")
Back to Journals » International Medical Case Reports Journal » Volume 14
Partial N Gene Sequencing for SARS-CoV-2 Verification and Pathway Tracing
Authors Lee SH , McGrath J , Connolly SP, Lambert J
Received 9 November 2020
Accepted for publication 25 December 2020
Published 11 January 2021 Volume 2021:14 Pages 1—10
DOI https://doi.org/10.2147/IMCRJ.S291166
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Sin Hang Lee,1 Jonathan McGrath,2 Stephen P Connolly,2 John Lambert2
1Milford Molecular Diagnostics Laboratory, Milford, CT, USA; 2Department of Infectious Diseases, Mater Misericordiae University Hospital, Dublin, Ireland
Correspondence: Sin Hang Lee
Milford Molecular Diagnostics Laboratory, 2044 Bridgeport Avenue, Milford, CT 06460, USA
Tel +1 203-878-1438
Fax +1 203-878-0109
Email [email protected]
Abstract: When SARS-CoV-2 prevalence is low, many RT-qPCR-positive test results are false positives. Sequencing of a 398-bp cDNA PCR amplicon derived from a highly conserved segment with single nucleotide polymorphisms of the nucleocapsid (N) gene in presumptive positive samples can verify true positives and differentiate at least 27 phylogenetically distinct strains of SARS-CoV-2 for helping track virus strain movement between individuals and across geographical areas. We report using this partial N gene sequencing method to confirm a case of mild COVID-19 disease. The patient was first seen on March 15, 2020, in the emergency department of the university hospital in Dublin, Ireland. RT-qPCR test on a nasopharyngeal swab sample was positive for SARS-CoV-2. Partial sequencing of the N gene in the residue of the tested RNA extract showed a characteristic set of 3-consecutive GGG-to-AAC mutations at positions 28881, 28882, 28883, which is known to first appear in samples collected in Continental Europe in February 2020. Using this sequencing-based method to re-test 9 reference nasopharyngeal swab samples supplied by the Connecticut State Department of Public Health Microbiology Laboratory revealed that 2 of the 9 positive samples had a single nucleotide mutation in the 398-base segment of the SARS-CoV-2 N gene. One of the 2 mutant samples showed a mutation at position 28821, which was first reported in a sample recently collected in the neighboring New York state. The other sample showed a novel frameshift nucleotide “A” insertion between position 29051 and position 29057, which co-existed with its wildtype parental virus in one sample. Routine sequencing of RT-qPCR-positive samples can minimize or eliminate false-positive SARS-CoV-2 test results that may cause unnecessary anxiety among the population and prevent false-positive tests from shutting down schools and workplaces unnecessarily as businesses try to resume normal operations in the community.
Keywords: false-positive, Sanger sequencing, phylogenetically distinct strains, COVID-19, SARS-CoV-2 verification, single nucleotide mutation, partial N gene sequencing, virus strain tracing
Introduction
According to the World Health Organization (WHO) Coronavirus disease (COVID-19) Weekly Epidemiological Update and Weekly Operational Update report, as of 13 December 2020, the COVID-19 cases and deaths continued to rise with 70 million cumulative cases and 1.6 million deaths globally since the start of the pandemic. The Regions of the Americas and Europe continue to shoulder the burden of the pandemic, accounting for 85% of new cases and 86% of new deaths globally. SARS-CoV-2 is the causative agent of the COVID-19 pandemic.1
Accurate diagnosis as well as identification and management of symptomatic and asymptomatic COVID-19 cases are important for proper patient care and for making appropriate public health policies.
The first genome sequence for SARS-CoV-2 was officially published in the GenBank database on January 12, 2020 (GenBank Sequence ID# MN908947.1). This allowed the rapid development of several real-time or quantitative polymerase chain reaction (RT-qPCR) assays, including the test kit panel developed by the US Centers of Disease Control and Prevention (CDC).2
However, the accuracy of these tests marketed under Emergency Use Authorization (EUA) remains unknown. A nasopharyngeal swab RT-qPCR test can be inaccurate in two ways. A false-positive result erroneously labels a person infected by SARS-CoV-2, with consequences including unnecessary quarantine and contact tracing, isolation of non-infected nursing home residents with COVID-19 patients in one room, and introduction of uncertainty in vaccine efficacy evaluation. The additional costs and consequences such results incur are inherently difficult to quantify; however, are estimated to be significant.3
False-negative results are more apparent, and were thought to be more consequential because infected persons who might be asymptomatic might not be isolated and could infect others.4 During the early months of the COVID-19 outbreak when hospitalized patients received the most attention, concerns were focused on the false-negative results of RT-qPCR tests. For example, in the month from January 6 to February 6, 2020, 308 of 413 (75%) patients admitted to a Wuhan teaching hospital in China who tested negative for SARS-CoV-2 by an RT-qPCR assay were found to have chest CT findings considered diagnostic of COVID-19 pneumonia.5 From March 23 to May 19, 2020, one specialized hospital in Italy admitted 16 patients with high suspicion of COVID-19 based on clinical and chest high-resolution-computed tomography (HRCT) findings for treatment despite negative test results of RT-qPCR on at least two consecutive nasopharyngeal swabs. During the treatment course in the hospital, only 3 of these 16 (18.7%) patients were found to be RT-qPCR positive upon re-testing.6
However, when the RT-qPCR assays were used to test the nasopharyngeal swabs of the patients with mild non-specific symptoms or asymptomatic persons with suspected infection, the problems of the false-positive test results began to surface. For one notable example, 77 positive SARS-CoV-2 test results on a group of football players in the United States all turned out to be false positives on repeat testing.7 The US Food and Drug Administration (FDA) has officially alerted clinical laboratory staff and health-care providers of an increased risk of false-positive results with some of these commercial test kits.8,9
With indiscriminate testing or screening in a population with low prevalence, many positive SARS-CoV-2 test results are in fact false positives. For example, using two different nucleic acid assays to re-test 52 first-time SARS-CoV-2-positive samples, Skittrall et al detected SARS-CoV-2 RNA in 29 (56%), but not in 23 (44%) of the 52 samples in the second-round testing for confirmation of true SARS-CoV-2 infection.10 It is of course questionable if the first positive tests, or the repeat negative tests on some of these samples with discordant results were correct. Elimination or minimization of false-positive SARS-CoV-2 RNA test results will reduce unnecessary anxiety among the population and prevent false-positive test results from shutting down schools and workplaces unnecessarily as businesses try to resume normal operations in the community.
