")
Back to Journals » Journal of Inflammation Research » Volume 14
PARP Inhibitors: An Innovative Approach to the Treatment of Inflammation and Metabolic Disorders in Sepsis
Received 6 January 2021
Accepted for publication 19 March 2021
Published 6 May 2021 Volume 2021:14 Pages 1827—1844
DOI https://doi.org/10.2147/JIR.S300679
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Weronika Wasyluk,1,2 Agnieszka Zwolak1
1Chair of Internal Medicine and Department of Internal Medicine in Nursing, Faculty of Health Sciences, Medical University of Lublin, Lublin, Poland; 2Doctoral School, Medical University of Lublin, Lublin, Poland
Correspondence: Weronika Wasyluk
Chair of Internal Medicine and Department of Internal Medicine in Nursing, Faculty of Health Sciences, Medical University of Lublin, Ul. Witolda Chodźki 7, Lublin, 20-093, Poland
Tel +48 81 448 7720
Email [email protected]
Abstract: Sepsis is not only a threat to the health of individual patients but also presents a serious epidemiological problem. Despite intensive research, modern sepsis therapy remains based primarily on antimicrobial treatment and supporting the functions of failing organs. Finding a cure for sepsis represents a great and as yet unfulfilled need in modern medicine. Research results indicate that the activity of poly (adenosine diphosphate (ADP)-ribose) polymerase (PARP) may play an important role in the inflammatory response and the cellular metabolic disorders found in sepsis. Mechanisms by which PARP-1 may contribute to inflammation and metabolic disorders include effects on the regulation of gene expression, impaired metabolism, cell death, and the release of alarmins. These findings suggest that inhibition of this enzyme may be a promising solution for the treatment of sepsis. In studies using experimental sepsis models, inhibition of PARP-1 has been shown to ameliorate the inflammatory response and increase survival. This action was described, among others, for olaparib, a PARP-1 inhibitor approved for use in oncology. While the results of current research are promising, the use of PARP inhibitors in non-oncological diseases raises some concerns, mainly related to the enzyme’s role in deoxyribonucleic acid (DNA) repair. However, the results of studies on experimental models indicate the effectiveness of even short-term PARP-1 inhibition and do not confirm concerns regarding its impact on the integrity of nuclear DNA. Current research presents PARP inhibition as a potential solution for the treatment of sepsis and indicates the need for further research.
Keywords: sepsis, septic shock, inflammation, metabolism, poly(ADP-ribose) polymerases, PARP inhibitors
Introduction
The beginning of this century brought results of epidemiological studies which indicate that sepsis is a global health threat. In a study published in 2001, Angus et al presented the results of a multicentre study conducted in the United States based on the analysis of hospital records of over 6 million patients, of whom 192,280 were diagnosed with severe sepsis(the term “severe sepsis” corresponds to the current definition of sepsis under SEPSIS-3)6. Of these, 51.1% received intensive care and 17.3% received ventilation at an Intermediate Care Unit or Coronary Care Unit. Patients with severe sepsis accounted for 11% of all patients admitted to the Intensive Care Unit (ICU), and in hospital mortality was 28.6%.1 In 2006, the results of a multicentre study conducted in 24 European countries were published. According to this study, 30% of patients admitted to the ICU had severe sepsis, with a mortality rate of 32.2%.2 The importance of these reports was confirmed by the recognition of sepsis as a Global Health Priority by the World Health Organization (WHO) in 2017.3
The concept of sepsis has evolved over time. At the turn of the 20th century, it was defined as the presence of bacteria in the blood and it was believed that the overall severity of illness resulted from the severity of the infection. The theories about the pathogenesis of sepsis did not start to resemble the current ones until the second half of the twentieth century when it was noticed that clinical symptoms are not always accompanied by the presence of microbes in the blood, and that the same pathogen may cause different responses. During the 20th century, endogenous mediators were discovered which were found to contribute to the induction of septic reactions.4 At the end of the 20th century, the definition of sepsis focused on the inflammatory response and sepsis was defined as a systemic inflammatory response syndrome in response to infection, and severe sepsis was defined as sepsis coexisting with organ failure.5 The current definition of sepsis, published in 2016, defines it as “life threatening organ dysfunction caused by a dysregulated host response to infection”.6 Therefore, over time, the perception of sepsis has evolved from an initial focus on infection to inflammation to systemic disturbance of homeostasis. One aspect of these disorders is metabolic dysregulation, including limitation of mitochondrial functions, changes in basic metabolic processes and limitation of the body’s metabolic capacity.7 Additionally, circulating cytokines and associated hormonal disruptions cause insulin resistance, and promote lipolysis and proteolysis, which alter the availability of nutrients for energy production and can lead to wasting.8
Although the metabolic disorders found in sepsis have been studied for a relatively long time,9 for many years the efforts of scientists to find a cure for sepsis have focused on combating inflammation. Despite intensive research, modern sepsis therapy remains based primarily on antimicrobial treatment and supporting failing organs. Since innovative solutions in the treatment of patients with sepsis constitute a great and so far unmet need for modern medicine, it seems justified to pay attention to components of the dysregulated host response to infection, including metabolism.10,11 Poly (ADP-ribose) polymerase (PARP) inhibitors are a promising group of drugs that could both suppress excessive inflammation and address some of the problems arising from metabolic disorders in sepsis. Therefore, the aim of this paper is to discuss the role of PARP in the inflammatory response and to review reports on the effectiveness of PARP inhibition in the treatment of sepsis.
Poly (ADP-Ribose) Polymerase
PARP is an evolutionarily conserved family of proteins involved in numerous biological processes such as deoxyribonucleic acid (DNA) repair, cell division and differentiation, but also in response to stress, immune response, and cell death. The first enzyme with poly (ADP-ribose) polymerase activity was described in 1963 and since then, seventeen human PARPs have been discovered which differ in their structural domains, function and cell location. The PARP superfamily catalyses the attachment of mono (ADP-ribose) (MAR) or poly (ADP-ribose) (PAR) to target proteins using nicotinamide adenine dinucleotide (NAD+) as a substrate. These processes are called MARylation and PARylation.12–14 Of the seventeen PARP found in humans, only a few catalyse PARylation (eg, PARP-1, PARP-2, PARP-5A, PARP-5B), while most catalyse MARylation.15–17 PARPs that catalyse MARylation have been shown to have amino acid substitutions at their catalytic centres that enable them to attach only MAR moieties to the target proteins.18,19 MARylation has been shown to play a role in intracellular signalling, including inflammatory and stress responses.19–21
The differences between the members of the PARP family, including the heterogeneity of the catalysed reactions, meant a new nomenclature for these compounds was proposed in 2010, taking into account their structural and biochemical features. Enzymes once classified as PARP are now called diphtheria toxin-like ADP-ribosyltransferases (ARTD) (Table 1). The name comes from a prototype bacterial toxin to which the proteins of the PARP family are structurally similar.18,22 Due to the numerous references to historic literature, the old nomenclature will be used in this review.
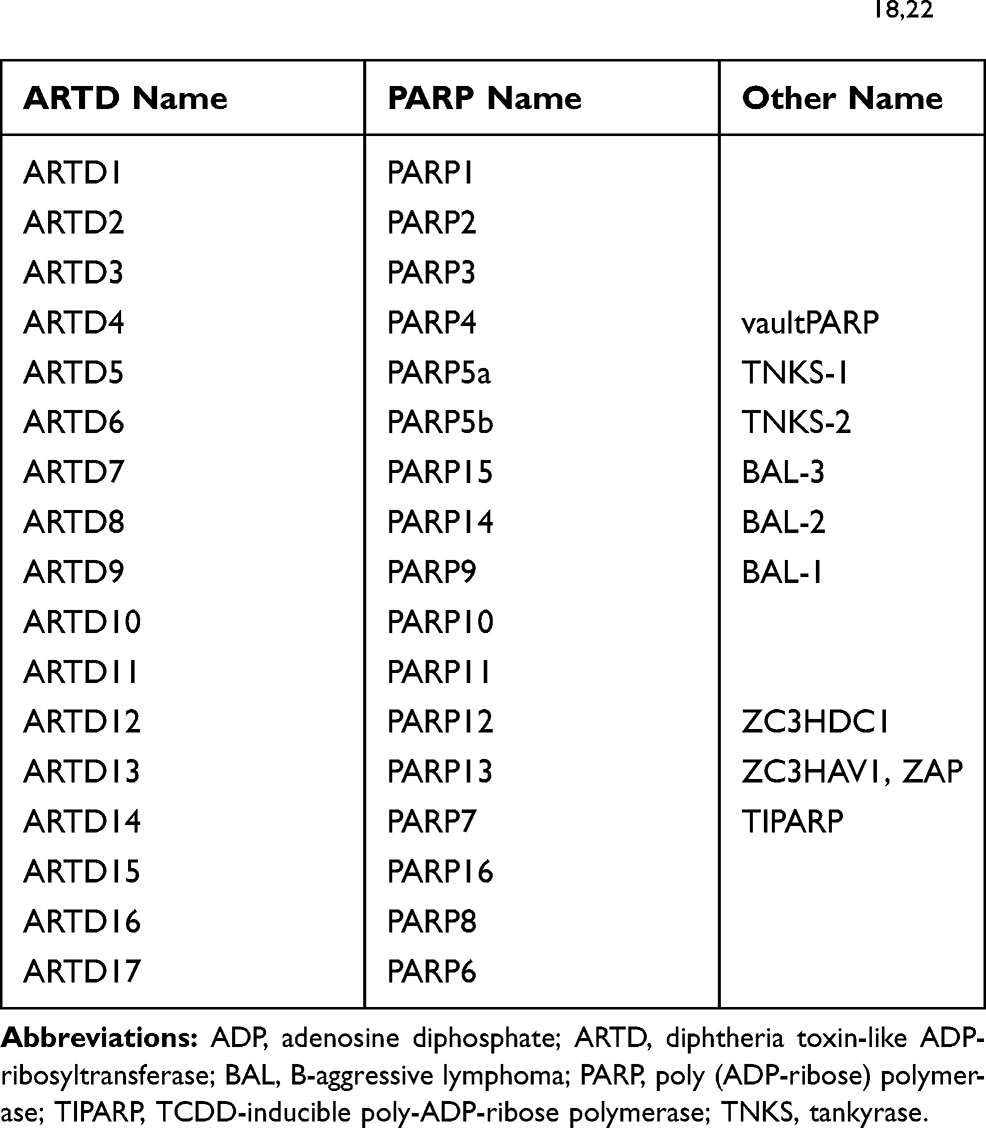 |
The majority of intracellular PARP activity is by PARP-1, which accounts for approximately 90% of NAD+ consumption by the PARP family.23,24 PARP-1 functions to add ADP-ribose moieties to target proteins and comprises an N-terminal, zinc-finger DNA binding domain (DBD), a central automodification domain (AMD), and a C-terminal, catalytic domain (CD).15,25 PAR acceptor proteins include DNA repair enzymes, histones, chromatin regulators, transcription factors, and PARP itself.13,14,26 The CD of PARP-1 is responsible for the addition of ADP-ribose groups, which catalyse three different enzymatic reactions: initiation, elongation and branching (Figure 1). Initiation is the attachment of the first ADP-ribose monomer to the amino acid residue of the acceptor protein. Elongation, the attachment of further monomers, takes place through the formation of a (2ʹ-1ʹʹ) ribose-ribose glycosidic bond. Branching involves the creation of a ribose-ribose bond between ADP-ribose (2ʹʹ-1ʹʹʹ) units.27–29 The dual specificity of PARP-1, allowing both elongation and branching, is unique among polymer-forming enzymes.30 Ruf et al have shown that the first of these reactions is preferred and proposed that branching may occur when the orientation of the bound polymer is reversed by a 180° rotation.30 More recent studies have shown that alongside the ability of PARP-1 to form branched PAR polymers, PARP-2 also has a significant role in the formation of side chains; catalysing additional PARylation at the top of existing PAR chains, resulting in the formation of branched PAR chains.31,32 Differences in the properties of the resulting polymer, such as length and branching structure, have an impact on the cellular response – it has been shown that different PAR reading proteins respond differently to specific features of the polymer structure, eg, some bind only linear or only branched PARs.32,33 Another noteworthy feature of PARP-1 is its autoPARylation ability, which inhibits its catalytic activity. PARP-1 may be PARylated by itself, PARP-2 and possibly other PARPs as well.34 While the above model of PAR polymer elongation by addition of new ADP-ribose units at the distal 2ʹ-OH end seems unchallenged in the context of PARylating another protein or a second PARP-1 molecule (intermolecular PARP-1 auto-modification), the mechanism of PARP-1 intramolecular automodification remains under discussion. This topic is beyond the scope of this paper, however has been recently reviewed by Alemasova and Lavrik35). The synthesis of PAR polymers can perform various functions in the regulation of cellular processes, including modification of the role of PARylated proteins, marking polymers for PAR readers, and in the case of autoPARylation; negatively regulate their own activity.13,14,26,36 PARP-1 plays a key role in numerous biological processes, and PARP-1 activation has been shown to be part of the pathophysiology of many inflammatory and neoplastic diseases, including cardiovascular disease, inflammatory lung diseases, diabetes, breast cancer and ovarian cancer.37 PARP-1 activity accounts for the vast majority of PARP activity in cells, and for this reason, PARP-1 is the most intensively researched and most extensively described PARP.37 Therefore, this review focuses primarily on PARP-1.
