Ranjan P, Tao C, et al. Rapid identification of pathogens associated with ventilator-associated pneumonia by Nanopore sequencing. Respir Res. 2021;22(1):310. doi:10.1186/s12931-021-01909-3
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.