Even at the early stage of the COVID-19 outbreak, both the WHO and the FDA had concerns about the potential flaws of the RT-qPCR tests being used for molecular detection of SARS-CoV-2 in clinical specimens.
In recognizing the fact that all nucleic acid-based tests depend on determination of the sequence of the nucleotides in the genome of the pathogen, the WHO guidance titled “WHO Laboratory testing for coronavirus disease (COVID-19) in suspected human cases-Interim guidance dated 19 March 2020” advises
Routine confirmation of cases of COVID-19 is based on detection of unique sequences of virus RNA by NAAT such as real-time reverse-transcription polymerase chain reaction (rRT-PCR) with confirmation by nucleic acid sequencing when necessary.11
Public records show that the FDA first issued a letter on February 4, 2020, authorizing emergency use of the CDC 2019-Novel Coronavirus (2019-nCoV, renamed as SARS-CoV-2) Real-Time Reverse Transcriptase (RT)-PCR Diagnostic Panel for the presumptive qualitative detection of nucleic acid from the 2019-nCoV in upper and lower respiratory specimens.12 Therefore, RT-qPCR tests for SARS-CoV-2 have been considered presumptive qualitative in nature by the FDA from the early days of the pandemic.
To resolve the problems caused by these inherently inaccurate tests, the FDA’s position is that false test results can be investigated using an additional EUA RT-qPCR assay, and/or Sanger sequencing.13 As another EUA RT-qPCR test may also generate false-negative or false-positive results, Sanger sequencing is the more reliable option.
Full-length genome sequencing can provide unquestionable evidence for SARS-CoV-2 detection,14 and has been used to identify phylogenetically distinct virus strains that are required for evaluation of COVID-19 reinfection cases as well as for tracing virus infection pathways and designing preventive strategies.15–17 However, routine sequencing of the almost 30,000-base full-length SARS-CoV-2 genome on every presumptive positive sample for diagnostic confirmation is costly and impractical in clinical practice.
According to the FDA guidance on molecular diagnosis of viral infection caused by human papillomavirus (HPV), a conventional PCR detection of genomic DNA followed by Sanger sequencing on both strands of the PCR amplicon (bi-directional sequencing) that contains a minimum of 100 contiguous bases is acceptable as valid diagnostics for HPV infection provided the sequence matches the reference or consensus sequence, eg with an Expected Value (E Value) <10−30 for the specific HPV DNA target based on a BLAST search of the GenBank database.18 Based on this FDA guidance, a protocol using the nested PCR amplicon of a 398-base SARS-CoV-2 N gene cDNA as the template for Sanger sequencing was developed for confirmatory diagnosis.19
We report our initial experience of using this partial N gene sequencing protocol for verification of a case of mild COVID-19 and discuss the possible use of the sequence data to reconstruct the possible immediate pathway of the infection. Routine analysis of the hypervariable regions of this highly conserved 398-base segment of the N gene on PCR-positive samples may identify many phylogenetically distinct strains for timely tracking SARS-CoV-2 strain movement between individuals and across geographical areas in a community.
Case Report and Partial Sequencing of Positive Samples
Case Report
On March 15, 2020, a 25-year-old female health-care worker with no significant past medical history presented to the Emergency Department of Mater Misericordiae University Hospital, Dublin, Ireland, with fever, sore throat, dry cough, fatigue and myalgia on a background of recent travel to France. A nasopharyngeal swab RT-qPCR testing for SARS-CoV-2 was positive (VIASURE SARS-CoV-2 Real Time PCR Detection Kit), with a Ct value 26.58. Vital signs were all within normal range. Laboratory investigations and Chest X-Ray were unremarkable. Following a 24-hour period of observation, the patient was discharged for a planned 14 days of self-isolation. The patient had an uneventful clinical course, reporting full resolution of symptoms, except for ongoing fatigue, and returning to work 19 days after initial symptom onset.