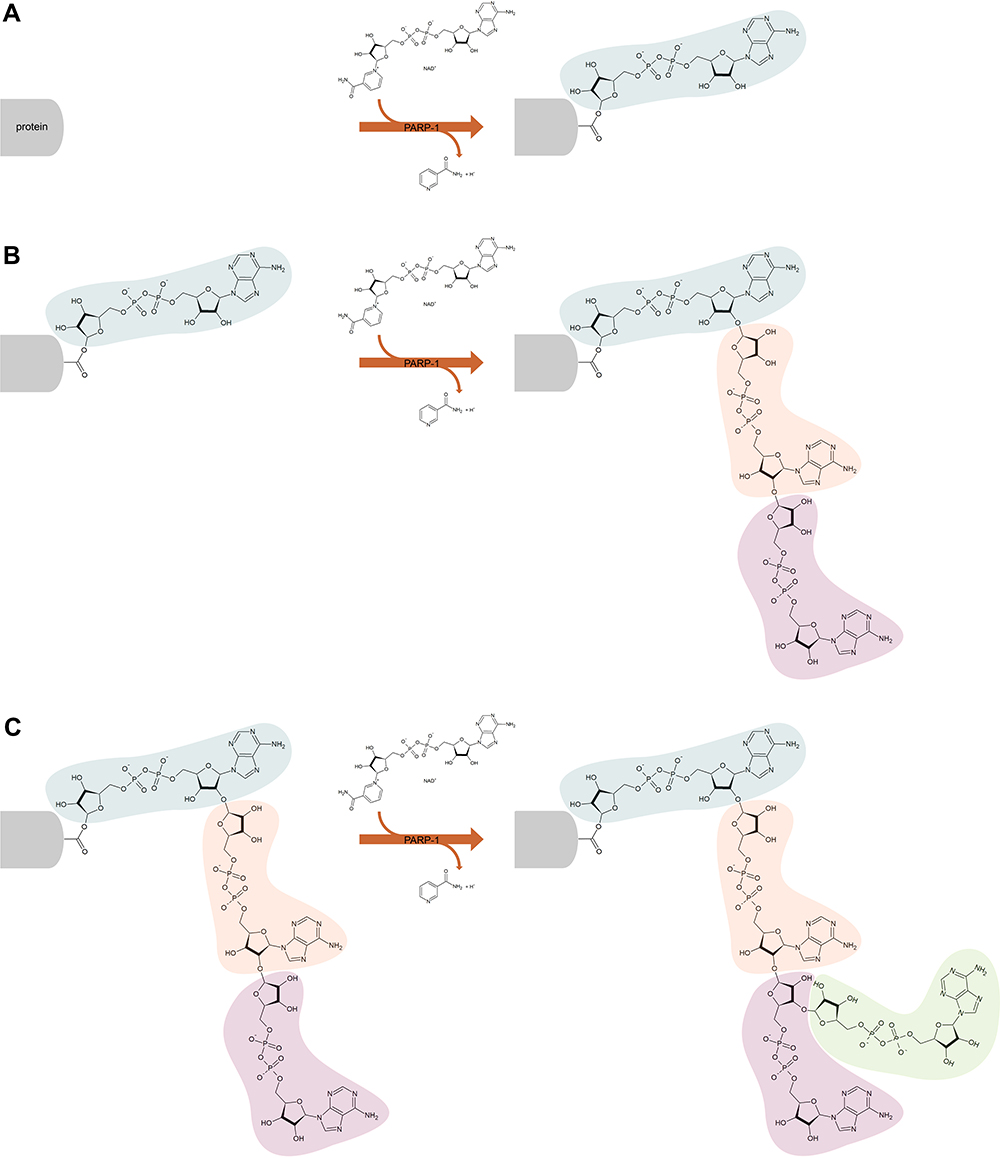 |
Figure 1 Scheme of poly (ADP-ribose) polymerase 1 (PARP-1) catalyzed poly (ADP-ribosylation) of the target protein. The PARP-1 catalytic domain attaches ADP-ribose polymers to proteins, catalyzing three different reactions – initiation (A), elongation (B) and branching (C). Initiation is the attachment of the first ADP-ribose monomer to the amino acid residue of the acceptor protein. Elongation, the attachment of further monomers, takes place through the formation of a (2ʹ-1ʹʹ) ribose-ribose glycosidic bond. Branching involves the creation of a ribose-ribose bond between ADP-ribose (2ʹʹ-1ʹʹʹ) units. |
The Role of PARP in Inflammation and Disorders of Cell Metabolism
There is ample evidence that PARP-1 is involved in the inflammatory response, with current knowledge summarized in Figure 2. PARP-1 is involved in the activation of innate immune cells (macrophages, neutrophils, dendritic cells and microglia), adaptive immune cells (lymphocytes) and the inflammatory response of non-immune cells (including fibroblasts, endothelial cells and astrocytes).38–41 One of the best described aspects of PARP’s involvement in the inflammatory response is the role of PARP-1 in macrophage activation. PARP-1 is involved in the mechanisms behind macrophage response to pathogen associated molecular patterns (PAMP), which include, eg, lipopolysaccharide (LPS), the most common bacterial factor that contributes to the development of sepsis. One of the elements of this is expression of pro-inflammatory cytokines.42–45 Contribution to the regulation of pro-inflammatory macrophage activation has also been demonstrated for other proteins in this family; PARP-2, PARP-9 and PARP-14. PARP-1 and PARP-2 have been shown to regulate a common set of inflammatory mediators (including interleukin-1β (IL-1β), tumour necrosis factor α (TNF-α) and inducible nitric oxide synthase (iNOS)), indicating that these enzymes can modulate inflammation through overlapping pathways. Furthermore, PARP-1 and PARP-2 can PARylate each other and form heterodimers.46–48 In addition to the NF-κB pathway described below, the involvement of other signalling pathways such as IFNγ-STAT1 and IL-4-STAT6 have been suggested.45,49
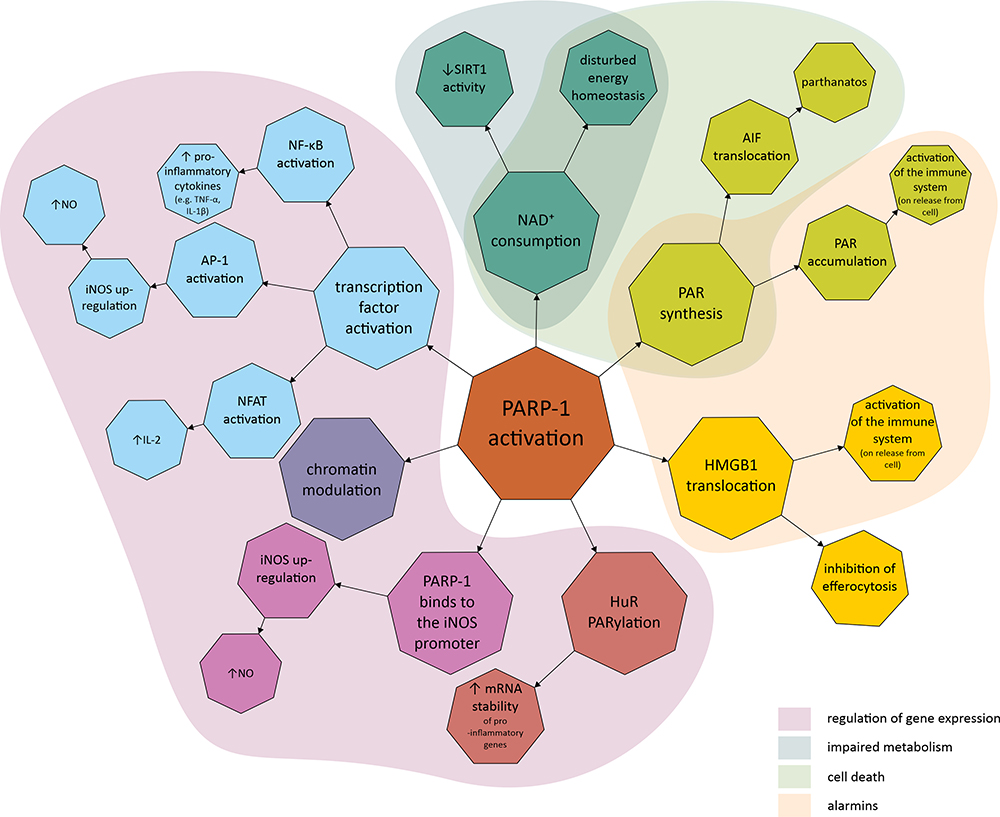 |
Figure 2 Molecular mechanisms linking poly (ADP-ribose) polymerase (PARP) with inflammatory response and disturbed cell metabolism. The first area covers the mechanisms involved in the regulation of gene expression, such as PARP-1 promotion and activation of transcription factors, chromatin modulation, PARP-1 binding to the promoter region of the gene, and regulation of post-transcriptional modification as exemplified by HuR PARylation. The second area covers the mechanisms related to metabolic impairment, which include a decrease in SIRT1 activity and cellular energy depletion, both of which are related to NAD+ depletion due to over-activation of PARP-1. The third area contains mechanisms related to cell death, including parthanatos, PARP-1 dependent and caspase-independent cell death related to PAR accumulation and their interaction with AIF. The final area covers the mechanisms leading to the production of alarmins, endogenous molecules that activate the innate immune system when released from the cell due to, eg, cellular damage. Abbreviations: AP-1, activator protein-1; HMGB1, high-mobility group box 1; HuR, human antigen R; IL, interleukin; iNOS, inducible nitric oxide synthase; mRNA, messenger ribonucleic acid; NAD+, nicotinamide adenine dinucleotide; NFAT, nuclear factor of activated T-cells; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NO, nitric oxide; PAR, poly (ADP-ribose); PARP-1, poly (ADP-ribose) polymerase 1; PARylation, poly ADP-ribosylation; SIRT1, sirtuin 1; TNF-α, tumour necrosis factor α. |
The mechanisms involved in the interaction between PARP-1 and the nuclear transcription factor NF-κB, which is crucial in the immune response, have been widely described.39,42–44,50 Research indicates that the interaction between PARP-1 and NF-κB can take place both enzymatically and non-enzymatically. Regarding the enzyme pathway, it has been proposed that in response to exposure to LPS or TNF-α, PARP-1 is phosphorylated by c-Abl tyrosine kinase at the conserved site Y829. Tyrosine-phosphorylated PARP-1 then catalyses the PARylation of the NF-κB p65/RelA subunit, which induces transcription of genes regulated by this transcription factor.44 Other studies have shown that PARylation supports the activation and nuclear retention of p65 NF-κB.50,51 As part of a non-enzymatic mechanism, PARP-1 also acts as a docking molecule, binding to the p65 and p50 subunits of NF-κB.14,52 When discussing the influence of PARP-1 on NF-κB, it is also worth noting that PARP-1 interacts with the histone acetyltransferase p300, which is a coactivator required for NF-κB-dependent gene transcription.53
In addition to NF-κB, PARP-1 modulates the cellular response to damage by regulating other transcription factors such as activator protein-1 (AP-1). AP-1 is a transcription factor involved in the cellular response to immune and oxidative stress.40 In animal studies, PARP-1 has been shown to contribute to acute lung injury in response to endotoxemia by regulating the iNOS pathway through the activation of AP-1, but not NF-κB.54 Another transcription factor regulated by PARP-1 is nuclear factor of activated T-cells (NFAT). PARylation of NFAT increases its capacity to bind to DNA, thereby facilitating the transcription of IL-2. A recent study has shown that the expression of NFAT-dependent cytokines (IL-2 and IL-4) was reduced in T cells genetically deficient in PARP-1 (PARP-1 (-/-)).55
In addition to affecting transcription factors, PARP-1 may also regulate the expression of genes related to inflammation by modulation of the chromatin structure, coregulation of the promoter, and modification of post-transcription ribonucleic acid (RNA).37,56,57 The ability of PARP-1 to PARylate histones and other chromatin-related proteins has been comprehensively described in the literature. This PARylation leads to a change in the spatial structure of these proteins and influences gene expression.58 PARP-1 can also elicit effects by directly binding to gene promoters, eg, binding to the iNOS promoter results in increased nitric oxide (NO) production.37,59