In mid-April, 36 days following the initial positive swab result and 40 days following the initial onset of symptoms, the patient re-presented to the Emergency Department reporting a 5-day history of mild dry cough, myalgia and 10 days of sore throat/coryzal symptoms. Vital signs showed O2 saturations via pulse oximetry of 99% on room air, a respiratory rate of 16 breaths per minute, heart rate of 122 beats per minute, blood pressure of 156/98 mmHg and a temperature 37.1°C. A chest radiograph revealed a focus of patchy opacification in the medial right lower zone. Repeat SARS-CoV-2 PCR testing was positive, with a Ct value of 30.72. A nasopharyngeal aspirate screening test reported negative for a broad range of other respiratory viruses. The severity of symptoms did not warrant admission and the patient was discharged for a further 14 days self-isolation. A subsequent SARS-CoV-2 PCR test performed 38 days post initial result (42 days post symptom onset) was positive with a Ct value 39.4 (Xpert® Xpress SARS CoV-2 kit). A negative result was obtained 40 days after first positive result and 44 days following illness onset. Anti-SARS-CoV-2 IgG was detected via point of care testing 58 days post original positive swab result. The patient made a full clinical recovery. An aliquot of the residue of the nasopharyngeal RNA extract tested positive on March 15, 2020, was sent on July 21, 2020, to Milford Molecular Diagnostics Laboratory for confirmatory DNA sequencing test according to a protocol previously published.19 A bi-directional sequencing of the heminested PCR amplicon of the cDNA showed a typical 398-base SARS-CoV-2 N gene sequence with 28881, 28882, 28883 GGG-to-AAC mutations as illustrated in Figure 1, panel a in comparison with panel b of the Wuhan-Hu-1 prototype sequence.
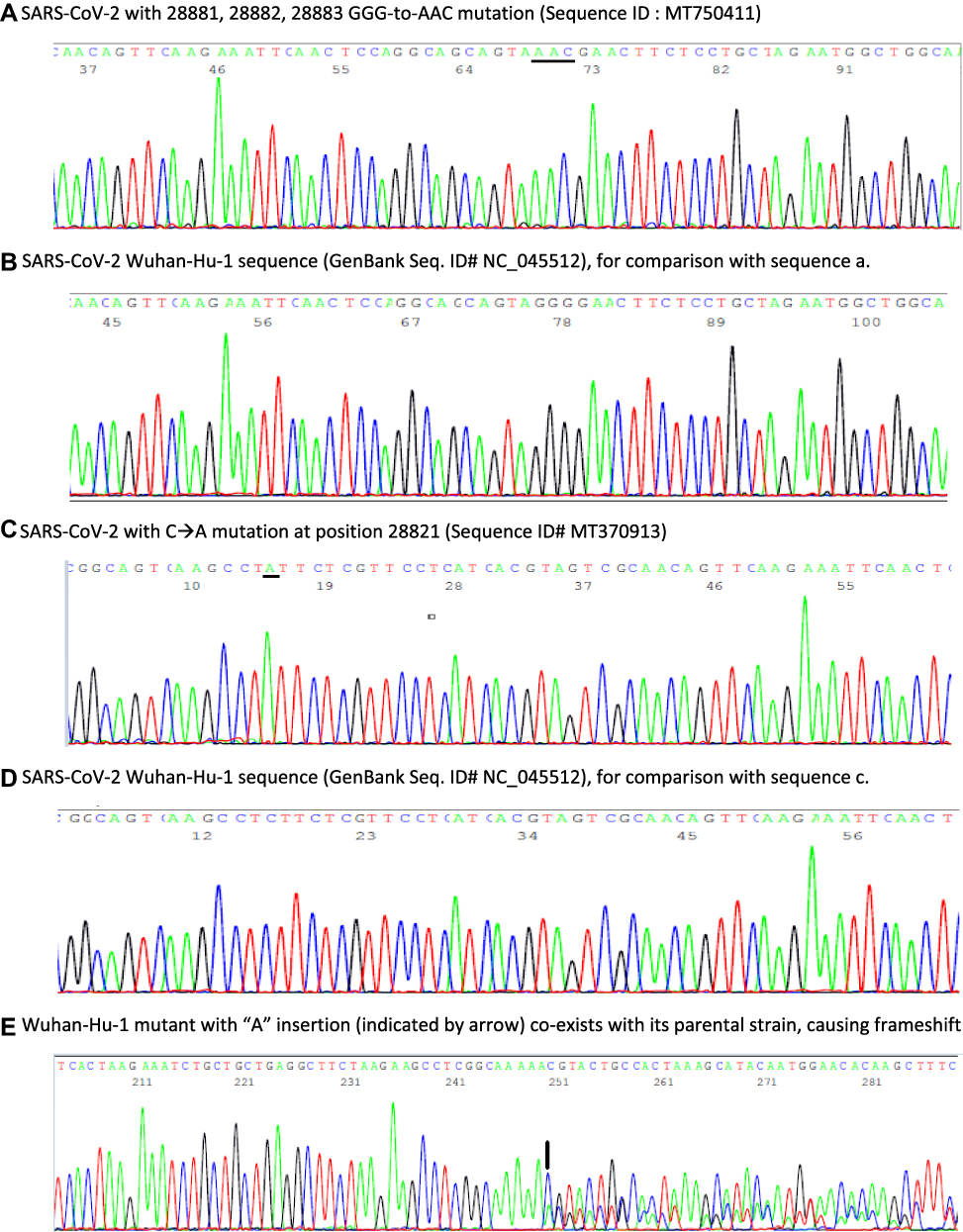 |
Figure 1 Panels of computer-generated base-calling DNA sequencing electropherograms, each excised from a 398-base highly conserved SARS-CoV-2 N gene sequence showing samples of prototype SARS-CoV-2 Wuhan-Hu-1 strain and 3 single nucleotide polymorphisms. (A) 3-consecutive 28881, 28882, 28883 GGG-to-AAC (underlined) mutations; (B) Prototype Wuhan-Hu-1 sequence of a; (C) 28821 C-to-A (underlined) mutation; (D) Prototype Wuhan-Hu-1 sequence of c; (E): A mutant sequence with a novel nucleotide “A” insertion between position 29051 and position 29057, resulting in an extra “A” at position 29057 (indicated by an arrow) and its wildtype parental sequence derived from one sample, causing a downstream frameshift in DNA sequencing with overlapping of two sets of base-calling peaks after position 29056. No SARS-CoV-2 mutant with a nucleotide “A” insertion between position 29051 and position 29057 was registered in the global database updated on October 30, 2020.21 |
Partial Sequencing of 9 Positive Samples
The partial N gene sequencing protocol was also used to re-test 9 SARS-CoV-2 RNA-positive human nasopharyngeal samples received from Connecticut State Department of Public Health Microbiology Laboratory on April 30, 2020. The results showed that 7 of the 9 RT-PCR-positive samples contained the prototype SARS-CoV-2 N gene sequence identical to that of the Wuhan-Hu-1 strain in this 398-base segment.
Two of the 9 positive samples showed a single nucleotide mutation within the 398-base sequence. One of the mutations occurred at position 28821 as illustrated in Figure 1C, in comparison with panel d of a prototype sequence. This C-to-A mutation (Sequence ID# MT370913) was first reported in a “nasal swab” specimen collected on March 17, 2020, in the State of New York (a contiguous State with Connecticut) according to information retrieved from the GenBank database updated in June 2020.
The other mutation detected was a single nucleotide “A” insertion, as shown in Figure 1E. This novel mutant co-infecting the host along with its wildtype parental virus caused a frameshift in Sanger sequencing. Frameshift nucleotide deletions in the SARS-CoV-2 genome have been observed occasionally.20 However, a SARS-CoV-2 mutant with frameshift nucleotide insertion in the genome, especially co-existing with its wildtype parental virus in one patient sample, has not been previously reported.20,21
It is generally assumed that all SARS-CoV-2 genomic nucleotide sequences shared one common ancestor towards the end of 2019 before undergoing numerous mutations as it spreads throughout the globe.21 The N gene appears to be the most conserved within the SARS-CoV-2 genome.22 Yet as of October 30, 2020, the total known number of single nucleotide mutations in the 1260-base N gene reached 948.21 However, the virus number with each specific mutation varies widely from 1 to >39,000. The most frequent 46 of the 948 single nucleotide mutation sites with ≥ 200 virus number21 (ie reported in at least 200 sequenced isolates) are summarized in Figure 2.
 |
Figure 2 SARS-CoV-2 Wuhan-Hu-1 N gene reference sequence (retrieved from the GenBank database-Sequence ID# NC_045512.2) with common single nucleotide mutation sites high-lighted green. According to the global variation annotation database published by China National Center for Bioinformation 2019 Novel Coronavirus Resource,21 the total worldwide reported virus number with single nucleotide variation reached 87,568 as of October 30, 2020. Within the SARS-CoV-2 N gene, there are 46 single nucleotide polymorphisms each of which has been reported for ≥200 times (>39,000 times at positions 28881, 28882 and 28883). These mutations are not evenly distributed in the N gene. As shown in Figure 2, 27 of the 46 frequent mutation sites (highlighted green) cluster in a 398-base segment of the 1260-base N gene. These 27 hypervariable nucleotides are flanked by two highly conserved sequences at both ends (typed in red), which serve as the nested PCR primer sites for amplification. As reference, the start codon (atg) and the stop codon (taa) of the N protein gene are highlighted yellow. The N1 sequence (28287–28358) and N2 sequence (29164–29230) targeted by the CDC RT-qPCR test kit for SARS-CoV-2 RNA detection are bold-faced and grey-shaded for location comparisons. |
Since 27 of the 46 most frequent single nucleotide mutations are located within a cluster of the 398-base SARS-CoV-2 N gene targeted for diagnostic confirmation (Figure 2), these single nucleotide polymorphisms (SNPs) or mutations, if present and demonstrated in the Sanger sequencing electropherogram, can also be used to identify phylogenetically distinct strains for tracking strain movement.