Gene expression associated with inflammatory response is regulated by both transcriptional and post-transcriptional mechanisms. Post-transcriptional mechanisms modify mRNA stability, which facilitates faster and more flexible control, and is especially important in coordinating the initiation and resolution of inflammation.60 PARP-1 may also influence the regulation of gene expression at the post-transcription level, modifying RNA binding proteins (RBP) such as human antigen R (HuR) by PARylation.61,62 Ke et al showed that the increased mRNA stability of pro-inflammatory genes in LPS exposed macrophages was abolished by PARP-1 inhibition. PARylation of HuR by PARP-1 has also been shown to enhance nucleocytoplasmic translocation, mRNA binding and promote mRNA stability.61
Increased activity of PARP-1 may also affect the regulation of metabolic homeostasis by indirectly affecting sirtuins. Sirtuins (SIRTs) are a group of enzymes that play important roles in cell metabolism. SIRT1, the most studied protein of the SIRT family, plays an important role in energy homeostasis by controlling the acetylation state and activity of many enzymes and transcription regulators (eg, peroxisome proliferator-activated receptor (PPAR)) that affects the expression of genes involved in carbohydrate, fat and protein metabolism and the development of inflammation.63,64 Since both SIRT1 and PARP-1 are NAD+ dependent, over-activation of PARP can reduce SIRT1 activity by depleting the NAD+ pool, which can disrupt cellular metabolic homeostasis.65 It has also been shown that under the influence of LPS stimulation, PARP-1 and SIRT1 physically interact. Interestingly, research results indicate that PARP-1 inhibition enhances the physical interaction between PARP-1 and SIRT1.66
Another theory related to the energy metabolism of cells concerns energy depletion caused by NAD+ consumption due to over-activation of PARP-1. NAD+ deficiency causes a reduction in the rate of glycolysis, electron transport chain and adenosine triphosphate (ATP) production, which can lead to impairment of mitochondrial and cellular function.67–70 In work by Khan et al, enterocytes of the Caco-2 cell line were immunostimulated with a cocktail of pro-inflammatory cytokines (cytomix) containing TNFα, IL-1β and interferon γ (INFγ). The immunostimulated cells were shown to consume oxygen at about half the rate of control cells. This effect was partially offset by the use of a PARP inhibitor. It was also shown that the decrease in oxygen uptake was associated with a decreased intracellular level of NAD+. Both the decrease in oxygen uptake and the decrease in intracellular NAD+ were completely abolished when liposome-encapsulated NAD+ was added to the immunostimulatory culture. The results of this experiment suggest that enterocytes exposed to pro-inflammatory cytokines consume less oxygen due to NAD+ depletion secondary to PARP activation by peroxynitrite or other oxidants.71 NAD+ depletion due to excessive PARP activation has also been proposed as a mechanism to explain cytopathic hypoxia, a phenomenon of decreased ATP production despite normal availability of oxygen in cells.9,72
Many studies show that PARP-1 also acts as a rheostat, promoting a different cellular response depending on the type, strength and duration of the harmful stimulus.68 Low intensity stressful stimuli primarily result in a transcriptional response and DNA repair, while excessive PARP-1 activity results in NAD+ depletion and PAR accumulation, which leads to serious disturbances in energy homeostasis or cell death.37,44,61
PARP-1 can induce cell death by apoptosis, necrosis, or parthanatos.68,73,74 Apoptosis or programmed cell death, does not usually cause inflammation. In contrast, necrosis is cell death associated with damage, including metabolic damage, eg, depletion of the ATP pool. Parthanatos is a PARP-1-dependent and caspase-independent cell death that has been described primarily in relation to neuronal cells.68,75
Research shows that the main cause of parthanatos is the accumulation of PARs, which interact with apoptosis inducing factor (AIF).37,76 AIF is a mitochondrial protein involved in energy production and cell death.77 Free PAR, generated in the cell nucleus due to PARP-1 activation, can be transferred to the cytoplasm. Binding of PAR by AIF allows AIF to be released from the mitochondria and translocated to the nucleus where it binds to DNA. Binding of AIF to DNA causes peripheral chromatin condensation and DNA fragmentation, which is characteristic of apoptosis; however, parthanatos also shows features typical of necrosis (eg, cell membrane disruption and energy depletion).68,77–80
PAR resulting from PARylation act not only intracellularly, they also play a role in intercellular communication. PAR when released out with the cell have the ability to stimulate macrophages, inducing the production of cytokines.81 Thus, it can be assumed that PAR released from the cell (eg, due to damage) can act as alarmins. Alarmins, or damaging molecular associated patterns (DAMP), are endogenous molecules released following cell damage that activate the innate immune system by interacting with pattern recognition receptors (PRRs). Another alarmin related to PARP-1 is high-mobility group box 1 (HMGB1), which has been shown to be an important mediator in the pathogenesis of sepsis.82,83 Research suggests that the PARP-1-dependent translocation of HMGB1 from the nucleus to the cytosol gives cells the ability to release this potent inflammatory mediator upon subsequent cell death.84 The translocation process of HMGB1 from the nucleus to the cytoplasm requires acetylation. PARP-1 catalyses the PARylation of HMGB1 which in turn facilitates acetylation.85,86 Furthermore, PARylated HMGB1 has been shown to reduce efferocytosis, which promotes maintenance of inflammation.87 Both released HMGB1 and free PAR are recognized by TLR4, which indicates the convergence of these pathways in distress signalling.81 In turn, the interaction of TLR4 with HMBG1 or LPS supports PARylation of HMGB1 (and therefore also its release), which forms a positive feedback loop, supporting inflammation.14 Interestingly, PARP-1 and HMGB1 are a target for some pathogens (eg, Streptococcus pyogenes which releases NAD+ glycohydrolase) as a strategy to avoid an immune response.88