Discussion
The patient described in this case report first presented to the Emergency Department with fever, sore throat, dry cough, fatigue and myalgia, and again with one focus of afebrile mild pneumonia 36 days later. This clinical picture is non-specific. The diagnosis of a “mild case” of COVID-1923 was supported by a positive VIASURE SARS-CoV-2 Real Time PCR test. The VIASURE SARS-CoV-2 (N1 + N2) Real Time PCR Detection Kit was adapted for the BD MAX System.24 However, the FDA has also cautioned all clinical laboratory staff and health-care providers that false-positive results can be generated with BD SARS-CoV-2 Reagents for the BD Max System.8
According to the FDA guidance, demonstration of a minimum of 100 unambiguous contiguous bases of a DNA sequence matching a unique reference genome sequence published in the GenBank database is acceptable as evidence for detection of HPV in a clinical sample.18 The Sanger sequencing data of a 398-bp cDNA amplicon of the N gene derived from the residue of the first sample RNA extract verified that this patient was indeed infected with a strain of SARS-CoV-2 beyond a reasonable doubt.
The SARS-CoV-2 detected in this case had a characteristic set of 3-consecutive GGG-to-AAC mutations at positions 28881, 28882, 28883 in its N gene (Figure 1A). These 3-consecutive nucleotide mutations were first observed in strains isolated in Continental Europe in February 2020.16,21 Based on the GenBank database, the first American strain of SARS-CoV-2 with GGG-to-AAC mutations at positions 28881, 28882, 28883 was discovered in a nasopharyngeal swab sample collected in California on April 15, 2020 (GenBank Sequence ID# MT750411). Therefore, the source of the SARS-CoV-2 found in Ireland on March 15, 2020, could not be linked to the United States.
The COVID-19 pandemic reached the Republic of Ireland on February 29, 2020.25 In March, the full-length genomes of 10 SARS-CoV-2 isolates in human specimens collected from March 3 to March 10, 2020, were sequenced and the nucleotide sequences deposited in the GISAID database.21 Nine of the 10 deposited sequences showed 4–9 SNPs in various genes. Only one isolate retained the prototype Wuhan-Hu-1 genome sequence. Among the 9 mutants, there were 5 SARS-CoV-2 sequences with the 3-consecutive 28881, 28882, 28883 GGG-to-AAC mutations. Chronologically, these latter 5 specimens were collected on March 6 in Dublin (GISAID Accession ID# EPI_ISL_418548), on March 6 in Tipperary (GISAID Accession ID# EPI_ISL_418516), on March 8 in Dublin (GISAID Accession ID# EPI_ISL_418581), on March 8 in Cord (GISAID Accession ID# EPI_ISL_418580), and on March 10 in Wicklow (GISAID Accession ID# EPI_ISL_418584),21 four southern-southeastern counties in Ireland. Since the patient in the current report apparently had her initial onset of symptoms on March 11, 2020, and the COVID-19 incubation period may range from 2 to 14 days,26 the source of her infection could be traced back to possible contacts with symptomatic or asymptomatic patients from these 4 counties if her recent trip to France was ended prior to February 25, 2020, ie 14 days before her initial onset of symptoms. This notwithstanding, the patient described in this report was not the first person carrying this mutant of SARS-CoV-2 into the Republic of Ireland, an island nation on the periphery of Europe.
Re-testing 9 SARS-CoV-2 positive samples collected in the state of Connecticut, USA showed 2 isolates with single nucleotide mutation in the 398-base N gene segment. One of them involved a recent mutation at position 28821 (Figure 1C). Based on the information retrieved from the GenBank database updated in June 2020, the first SARS-CoV-2 N gene C-to-A mutation at position 28821 was discovered in a specimen collected in New York State on March 17, 2020. Confirmation of this viral strain with this SNP in a sample supplied by the Connecticut State Department of Public Health Microbiology Laboratory on April 30 suggests that this virus strain probably spread from the neighboring New York State after March 17. The reported SARS-CoV-2 virus number with single nucleotide mutation at position 28821 reached 732 worldwide on November 4, 2020, from zero in early March 2020.21 If routine sequencing of this 398-bp cDNA had been performed immediately on all positive SARS-CoV-2 samples in New York and Connecticut, the infection pathways of many phylogenetically distinct virus strains could have been tracked to assist public health policy development.
The finding of a SARS-CoV-2 mutant with frameshift nucleotide “A” insertion between position 29051 and position 29057, which co-existed with its wildtype parental virus in one sample (Figure 1E), was unexpected because frameshift nucleotide insertion in the N gene was not reported and entered into the global database. During genome replication within the host, viruses often acquire genome mutations. The rates of spontaneous mutation vary widely among viruses. RNA viruses mutate faster than DNA viruses. However, only when those mutations can be passed down to descendant viruses in subsequently infected individuals,27 the mutations can be observed, documented, and reported to the GenBank. If the frameshift nucleotide “A” insertion was detrimental to the virus,27 this mutation could not be passed to the viral progeny and would not have been recorded in the GenBank database. Since the cDNA of the mutated sequence was co-amplified along with the cDNA of its wildtype parental sequence during the nested PCR process, based on evaluation of the electropherogram of the overlapped sequences (Figure 1E) the viral load of the mutant in the clinical sample was probably comparable to that of its wildtype parental virus. The biological significance of such a frameshift nucleotide insertion on the host is unknown.