PARP Inhibitors – Opportunities and Threats
While the first enzyme with poly (ADP-ribose) polymerase activity was described in 1963, the potential benefits of its blockade were not discovered until almost 20 years later.12,89 The first study was on DNA repair and used NAD+-like compounds that were found to bind to the catalytic site of PARP.89,90 Due to the role of PARP-1 in the detection and repair of DNA damage, the search for pharmacological inhibitors remains the subject of intense research. Preclinical data showing the efficacy of PARP inhibitors in the suppression of growth of BRCA mutant cells was first published in 2005.91,92 The first clinical trial using olaparib as monotherapy was reported in 2009 and showed anti-tumour activity in cancers associated with BRCA1 or BRCA2 mutations.93 In 2014, this drug was approved in Europe for the treatment of patients with platinum-sensitive serous ovarian cancer with BRCA1/2 mutations.94
Structurally, all PARP inhibitors that have been clinically tested to date contain motifs that mimic the nicotinamide component of NAD+ and bind to the catalytic site of PARP (Figure 3). Nicotinamide itself is also a natural inhibitor of this enzyme.95 The discovery of the NAD+-dependent PARP-1 activation pathway allowed for the design of many PARP inhibitors, ranging from first-generation inhibitors with millimolar levels of activity, including nicotinamide and 3-aminobenzamide (3-AB), to the current, third-generation inhibitors with efficacy in nanomolar concentrations (Figure 4).14 Since PARP inhibitors act by a competitive inhibition mechanism, competing with NAD+ at the active site of the enzyme, they have the potential to inhibit other NAD+ utilizing enzymes. This is an example of polypharmacology. There are three types of polypharmacology demonstrated by PARP-1 inhibitors; intra-family polypharmacology, inter-family polypharmacology and multi-signaling polypharmacology.96 Intra-family polypharmacology is defined as non-selective activity against targets of the same family,96 and has been demonstrated for many PARP inhibitors. Wahlberg et al assessed a series of 185 small molecule PARP inhibitors (including research reagents and clinically tested compounds), investigating their ability to bind to the catalytic domains of 13 out of 17 members of the PARP family. The most polypharmacological profile was found in rucaparib, which bound to 9 of the 13 PARPs assessed. The study also assessed PJ34, olaparib and 3-AB, mentioned in this review, for which binding to 8, 7 and 1 out of the 13 evaluated PARPs was shown, respectively.97 Inter-family polypharmacology of PARP-1 inhibitors, ie, activity against targets from different families,96 has been demonstrated for sirtuins and mono-ADP-ribosyltransferases (ARTs).98,99 Multi-signaling polypharmacology, ie, multi-signaling activity mediated by the same target, includes the trapping mechanism (autoPARylation of PARP-1 has been shown to allow the enzyme to break free from DNA, and inhibition of this process causes PARP trapping), which was described, among others, in the context of olaparib.14,96 Due to the polypharmacological phenomenon of PARP-1 inhibitors, scientific reports should be carefully analysed as there are many studies available which have found that the results obtained reflect PARP-1 inhibition, without considering the effect of the pharmacological intervention on other enzymes. A potential solution to this problem in scientific research may be to include a control model using PARP-1 genetic knockout, or to use PARP-1 short interfering RNA (siRNA), which would allow assessment of the relationship between the observed effects and PARP-1 activity.98,100 There are also studies on the development of PARP inhibitors with an alternative mechanism of action other than similarity to NAD+, which may show greater selectivity and lower cytotoxicity.101,102
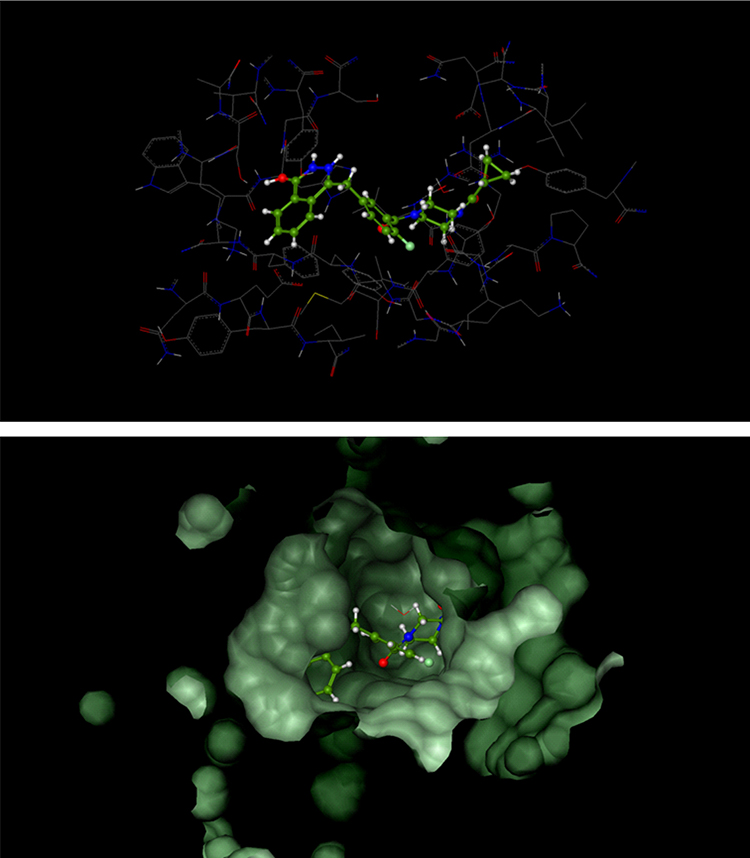 |
Figure 3 Catalytic domain of poly (ADP-ribose) polymerase 1 (PARP-1) in complex with olaparib. |
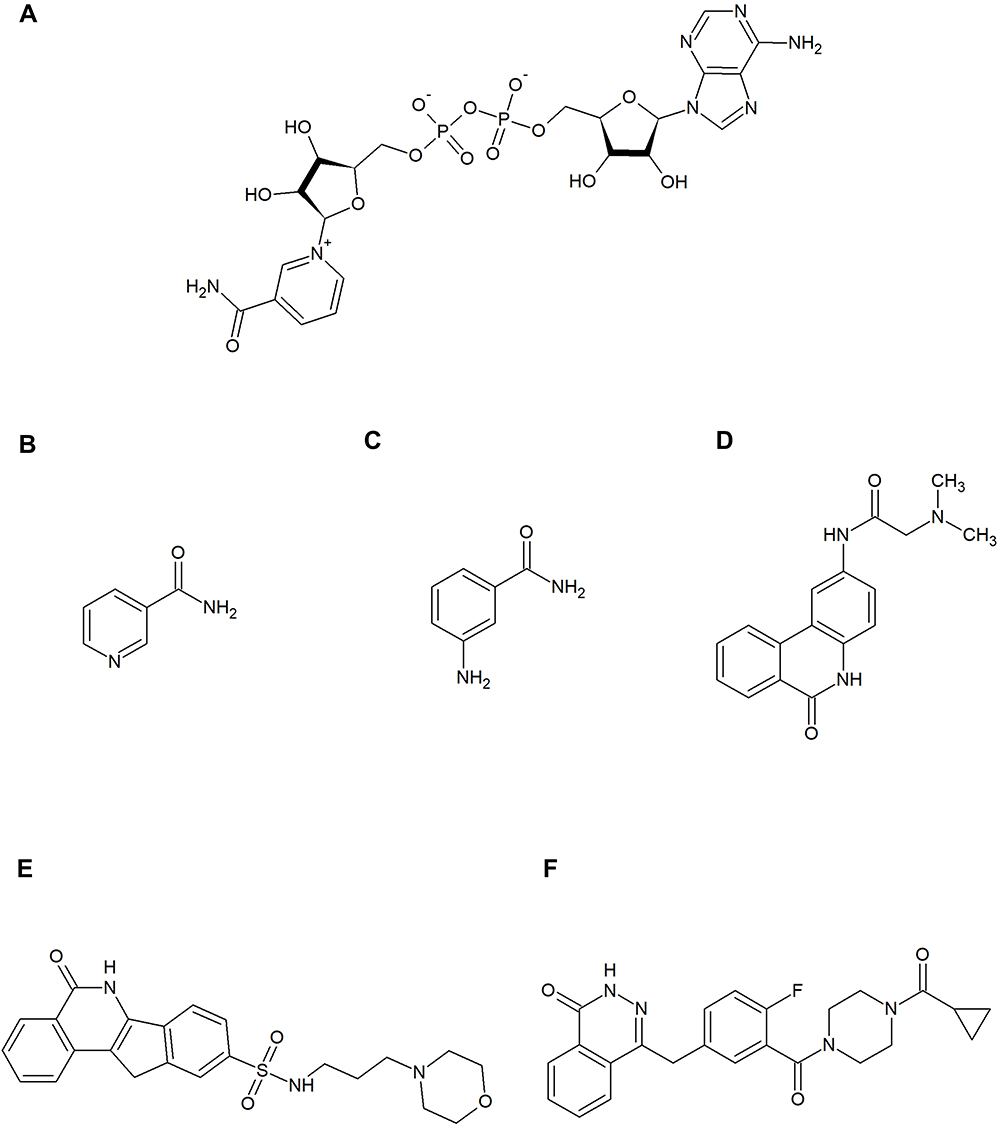 |
Figure 4 Nicotinamide adenine dinucleotide (NAD+) and poly (ADP-ribose) polymerase (PARP) inhibitors. (A) shows the substrate of the PARP family enzymes – NAD+. (B–F) show the PARP inhibitors mentioned in this review: nicotinamide (B), 3-aminobenzamide (C), PJ34 (D), and INO-1001 (E), olaparib (F). These contain motifs that mimic the nicotinamide component of NAD+ and bind to the catalytic site of PARP. Nicotinamide itself is also a natural inhibitor of this enzyme. |
Although the development of PARP inhibitors was initially related to oncology, their efficacy has been demonstrated in many models (in vitro and in vivo) of inflammatory diseases. There have been studies reporting efficacy of olaparib in alleviating sepsis,103,104 as well as studies in asthma,105 liver disease106,107 and transient cerebral ischemia.108
The effectiveness of PARP inhibition in inflammatory diseases, including sepsis, may result from the inhibition of the PARP-1-mediated components of the inflammatory response presented above. PARP inhibition facilitates a reduction in NAD+ consumption and also affects cellular energy homeostasis. Inhibition of PARP also results in a reduction in NF-κB activation and inflammatory cytokine expression, and reduces the formation of PAR which are involved in AIF release from mitochondria. It is possible that it also contributes to the reduction of genotoxicity caused by oxidative stress and inflammation. Interestingly, the effective doses of PARP inhibitors in non-oncological studies are lower than those required in oncology, which may be related to the synergistic combination of the above effects.14,109
Since PARP-1 is an enzyme also associated with the recognition of DNA damage and the recruitment of repair enzymes, its inhibition in inflammatory diseases is of concern. There is some evidence that PARP inhibition is associated with an increased risk of genotoxic effects and there are possible side effects of long-term treatment (which in the case of sepsis is not such a significant problem).14 However, other in vitro studies have demonstrated no genotoxic effects of PARP inhibition. The situation in vivo is more complex and there is a possibility of indirect protection against DNA damage caused by oxidative reactions.14,104,110 This issue undoubtedly requires further research.