When there are proven false-positive SARS-CoV-2 RT-qPCR tests, regulators and literature reviewers tend to lay blame on technical errors, such as reagent or sample cross contaminations,3,28 or lack of quality control for manufacturing a specific test kit,8,9 for the cause of the false positives. However, the inherent flaw of PCR, especially qPCR, as a diagnostic tool for infectious diseases warrants discussion.
Real-time or quantitative PCR (qPCR) was first described in 1993 to monitor the accumulation of double-stranded DNA (dsDNA) being generated in each PCR using the increase in the fluorescence of ethidium bromide (EtBr) that results from its binding to dsDNA as the PCR products. The kinetics of fluorescence accumulation during thermocycling are directly related to the starting number of DNA copies. The basic principle dictates that the fewer cycles necessary to produce a detectable fluorescence, the greater the number of target sequences in the sample being tested. Results obtained with this approach indicate that a kinetic approach to PCR analysis can quantitate DNA.29 This is referred to as dye-based qPCR for quantitation of small amounts of target DNA known to exist in a sample. It was not designed to determine if a target DNA is present in the sample being tested.
When qPCR is adapted into a “plus/minus” or a “yes/no” assay for the purpose of making diagnosis of an infectious disease, the dye-based qPCR is converted to a probe-based qPCR. Instead of a free dye like EtBr, a target-specific probe that is an oligonucleotide (ssDNA) of about 25 bases long, complementary to a segment of the target DNA sequence, is introduced into the probe-based qPCR in addition to the PCR primers. The most common probe type is a hydrolysis probe, which incorporates a fluorophore attached to the 5ʹ end and a quencher attached to the 3ʹ end of the probe, as the TaqMan® probes commonly used in the SARS-CoV-2 RT-qPCR test kits.2
Fluorescence resonance energy transfer (FRET) prevents fluorescence emission of the fluorophore due to proximity of the quencher while the probe is intact. If a target DNA template is present in the PCR mixture, the probe is hydrolyzed during enzymatic primer extension and amplification of the specific sequence to which the primer is bound. The cleavage and degradation of the probe by the 5ʹ-3ʹ exonuclease activity of the Taq polymerase separate the fluorophore from the quencher, allowing fluorescence of the fluorophore and resulting in an amplification-dependent increase in fluorescence. In other words, diagnostic qPCR actually uses the PCR process to test if DNA/DNA binding (hybridization) has taken place between a set of known oligonucleotides (primers and probe) and a DNA molecule in the sample. It assumes the primers and the probe were all bound to their respective segments of a target ssDNA with fully matching bases before a fluorescence signal was emitted as the result of PCR amplification-dependent degradation of the probe.
In reality, however, this assumption is not always valid. In the nasopharyngeal swab samples taken from the patients, there are numerous human cells, bacteria, viruses, plasmids and fungi all of which can contribute nucleic acids, namely DNAs and RNAs, to the sample extract being tested even when there is no SARS-CoV-2 RNA in the specimen. In the absence of fully matching SARS-CoV-2 genomic RNA or cDNA as the preferred target template, the PCR primers and the probe can bind to partially matched DNA and initiate enzymatic primer extensions and probe degradation. A minimum of only 6 nucleotides matching the sequence of any DNA at the 3′ end of a primer is required to initiate enzymatic primer extension.30 PCR amplification may take place if there is a nontarget DNA with two segments of sequences partially matching those of the primer pair in the reaction mixture to initiate the first PCR cycle. Exponential primer-defined PCR amplification of non-target DNA will proceed after the first PCR cycle is completed.
If such an undesirable PCR should take place and if the interprimer region of the PCR product also had a sequence matching part of the probe, the probe would attach to the PCR product and undergo hydrolysis by the action of the DNA polymerase during PCR amplification, leading to separation of the fluorophore from its quencher cycle after cycle, and eventually to a false-positive result.
PCR amplification of undesirable DNA in clinical diagnostic work is a well-known phenomenon although it is only infrequently reported in the world literature. For examples, PCR amplification of unintended DNA from Pusillimonas, an environmental bacterial species often contaminating patient blood samples, by a pair of specific primers designed for Borrelia burgdorferi DNA amplification,31 PCR amplification of human genomic DNA by PCR primers designed for human papillomavirus L1 gene DNA amplification,32 and unexpected PCR amplification of Homo sapiens BAC clone RP11-154F14 by the CDC’s primers designed for human RNase P gene19 have been confirmed by DNA sequencing and reported in peer-reviewed journals. These Sanger sequencing-proven nonspecific PCR amplifications observed during testing clinical samples, all due to partial base-matching between primer and unintended DNA, have provided a mechanism for false positives in PCR-based SARS-CoV-2 tests.
Diagnostic qPCR has been known to be associated with high frequency of false-positives.33 Since its success depends on a high ratio of template/non-template DNA in the reaction mixture,34 qPCR is not suitable for diagnosis of spirochetemia in Lyme disease patients due to the presence of an overwhelming amount of human genomic DNA in the blood sample. A group of researchers concluded in 2001 that until the specificity of qPCR techniques is determined, the clinical utility of such testing relative to other testing modalities for Lyme disease will remain uncertain.35 The same conclusion should apply to the use of RT-qPCR for the diagnosis of COVID-19, a more pressing and pivotal public health at present than Lyme disease.
Conclusion
In the setting of low SARS-CoV-2 prevalence, routine sequencing of a unique 398-base highly conserved segment of the N gene with clustered single nucleotide polymorphisms can confirm PCR-based true-positive test results and eliminate false positives beyond a reasonable doubt and may provide valuable information for tracing virus strain movement in the community. As businesses and schools attempt to return to normal operations, extremely accurate molecular tests for SARS-CoV-2 with 100% specificity must be implemented for timely verification of true asymptomatic or mild cases of COVID-19 for making appropriate public health policies, thus mitigating the unnecessary panic in a population and negative impacts on local economies resulting from false-positive results.
Ethics and Consent
Institutional approval was not required for a case report. Written informed consent has been provided by the patient to have the case details published.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding was received for this case report.
Disclosure
SHL is the director of a CLIA-certified high-complexity molecular diagnostic laboratory and reports no other potential conflicts of interest for this work. The other authors declare that they have no conflicts of interest for this work.
References
1. WHO Coronavirus disease (COVID-19) weekly epidemiological update and weekly operational update; December 15, 2020. Available from: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports.
2. CDC. 2019-Novel Coronavirus (2019-nCoV) real-time RT-PCR diagnostic panel. Instructions for use. Available from: https://www.fda.gov/media/134922/download.
3. Surkova E, Nikolayevskyy V, Drobniewski F. False-positive COVID-19 results: hidden problems and costs. Lancet Respir Med. 2020;8:1167–1168. doi:10.1016/S2213-2600(20)30453-7
4. Woloshin S, Patel N, Kesselheim AS. False negative tests for SARS-CoV-2 infection - challenges and implications. N Engl J Med. 2020;383:e38. doi:10.1056/NEJMp2015897
5. Ai T, Yang Z, Hou H, et al. Correlation of chest CT and RT-PCR testing for coronavirus disease 2019 (COVID-19) in China: a report of 1014 cases. Radiology. 2020;296(2):E32–E40. doi:10.1148/radiol.2020200642
6. Di Paolo M, Iacovelli A, Olmati F, et al. False-negative RT-PCR in SARS-CoV-2 disease: experience from an Italian COVID-19 unit. ERJ Open Res. 2020;6:00324–2020. PMID: 32685435; PMCID: PMC7357270. doi:10.1183/23120541.00324-2020
7. Patra K. Around the NFL- All 77 false-positive COVID-19 tests come back negative upon reruns; August 24, 2020. Available from: https://www.nfl.com/news/all-77-false-positive-covid-19-tests-come-back-negative-upon-reruns.
8. FDA. False positive results with BD SARS-CoV-2 reagents for the BD max system - letter to clinical laboratory staff and health care providers. Available from: https://www.fda.gov/medical-devices/letters-health-care-providers/false-positive-results-bd-sars-cov-2-reagents-bd-max-system-letter-clinical-laboratory-staff-and.
9. FDA. Risk of inaccurate results with thermo fisher scientific TaqPath COVID-19 combo kit - letter to clinical laboratory staff and health care providers. Available from: https://www.fda.gov/medical-devices/letters-health-care-providers/risk-inaccurate-results-thermo-fisher-scientific-taqpath-covid-19-combo-kit-letter-clinical?utm_campaign=2020-08-17%20Risk%20of%20Inaccurate%20Results%20with%20Thermo%20Fisher%20Scientific%20TaqPath&utm_medium=email&utm_source=Eloqua.
10. Skittrall JP, Wilson M, Smielewska AA, et al. Specificity and positive predictive value of SARS-CoV-2 nucleic acid amplification testing in a low prevalence setting. Clin Microbiol Infect. 2020. doi:10.1016/j.cmi.2020.10.003
11. WHO. Laboratory testing for coronavirus disease (COVID-19) in suspected human cases-Interim guidance 19 March 2020. Available from: https://www.who.int/publications/i/item/10665-331501.
12. Open letter from FDA to Robert R. Redfield, MD, Director, Centers for Disease Control and Prevention; March 15, 2020. Available from: https://www.fda.gov/media/134919/download.
13. FDA. Molecular diagnostic template for laboratories. Policy for coronavirus disease-2019 tests during the public health emergency (Revised) Available from: https://www.fda.gov/media/135659/download.