PARP Inhibition in Experimental Models of Sepsis
Many reports have demonstrated the benefits of PARP inhibition in experimental models of sepsis (Table 2). The benefits of PARP inhibition are well illustrated by studies using genetically determined PARP-1 deficient mice (PARP-1 (-/-)), for example, Oliver et al observed that LPS administration resulted in rapid activation of the NF-κB pathway in mouse PARP-1 (+/+) macrophages, which was not observed in PARP-1 (-/-) mice. These authors also described resistance of PARP-1 (-/-) mice to LPS-induced septic shock.111 Moreover, Soriano et al showed that PARP-1 (-/-) mice that underwent cecal ligation and puncture (CLP) had significantly lower plasma levels of TNF-α, IL-6 and IL-10, showed reduced organ inflammation, and had a higher survival compared to PARP-1 mice (+/+).112
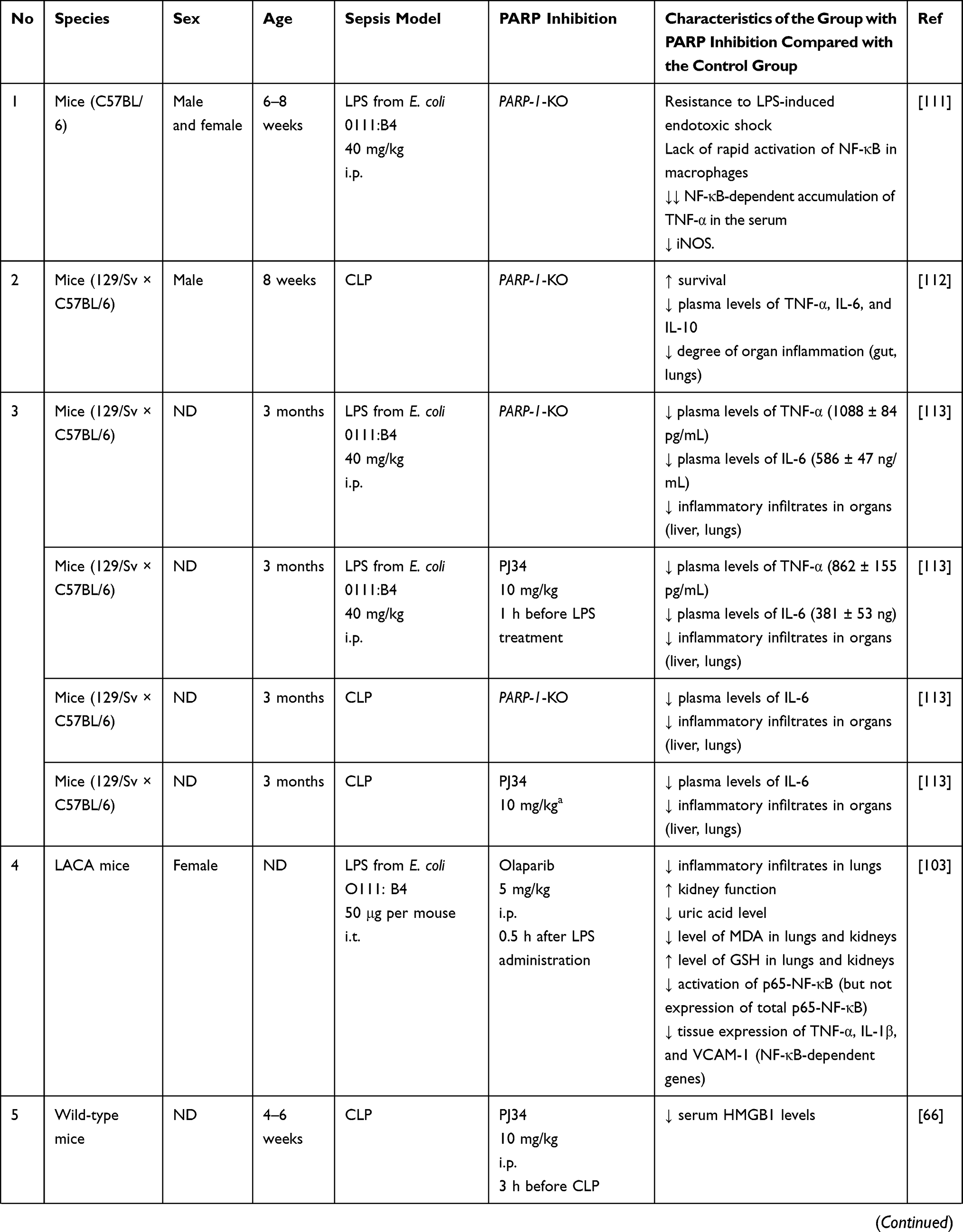 | 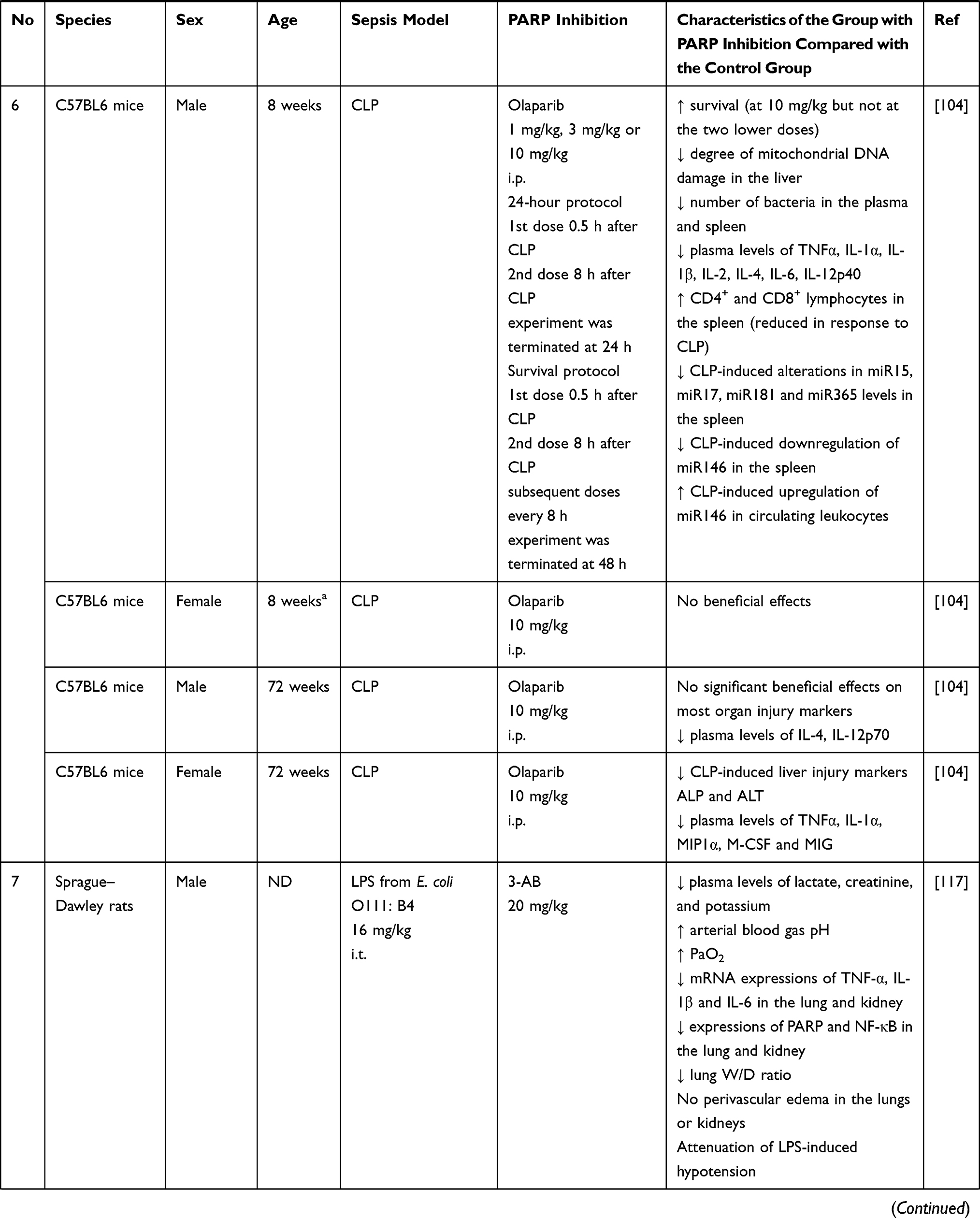 | 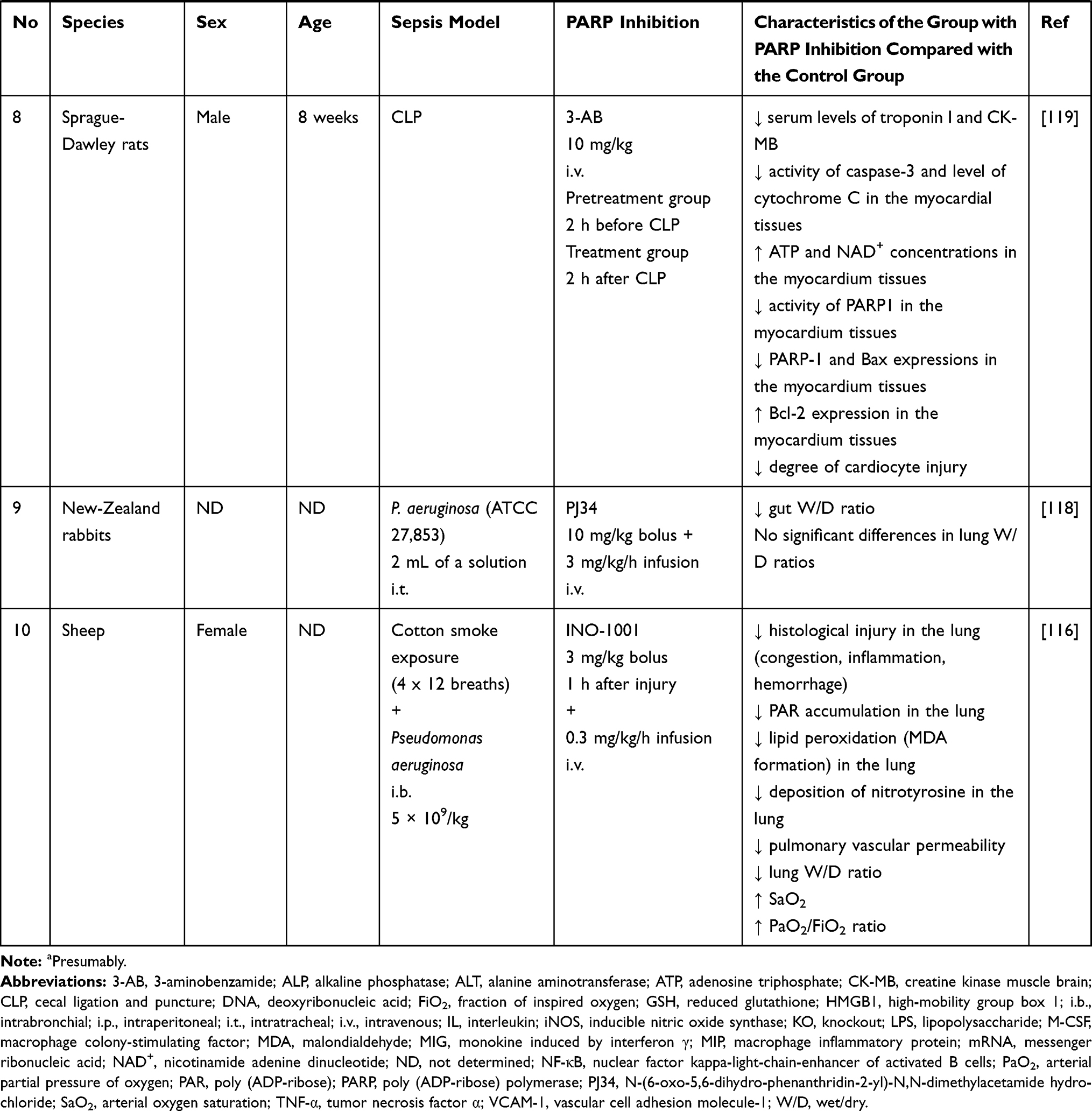 |
Table 2 Summary of the Effects of Inhibition of Poly (ADP-Ribose) Polymerase (PARP) in Experimental Models of Sepsis |
In 2005, Corral et al conducted a study in which they used two murine models of endotoxemia using CLP and LPS injection, as well as two methods of endotoxemia resistance using genetic blockade using PARP-1 (-/-) mice and pharmacological inhibition of PARP-1 using PJ34. This study revealed that levels of pro-inflammatory mediators were increased, and intensity of mononuclear cell infiltration and organ degeneration were more profound in animals in which PARP-1 was not blocked; however, impaired blood coagulation was seen in all groups of animals.113 Moreover, it was observed in the study that although both PARP-1 (-/-) and PARP-1 (+/+) mice treated with PJ34 showed a reduction in the level of proinflammatory mediators (TNF-α and IL-6) compared to the control group, this decrease was more pronounced in the group of mice with pharmacological inhibition of PARP (Table 2).113 These differences may result from the polypharmacological action of PJ34, which affects a particularly wide group of targets.96 It has been shown that PJ34 strongly binds to PARP-1, PARP-2 and PARP-3, and has activity against PARP-4, TNKS-1, TNKS-2, PARP-14 and PARP-16.97 In addition, the phenomenon of inter-family polypharmacology was described in relation to the compound, including inhibition of matrix metalloproteinase 2 (MMP2).114 Another interesting phenomenon described for PJ34 is the inhibition of ADP-induced platelet aggregation, probably through competitive P2Y12 antagonism.96,115 The polypharmacology of PJ34 may be responsible for the described differences in the concentration of pro-inflammatory mediators in the compared groups and should also be taken into account when interpreting other studies using this compound.