14. Paden CR, Tao Y, Queen K, et al. Rapid, sensitive, full-genome sequencing of severe acute respiratory syndrome coronavirus 2. Emerg Infect Dis. 2020;26:2401–2405. PMID: 32610037; PMCID: PMC7510745. doi:10.3201/eid2610.201800
15. To KK, Hung IF, Ip JD, et al. COVID-19 re-infection by a phylogenetically distinct SARS-coronavirus-2 strain confirmed by whole genome sequencing [published online ahead of print, 2020 Aug 25]. Clin Infect Dis. 2020:ciaa1275. doi:10.1093/cid/ciaa1275.
16. Van Elslande J, Vermeersch P, Vandervoort K, et al. Symptomatic SARS-CoV-2 reinfection by a phylogenetically distinct strain. Clin Infect Dis. 2020:ciaa1330. doi:10.1093/cid/ciaa1330.
17. Forster P, Forster L, Renfrew C, Forster M. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc Natl Acad Sci U S A. 2020;117:9241–9243. doi:10.1073/pnas.2004999117
18. FDA. Establishing the performance characteristics of in vitro diagnostic devices for the detection or detection and differentiation of human papillomaviruses. Available from: https://www.fda.gov/media/92930/download.
19. Lee SH. Testing for SARS-CoV-2 in cellular components by routine nested RT-PCR followed by DNA sequencing. Int J Geriatr Rehabil. 2020;2:69–96.
20. Koyama T, Platt D, Parida L. Variant analysis of SARS-CoV-2 genomes. Bull World Health Organ. 2020;98:495–504. PMID: 32742035; PMCID: PMC7375210. doi:10.2471/BLT.20.253591
21. China National Center for Bioinformation 2019 Novel Coronavirus Resource (2019nCoVR). Available from: https://bigd.big.ac.cn/ncov/variation/annotation.
22. Grifoni A, Sidney J, Zhang Y, Scheuermann RH, Peters B, Sette A. Candidate targets for immune responses to 2019-novel coronavirus (nCoV): sequence homology- and bioinformatic-based predictions. SSRN. 2020:3541361. PMID: 32714104; PMCID: PMC7366807. doi:10.2139/ssrn.3541361
23. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese Center for disease control and prevention. JAMA. 2020;323:1239–1242. PMID: 32091533. doi:10.1001/jama.2020.2648
24. Becton, Dickinson and Company. CerTest Biotec and BD announce COVID-19 diagnostic test. Available from: https://news.bd.com/2020-03-10-CerTest-Biotec-and-BD-Announce-COVID-19-Diagnostic-Test.
25. BBC News. Coronavirus: first case confirmed in Republic of Ireland; February 29, 2020. Available from: https://www.bbc.com/news/world-europe-51693259.
26. CDC. Coronavirus disease 2019 (COVID-19). Available from: https://www.cdc.gov/coronavirus/2019-ncov/symptoms-testing/symptoms.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fcoronavirus%2F2019-ncov%2Fabout%2Fsymptoms.html.
27. Sanjuán R, Domingo-Calap P. Mechanisms of viral mutation. Cell Mol Life Sci. 2016;73:4433–4448. PMID: 27392606; PMCID: PMC5075021. doi:10.1007/s00018-016-2299-6
28. Kelly M, Cahlan S, Samuels E. What went wrong with coronavirus testing in the US. Washington Post; March 30, 2020. Available from: https://www.washingtonpost.com/politics/2020/03/30/11-100000-what-went-wrong-withcoronavirus-testing-us/.
29. Higuchi R, Fockler C, Dollinger G, Watson R. Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology. 1993;11:1026–1030. PMID: 7764001. doi:10.1038/nbt0993-1026
30. Ryu KH, Choi SH, Lee JS. Restriction primers as short as 6-mers for PCR amplification of bacterial and plant genomic DNA and plant viral RNA. Mol Biotechnol. 2000;14:1–3. PMID: 10911610. doi:10.1385/MB:14:1:01
31. Lee SH, Vigliotti VS, Vigliotti JS, Jones W, Williams J, Walshon J. Early Lyme disease with spirochetemia - diagnosed by DNA sequencing. BMC Res Notes. 2010;3:273. PMID: 21040573; PMCID: PMC2984391. doi:10.1186/1756-0500-3-273
32. Lee SH. Detection of human papillomavirus L1 gene DNA fragments in postmortem blood and spleen after Gardasil® vaccination—A case report. Adv Biosci Biotechnol. 2012;3:1214–1224. doi:10.4236/abb.2012.38148
33. Nowrouzian FL, Adlerberth I, Wold AE. High frequency of false-positive signals in a real-time PCR-based “Plus/Minus” assay. APMIS. 2009;117:68–72. doi:10.1111/j.1600-0463.2008.00010.x
34. Ruiz-Villalba A, van Pelt-verkuil E, Gunst QD, Ruijter JM, van den Hoff MJ. Amplification of nonspecific products in quantitative polymerase chain reactions (qPCR). Biomol Detect Quantif. 2017;14:7–18. doi:10.1016/j.bdq.2017.10.001
35. Nowakowski J, Schwartz I, Liveris D, et al.; Lyme Disease Study Group. Laboratory diagnostic techniques for patients with early Lyme disease associated with erythema migrans: a comparison of different techniques. Clin Infect Dis. 2001;33(12):2023–2027. doi:10.1086/324490
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.