There are also reports focusing on specific organs. In studies evaluating the usefulness of PARP inhibition in the prevention of lung damage, endotracheal administration of pathogens or LPS is often used. Murakami et al showed that a PARP inhibitor (INO-1001) reduces lung damage in an ovine model of acute lung injury (ALI).116 Khin Hnin Si et al described an experiment in which rats were administered intratracheal LPS, which induced metabolic acidosis, hypotension, hypoxemia, and increased levels of creatinine and potassium in the plasma. Increased expression of cytokine mRNA in lung and kidney has also been found, which has been shown to be associated with strong expression of PARP and NF-κB. It has been shown that administration of PARP inhibitor (3-AB) to animals significantly alleviated metabolic acidosis and other biochemical disorders. PARP inhibition also reduced cytokine mRNA expression as well as PARP and NF-κB expression.117 Kapoor et al used the modern PARP inhibitor olaparib, and demonstrated that it alleviated acute lung and kidney damage following endotracheal LPS administration in mice. Use of olaparib 30 minutes after endotracheal LPS administration reduced the total number of inflammatory cells, particularly neutrophils, in the lungs. There was also a reduction in pulmonary edema and a lower protein content of the bronchoalveolar fluid. Olaparib has also been shown to provide protection against secondary kidney damage. In addition, RT-PCR results show that olaparib downregulates LPS-induced expression of NF-κB dependent genes (TNF-α, IL-1β, and VCAM-1) in the lung, without altering the expression of total p65NF-κB.103
Lobo et al assessed intestinal damage in sepsis caused by administering Pseudomonas aeruginosa to rabbits and evaluated the effect of a PARP inhibitor (PJ34) on gut health. This study showed that PARP inhibition reduces intestinal inflammation and may reduce bacterial translocation.118
Zhang et al showed that the PARP inhibitor, 3-AB, in a rat model of sepsis had a cardioprotective effect. PARP inhibition by 3-AB was associated with an increase in ATP, NAD+ and Bcl-2 levels, a decrease in PARP and caspase 3 activation, and a decrease in malondialdehyde (MDA), cytochrome C, creatine kinase muscle brain (CK-MB) and troponin I.119 Myocardial dysfunction was also investigated in two patient studies in which PARP inhibition was not used, but PARP-1 activity was assessed. In 2006, the results of a study by Soriano et al involving 25 patients with sepsis or septic shock showed that the heart tissue of patients with sepsis and impaired cardiac function had significantly elevated levels of PARP activation. They also described a positive correlation between troponin I levels and PAR staining densitometry.120 In 2014, the results of a study by Li et al evaluating 64 patients in septic shock showed that the activity of PARP-1 in circulating mononuclear cells in non-surviving patients was significantly higher than in surviving patients. In addition, this study showed that PARP-1 activity in these cells was an independent risk factor for myocardial dysfunction, indicating a potential prognostic significance.121
In 2014, Walko et al, using a mouse model of sepsis, showed that the administration of PJ-34, a specific PARP-1 inhibitor, reduced the concentration of HMGB1 in the serum of mice undergoing CLP compared to a control group. Reduction of HMGB1 acetylation induced by PARP inhibition has been proposed as a likely mechanism.66 Two interesting studies on PARP inhibition were published in 2019. In the first one, Ahmad et al demonstrated the beneficial effects of the PARP inhibitor olaparib in both mice undergoing CLP and cells under oxidative stress. Interestingly, in the case of the animal studies, the effect was related to their sex and age. Olaparib was shown to be protective and anti-inflammatory in the organs of young adult male mice, while its beneficial effects were limited in older male and female mice and not seen in young female mice.104 These differences in the efficacy of olaparib may be due to the physiological inhibitory effect of estrogen on PARP activation.69,109 Olaparib (in the dose and concentration range evaluated) has also been shown to have no adverse effects on nuclear DNA integrity and has a protective effect on mitochondrial DNA and mitochondrial function.104 In another new study, Zhang et al, using a model of human aortic endothelial cells (HAECs) affected by LPS-induced inflammation, demonstrated that remifentanil, a potent opioid analgesic, could reduce the LPS-induced inflammatory response through the PARP-1/NF-κB signalling pathway. Cells pretreated with remifentanil showed a decrease in iNOS, ICAM-1, PARP-1 and PAR expression compared to the control group. Furthermore, the use of remifentanil reduced the production of superoxide anions and DNA damage. The effect of remifentanil on the inhibition of PARP-1 expression by small interfering RNAs (siRNAs) was also compared. Both methods inhibited LPS-induced NF-κB p65 expression and translocation to the nucleus, suggesting that remifentanil reduces the LPS-induced inflammatory response via the PARP-1/NF-κB signalling pathway. Although there is no confirmation of these reports in animal models or clinical trials, these results appear to be promising for use of analgesia in patients with sepsis, and the issue requires further research.122
Conclusions
Despite intensive research, modern sepsis therapy is based primarily on antimicrobial treatment and supporting failing organs. Sepsis poses a serious threat to the health of patients; therefore, innovative solutions are necessary for the treatment of inflammation and metabolic disorders. As excess PARP activity appears to be an important component of the inflammatory response and promotion of metabolic disorders at a cellular level, inhibition of this enzyme may be a promising solution in the treatment of sepsis. Over the past two decades, PARP-1 inhibition has been repeatedly shown to ameliorate the inflammatory response and increase survival using experimental sepsis models. Such action is described, among others, for olaparib, a PARP-1 inhibitor approved for the treatment of selected oncology patients. Despite the promising effects, the use of PARP-1 inhibitors in non-oncological diseases raises some concerns, mainly related to the role of PARP-1 in DNA repair, and concerning the risk of mutagenesis and oncogenesis. However, the results of studies on experimental models indicate the effectiveness of even short-term PARP-1 inhibition and do not confirm such concerns about its impact on the integrity of nuclear DNA. The collected data so far indicate that inhibition of PARP-1 may be a sought after and innovative solution in the treatment of sepsis.
Acknowledgments
We would like to thank Professor Tomasz Plech from the Medical University of Lublin for his help in creating the figures.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–1310. doi:10.1097/00003246-200107000-00002
2. Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med. 2006;34(2):344–353. doi:10.1097/01.CCM.0000194725.48928.3A
3. Reinhart K, Daniels R, Kissoon N, Machado FR, Schachter RD, Finfer S. Recognizing sepsis as a global health priority — a WHO resolution. N Engl J Med. 2017;377(5):414–417. doi:10.1056/NEJMp1707170
4. Kübler A. Definition. In: Kübler A, ed. Sepsis. Wrocław: Edra Urban & Partner; 2017:9–22.
5. Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest. 1992;101:1644–1655. doi:10.1378/chest.101.6.1644
6. Singer M, Deutschman CS, Seymour C, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA - J Am Med Assoc. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
7. De Waele E, Malbrain MLNG, Spapen H. Nutrition in sepsis: a bench-to-bedside review. Nutrients. 2020;12:2. doi:10.3390/nu12020395
8. Ingels C, Gunst J, Van den Berghe G. Endocrine and metabolic alterations in sepsis and implications for treatment. Crit Care Clin. 2018;34(1):81–96. doi:10.1016/j.ccc.2017.08.006
9. Fink M. Cytopathic hypoxia in sepsis. Acta Anaesthesiol Scand Suppl. 1997;110:87–95. doi:10.1111/j.1399-6576.1997.tb05514.x
10. Cohen J, Vincent JL, Adhikari NKJ, et al. Sepsis: a roadmap for future research. Lancet Infect Dis. 2015;15(5):581–614. doi:10.1016/S1473-3099(15)70112-X
11. Kenig A, Ilan Y. A personalized signature and chronotherapy-based platform for improving the efficacy of sepsis treatment. Front Physiol. 2019;10:1542. doi:10.3389/fphys.2019.01542
12. Chambon P, Weill JD, Mandel P. Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963;11(1):39–43. doi:10.1016/0006-291X(63)90024-X
13. Bai P. Biology of poly(ADP-ribose) polymerases: the factotums of cell maintenance. Mol Cell. 2015;58(6):947–958. doi:10.1016/j.molcel.2015.01.034
14. Pazzaglia S, Pioli C. Multifaceted role of PARP-1 in DNA repair and inflammation: pathological and therapeutic implications in cancer and non-cancer diseases. Cells. 2019;9:1. doi:10.3390/cells9010041
15. Gupte R, Liu Z, Kraus WL. Parps and adp-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 2017;31(2):101–126. doi:10.1101/gad.291518.116
16. Hottiger MO. Nuclear ADP-ribosylation and its role in chromatin plasticity, cell differentiation, and epigenetics. Annu Rev Biochem. 2015;84:227–263. doi:10.1146/annurev-biochem-060614-034506
17. Ryu KW, Kim DS, Kraus WL. New facets in the regulation of gene expression by ADP-ribosylation and poly(ADP-ribose) polymerases. Chem Rev. 2015;115(6):2453–2481. doi:10.1021/cr5004248
18. Hottiger MO, Hassa PO, Lüscher B, Schüler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010;35(4):208–219. doi:10.1016/j.tibs.2009.12.003
19. Di Girolamo M, Fabrizio G. The ADP-Ribosyl-Transferases Diphtheria Toxin-Like (ARTDs) family: an overview. Challenges. 2018;9:1. doi:10.3390/challe9010024
20. Feijs KLH, Verheugd P, Lüscher B. Expanding functions of intracellular resident mono-ADP-ribosylation in cell physiology. FEBS J. 2013;280(15):3519–3529. doi:10.1111/febs.12315
21. Fabrizio G, Scarpa ES, Di Girolamo M. State of the art of protein mono-ADP-ribosylation: biological role and therapeutic potential. Front Biosci. 2015;20:405–430. doi:10.2741/4316
22. Kuny CV, Sullivan CS, Spindler KR. Virus–host interactions and the ARTD/PARP family of enzymes. PLoS Pathog. 2016;12(3):e1005453. doi:10.1371/journal.ppat.1005453
23. Bai P, Cantó C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012;16(3):290–295. doi:10.1016/j.cmet.2012.06.016
24. Xie N, Zhang L, Gao W, et al. NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct Target Ther. 2020;5:1. doi:10.1038/s41392-020-00311-7
25. Kameshita I, Matsuda Z, Taniguchi T, Shizuta Y. Poly (ADP-ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J Biol Chem. 1984;259(8):4770–4776. doi:10.1016/S0021-9258(17)42913-9
26. Tao Z, Gao P, Liu HW. Identification of the ADP-ribosylation sites in the PARP-1 automodification domain: analysis and implications. J Am Chem Soc. 2009;131(40):14258–14260. doi:10.1021/ja906135d
27. Rolli V, O’Farrell M, Ménissier-de Murcia J, de Murcia G. Random mutagenesis of the poly(ADP-ribose) polymerase catalytic domain reveals amino acids involved in polymer branching. Biochemistry. 1997;36(40):12147–12154. doi:10.1021/bi971055p
28. Leung AKL. Poly(ADP-ribose): an organizer of cellular architecture. J Cell Biol. 2014;205(5):613–619. doi:10.1083/jcb.201402114
29. Aberle L, Krüger A, Reber JM, et al. PARP1 catalytic variants reveal branching and chain length-specific functions of poly(ADP-ribose) in cellular physiology and stress response. Nucleic Acids Res. 2020;48(18):10015–10033. doi:10.1093/nar/gkaa590
30. Ruf A, Rolli V, de Murcia G, Schulz GE. The mechanism of the elongation and branching reaction of poly(ADP-ribose) polymerase as derived from crystal structures and mutagenesis. J Mol Biol. 1998;278(1):57–65. doi:10.1006/jmbi.1998.1673
31. Kistemaker HAV, Overkleeft HS, van der Marel GA, Filippov DV. Branching of poly(ADP-ribose): synthesis of the core motif. Org Lett. 2015;17(17):4328–4331. doi:10.1021/acs.orglett.5b02143
32. Chen Q, Kassab MA, Dantzer F, Yu X. PARP2 mediates branched poly ADP-ribosylation in response to DNA damage. Nat Commun. 2018;9(1):3233. doi:10.1038/s41467-018-05588-5
33. Pourfarjam Y, Ventura J, Kurinov I, Cho A, Moss J, Kim I-K. Structure of human ADP-ribosyl-acceptor hydrolase 3 bound to ADP-ribose reveals a conformational switch that enables specific substrate recognition. J Biol Chem. 2018;293(32):12350–12359. doi:10.1074/jbc.RA118.003586
34. Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39(1):8–24. doi:10.1016/j.molcel.2010.06.017
35. Alemasova EE, Lavrik OI. Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019;47(8):3811–3827. doi:10.1093/nar/gkz120
36. Kamaletdinova T, Fanaei-Kahrani Z, Wang Z-Q. The enigmatic function of PARP1: from PARylation activity to PAR readers. Cells. 2019;8(12). doi:10.3390/cells8121625
37. Ke Y, Wang C, Zhang J, et al. The role of PARPs in inflammation-and metabolic-related diseases: molecular mechanisms and beyond. Cells. 2019;8(9). doi:10.3390/cells8091047
38. Bai P, Virág L. Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Lett. 2012;586(21):3771–3777. doi:10.1016/j.febslet.2012.09.026
39. Kunze FA, Hottiger MO. Regulating immunity via ADP-ribosylation: therapeutic implications and beyond. Trends Immunol. 2019;40(2):159–173. doi:10.1016/j.it.2018.12.006
40. Andreone TL, O’Connor M, Denenberg A, Hake PW, Zingarelli B. Poly(ADP-ribose) polymerase-1 regulates activation of activator protein-1 in murine fibroblasts. J Immunol. 2003;170(4):2113–2120. doi:10.4049/jimmunol.170.4.2113
41. Carrillo A, Monreal Y, Ramírez P, et al. Transcription regulation of TNF-α-early response genes by poly(ADP-ribose) polymerase-1 in murine heart endothelial cells. Nucleic Acids Res. 2004;32(2):757–766. doi:10.1093/nar/gkh239
42. Hassa PO, Haenni SS, Buerki C, et al. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-κB-dependent transcription. J Biol Chem. 2005;280(49):40450–40464. doi:10.1074/jbc.M507553200
43. Liu L, Ke Y, Jiang X, et al. Lipopolysaccharide activates ERK-PARP-1-RelA pathway and promotes nuclear factor-κB transcription in murine macrophages. Hum Immunol. 2012;73(5):439–447. doi:10.1016/j.humimm.2012.02.002
44. Bohio AA, Sattout A, Wang R, et al. c-Abl–mediated tyrosine phosphorylation of PARP1 is crucial for expression of proinflammatory genes. J Immunol. 2019;203(6):1521–1531. doi:10.4049/jimmunol.1801616
45. Fehr AR, Singh SA, Kerr CM, Mukai S, Higashi H, Aikawa M. The impact of PARPs and ADP-ribosylation on inflammation and host-pathogen interactions. Genes Dev. 2020;34(5):341–359. doi:10.1101/gad.334425.119
46. Rosado MM, Bennici E, Novelli F, Pioli C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology. 2013;139(4):428–437. doi:10.1111/imm.12099
47. Szántó M, Brunyánszki A, Kiss B, et al. Poly(ADP-ribose) polymerase-2: emerging transcriptional roles of a DNA-repair protein. Cell Mol Life Sci. 2012;69(24):4079–4092. doi:10.1007/s00018-012-1003-8
48. Popoff I, Jijon H, Monia B, et al. Antisense oligonucleotides to poly(ADP-ribose) polymerase-2 ameliorate colitis in interleukin-10-deficient mice. J Pharmacol Exp Ther. 2002;303(3):1145–1154. doi:10.1124/jpet.102.039768
49. Iwata H, Goettsch C, Sharma A, et al. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat Commun. 2016;7. doi:10.1038/ncomms12849
50. Zerfaoui M, Errami Y, Naura AS, et al. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-κB upon TLR4 stimulation. J Immunol. 2010;185(3):1894–1902. doi:10.4049/jimmunol.1000646
51. Stilmann M, Hinz M, Arslan SÇ, Zimmer A, Schreiber V, Scheidereit C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IκB kinase activation. Mol Cell. 2009;36(3):365–378. doi:10.1016/j.molcel.2009.09.032
52. Hassa PO, Covic M, Hasan S, Imhof R, Hottiger MO. The enzymatic and DNA binding activity of PARP-1 are not required for NF-κB coactivator function. J Biol Chem. 2001;276(49):45588–45597. doi:10.1074/jbc.M106528200
53. Hassa PO, Buerki C, Lombardi C, Imhof R, Hottiger MO. Transcriptional coactivation of nuclear factor-κB-dependent gene expression by p300 is regulated by Poly(ADP)-ribose polymerase-1. J Biol Chem. 2003;278(46):45145–45153. doi:10.1074/jbc.M307957200
54. Kiefmann R, Heckel K, Doerger M, et al. Role of PARP on iNOS pathway during endotoxin-induced acute lung injury. Intensive Care Med. 2004;30(7):1421–1431. doi:10.1007/s00134-004-2301-x
55. Olabisi OA, Soto-Nieves N, Nieves E, et al. Regulation of transcription factor NFAT by ADP-ribosylation. Mol Cell Biol. 2008;28(9):2860–2871. doi:10.1128/mcb.01746-07
56. Kraus WL. Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr Opin Cell Biol. 2008;20(3):294–302. doi:10.1016/j.ceb.2008.03.006
57. Caiafa P, Guastafierro T, Zampieri M. Epigenetics: poly(ADP-ribosyl)ation of PARP-1 regulates genomic methylation patterns. FASEB J. 2009;23(3):672–678. doi:10.1096/fj.08-123265
58. Posavec Marjanović M, Crawford K, Ahel I. PARP, transcription and chromatin modeling. Semin Cell Dev Biol. 2017;63:102–113. doi:10.1016/j.semcdb.2016.09.014
59. Yu Z, Kuncewicz T, Dubinsky WP, Kone BC. Nitric oxide-dependent negative feedback of PARP-1 trans-activation of the inducible nitric-oxide synthase gene. J Biol Chem. 2006;281(14):9101–9109. doi:10.1074/jbc.M511049200
60. Anderson P. Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat Rev Immunol. 2010;10(1):24–35. doi:10.1038/nri2685
61. Ke Y, Han Y, Guo X, et al. PARP1 promotes gene expression at the post-transcriptional level by modulating the RNA-binding protein HuR. Nat Commun. 2017;8(1):1–16. doi:10.1038/ncomms14632
62. Ji Y, Tulin AV. Post-transcriptional regulation by poly(ADP-ribosyl)ation of the RNA-binding proteins. Int J Mol Sci. 2013;14(8):16168–16183. doi:10.3390/ijms140816168
63. Bai P, Cantó C, Oudart H, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13(4):461–468. doi:10.1016/j.cmet.2011.03.004
64. Cantó C, Auwerx J. Targeting sirtuin 1 to improve metabolism: all you need is NAD +? Pharmacol Rev. 2012;64(1):166–187. doi:10.1124/pr.110.003905
65. Brady PN, Goel A, Johnson MA. Poly(ADP-ribose) polymerases in host-pathogen interactions, inflammation, and immunity. Microbiol Mol Biol Rev. 2018;83(1). doi:10.1128/mmbr.00038-18
66. Walko TD, Di Caro V, Piganelli J, Billiar TR, Clark RSB, Aneja RK. Poly(ADP-ribose) polymerase 1-sirtuin 1 functional interplay regulates LPS-mediated high mobility group box 1 secretion. Mol Med. 2014;20(1):612–624. doi:10.2119/molmed.2014.00156
67. Zhang D, Hu X, Li J, et al. DNA damage-induced PARP1 activation confers cardiomyocyte dysfunction through NAD + depletion in experimental atrial fibrillation. Nat Commun. 2019;10:1. doi:10.1038/s41467-019-09014-2
68. Luo X, Lee kraus W. On par with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012;26(5):417–432. doi:10.1101/gad.183509.111
69. Szabó C. Role of poly(ADP-ribose) polymerase activation in the pathogenesis of inflammation and circulatory shock. In: Bürkle A, ed. Poly(ADP-Ribosyl)ation. Vol. 280. US: Springer; 2006:184–202. doi:10.1007/0-387-36005-0_16
70. Evgenov O, Liaudet L. Role of nitrosative stress and activation of poly(ADP-ribose) polymerase-1 in cardiovascular failure associated with septic and hemorrhagic shock. Curr Vasc Pharmacol. 2005;3(3):293–299. doi:10.2174/1570161054368580
71. Khan AU, Delude RL, Han YY, et al. Liposomal NAD + prevents diminished O2 consumption by immunostimulated Caco-2 cells. Am J Physiol - Lung Cell Mol Physiol. 2002;282(5):L1082–L1091. doi:10.1152/ajplung.00358.2001
72. Fink MP. Bench-to-bedside review: cytopathic hypoxia. Crit Care. 2002;6(6):491–499. doi:10.1186/cc1824
73. Yu SW, Wang H, Poitras MF, et al. Mediation of poly(ADP-ribose) polymerase-1 - dependent cell death by apoptosis-inducing factor. Science. 2002;297(5579):259–263. doi:10.1126/science.1072221
74. Du L, Zhang X, Han YY, et al. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278(20):18426–18433. doi:10.1074/jbc.M301295200
75. Koh DW, Dawson TM, Dawson VL. Mediation of cell death by poly(ADP-ribose) polymerase-1. Pharmacol Res. 2005;52(1):5–14. doi:10.1016/j.phrs.2005.02.011
76. Palazzo L, Ahel I. PARPs in genome stability and signal transduction: implications for cancer therapy. Biochem Soc Trans. 2018;46(6):1681–1695. doi:10.1042/BST20180418
77. Pospisilik JA, Knauf C, Joza N, et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131(3):476–491. doi:10.1016/j.cell.2007.08.047
78. Andrabi SA, Umanah GKE, Chang C, et al. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A. 2014;111(28):10209–10214. doi:10.1073/pnas.1405158111
79. Andrabi SA, Dawson TM, Dawson VL. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann N Y Acad Sci. 2008;1147(1):233–241. doi:10.1196/annals.1427.014
80. Wang Y, Kim NS, Haince J-F, et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci Signal. 2011;4(167):167. doi:10.1126/scisignal.2000902
81. Krukenberg KA, Kim S, Tan ES, Maliga Z, Mitchison TJ. Extracellular poly(ADP-ribose) Is a pro-inflammatory signal for macrophages. Chem Biol. 2015;22(4):446–452. doi:10.1016/j.chembiol.2015.03.007
82. Czura CJ, Yang H, Amella CA, Tracey KJ. HMGB1 in the immunology of sepsis (not septic shock) and arthritis. Adv Immunol. 2004;84:181–200. doi:10.1016/S0065-2776(04)84005-7
83. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 2018;18(4):4. doi:10.4110/in.2018.18.e27
84. Ditsworth D, Zong WX, Thompson CB. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J Biol Chem. 2007;282(24):17845–17854. doi:10.1074/jbc.M701465200
85. Yang Z, Li L, Chen L, et al. PARP-1 mediates LPS-induced HMGB1 release by macrophages through regulation of HMGB1 acetylation. J Immunol. 2014;193(12):6114–6123. doi:10.4049/jimmunol.1400359
86. Bonaldi T, Talamo F, Scaffidi P, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22(20):5551–5560. doi:10.1093/emboj/cdg516
87. Davis K, Banerjee S, Friggeri A, Bell C, Abraham E, Zerfaoui M. Poly(ADP-ribosyl)ation of high mobility group box 1 (HMGB1) protein enhances inhibition of efferocytosis. Mol Med. 2012;18(3):359–369. doi:10.2119/molmed.2011.00203
88. Chandrasekaran S, Caparon MG. The streptococcus pyogenesNAD+ glycohydrolase modulates epithelial cell PARylation and HMGB1 release. Cell Microbiol. 2015;17(9):1376–1390. doi:10.1111/cmi.12442
89. Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair [23]. Nature. 1980;283(5747):593–596. doi:10.1038/283593a0
90. Yuan Y, Liao YM, Hsueh CT, Mirshahidi HR. Novel targeted therapeutics: inhibitors of MDM2, ALK and PARP. J Hematol Oncol. 2011;4:16. doi:10.1186/1756-8722-4-16
91. Farmer H, McCabe H, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi:10.1038/nature03445
92. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi:10.1038/nature03443
93. Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi:10.1056/nejmoa0900212
94. Mądry R, Stanisławiak-Rudowicz J. Management of adverse reactions during maintenance therapy with olaparib in ovarian cancer patients. Onkol Prakt Klin Edu. 2018;4(3):167–178.
95. Wiśnik E, Ryksa M, Koter-Michalak M. Inhibitory PARP1: współczesne próby zastosowania w terapii przeciwnowotworowej i perspektywy na przyszłość. Postepy Hig Med Dosw. 2016;70:280–294. doi:10.5604/17322693.1199303
96. Passeri D, Camaioni E, Liscio P, et al. Concepts and molecular aspects in the polypharmacology of PARP-1 inhibitors. ChemMedChem. 2016;11(12):1219–1226. doi:10.1002/cmdc.201500391
97. Wahlberg E, Karlberg T, Kouznetsova E, et al. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat Biotechnol. 2012;30(3):283–288. doi:10.1038/nbt.2121
98. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10(4):293–301. doi:10.1038/nrc2812
99. Hassa PO. The molecular “Jekyll and Hyde” duality of PARP1 in cell death and cell survival. Front Biosci 2009;14:72–111. doi:10.2741/3232
100. Giansanti V, Donà F, Tillhon M, Scovassi AI. PARP inhibitors: new tools to protect from inflammation. Biochem Pharmacol. 2010;80(12):1869–1877. doi:10.1016/j.bcp.2010.04.022
101. Jain PG, Patel BD. Medicinal chemistry approaches of poly ADP-ribose polymerase 1 (PARP1) inhibitors as anticancer agents - A recent update. Eur J Med Chem. 2019;165:198–215. doi:10.1016/j.ejmech.2019.01.024
102. Thomas C, Ji Y, Lodhi N, et al. Non-NAD-like poly(ADP-ribose) polymerase-1 inhibitors effectively eliminate cancer in vivo. EBioMedicine. 2016;13:90–98. doi:10.1016/j.ebiom.2016.10.001
103. Kapoor K, Singla E, Sahu B, Naura AS. PARP inhibitor, olaparib ameliorates acute lung and kidney injury upon intratracheal administration of LPS in mice. Mol Cell Biochem. 2014;400(1–2):153–162. doi:10.1007/s11010-014-2271-4
104. Ahmad A, de Camargo Vieira J, de Mello AH, et al. The PARP inhibitor olaparib exerts beneficial effects in mice subjected to cecal ligature and puncture and in cells subjected to oxidative stress without impairing DNA integrity: a potential opportunity for repurposing a clinically used oncological drug for the experimental therapy of sepsis. Pharmacol Res. 2019;145. doi:10.1016/j.phrs.2019.104263
105. Ghonim MA, Pyakurel K, Ibba SV, et al. PARP is activated in human asthma and its inhibition by olaparib blocks house dust mite-induced disease in mice. Clin Sci. 2015;129(11):951–962. doi:10.1042/CS20150122
106. Gariani K, Ryu D, Menzies KJ, et al. Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease. J Hepatol. 2017;66(1):132–141. doi:10.1016/j.jhep.2016.08.024
107. Mukhopadhyay P, Horváth B, Rajesh M, et al. PARP inhibition protects against alcoholic and non-alcoholic steatohepatitis. J Hepatol. 2017;66(3):589–600. doi:10.1016/j.jhep.2016.10.023
108. Teng F, Zhu L, Su J, et al. Neuroprotective effects of poly(ADP-ribose)polymerase inhibitor olaparib in transient cerebral ischemia. Neurochem Res. 2016;41(7):1516–1526. doi:10.1007/s11064-016-1864-6
109. Berger NA, Besson VC, Boulares AH, et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br J Pharmacol. 2018;175(2):192–222. doi:10.1111/bph.13748
110. Bao Z, Xiong J, Li W, Chen Z, Shen H, Ying S. Genomic instability in chronic airway inflammatory diseases. Biomed J. 2015;38(2):117–124. doi:10.4103/2319-4170.143478
111. Oliver FJ, Ménissier-de Murcia J, Nacci C, et al. Resistance to endotoxic shock as a consequence of defective NF-κB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999;18(16):4446–4454. doi:10.1093/emboj/18.16.4446
112. Soriano FG, Liaudet L, Szabó É, et al. Resistance to acute septic peritonitis in poly(ADP-ribose) polymerase-1-deficient mice. Shock. 2002;17(4):286–292. doi:10.1097/00024382-200204000-00008
113. Corral J, Yélamos J, Hernández-Espinosa D, et al. Role of lipopolysaccharide and cecal ligation and puncture on blood coagulation and inflammation in sensitive and resistant mice models. Am J Pathol. 2005;166(4):1089–1098. doi:10.1016/S0002-9440(10)62329-2
114. Nicolescu AC, Holt A, Kandasamy AD, Pacher P, Schulz R. Inhibition of matrix metalloproteinase-2 by PARP inhibitors. Biochem Biophys Res Commun. 2009;387(4):646–650. doi:10.1016/j.bbrc.2009.07.080
115. Lechaftois M, Dreano E, Palmier B, et al. Another “string to the bow” of PJ34, a potent poly(ADP-ribose)polymerase inhibitor: an antiplatelet effect through P2Y12 antagonism? PLoS One. 2014;9(10):e110776–e110776. doi:10.1371/journal.pone.0110776
116. Murakami K, Enkhbaatar P, Shimoda K, et al. Inhibition of poly (ADP-ribose) polymerase attenuates acute lung injury in an ovine model of sepsis. Shock. 2004;21(2):126–133. doi:10.1097/01.shk.0000108397.56565.4a
117. Khin Hnin Si M, Mitaka C, Tulafu M, et al. Inhibition of poly (adenosine diphosphate-ribose) polymerase attenuates lung-kidney crosstalk induced by intratracheal lipopolysaccharide instillation in rats. Respir Res. 2013;14(1):126. doi:10.1186/1465-9921-14-126
118. Lobo SM, Orrico SRP, Queiroz MM, et al. Pneumonia-induced sepsis and gut injury: effects of a poly-(ADP-ribose) polymerase inhibitor. J Surg Res. 2005;129(2):292–297. doi:10.1016/j.jss.2005.05.018
119. Zhang L, Yao J, Wang X, Li H, Liu T, Zhao W. Poly (ADP-ribose) synthetase inhibitor has a heart protective effect in a rat model of experimental sepsis. Int J Clin Exp Pathol. 2015;8(9):9824–9835.
120. Soriano FG, Nogueira AC, Caldini EG, et al. Potential role of poly(adenosine 5′-diphosphate-ribose) polymerase activation in the pathogenesis of myocardial contractile dysfunction associated with human septic shock. Crit Care Med. 2006;34(4):1073–1079. doi:10.1097/01.CCM.0000206470.47721.8D
121. Li L, Hu BC, Gong SJ, Yu YH, Dai HW, Yan J. Association of poly(ADP-ribose) polymerase activity in circulating mononuclear cells with myocardial dysfunction in patients with septic shock. Chin Med J (Engl). 2014;127(15):2775–2778. doi:10.3760/cma.j.issn.0366-6999.20140378
122. Zhang JN, Ma Y, Wei XY, et al. Remifentanil protects against lipopolysaccharide-induced inflammation through PARP-1/NF- κ B signaling pathway. Mediators Inflamm. 2019;2019. doi:10.1155/2019/3013716
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.