")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 6
Palbociclib: an evidence-based review of its potential in the treatment of breast cancer
Authors Cadoo K, Gucalp A, Traina T
Received 5 March 2014
Accepted for publication 17 April 2014
Published 4 August 2014 Volume 2014:6 Pages 123—133
DOI https://doi.org/10.2147/BCTT.S46725
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Video abstract presented by Karen A Cadoo.
Views: 901
Karen A Cadoo, Ayca Gucalp, Tiffany A Traina
Breast Cancer Medicine Service, Memorial Sloan Kettering Cancer Center and Weill Medical College of Cornell University, New York, NY, USA
Abstract: Cellular proliferation, growth, and division following DNA (deoxyribonucleic acid) damage are tightly controlled by the cell-cycle regulatory machinery. This machinery includes cyclin-dependent kinases (CDKs) which complex with their cyclin partners, allowing the cell cycle to progress. The cell-cycle regulatory process plays a critical role in oncogenesis and in the development of therapeutic resistance; it is frequently disrupted in breast cancer, providing a rational target for therapeutic development. Palbociclib is a potent and selective inhibitor of CDK4 and -6 with significant activity in breast cancer models. Furthermore, it has been shown to significantly prolong progression-free survival when combined with letrozole in the management of estrogen receptor-positive metastatic breast cancer. In this article we review the cell cycle and its regulatory processes, their role in breast cancer, and the rationale for CDK inhibition in this disease. We describe the preclinical and clinical data relating to the activity of palbociclib in breast cancer and the plans for the future development of this agent.
Keywords: cell-cycle regulation, cyclin-dependent kinases, CDK4/6 inhibition
Introduction
The addition of targeted agents to our armamentarium has provided the potential for significant clinical benefit for patients with metastatic breast cancer (MBC).1 Extensive efforts are underway to design novel therapies, including those that target vulnerabilities within the cancer cell cycle. Aberrations of the cell cycle are ubiquitous in cancer,2 and there is increasing recognition of the role of cell-cycle regulators in intrinsic and acquired resistance to therapy.3 Cellular proliferation and growth, in addition to division following deoxyribonucleic acid (DNA) damage, are tightly controlled by the cell-cycle regulatory machinery. Cell-cycle transition and the commitment to cell division are coordinated by cyclin-dependent protein kinases (CDKs) (Figure 1). These serine/threonine kinases rely on a regulatory cyclin partner to form CDK–cyclin heterodimer complexes that control cell cycling.2 Palbociclib is a potent and selective inhibitor of CDK4 and -6, which are critical components of the cell-cycle regulatory machinery. This review will discuss the role of the cell cycle in breast cancer and the data relating to palbociclib as a therapeutic strategy.
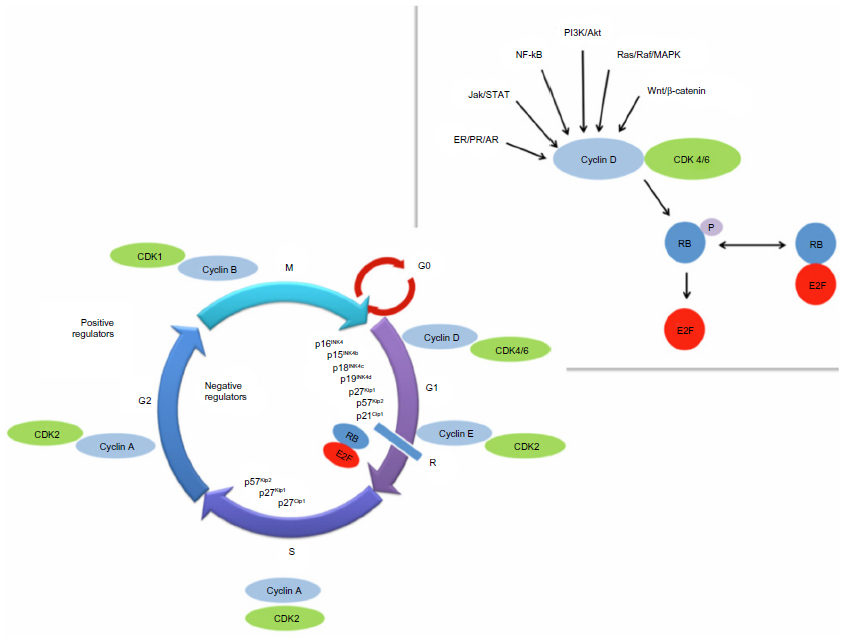 | Figure 1 The cell cycle and regulatory process. |
The cell cycle
Each individual cell receives a number of external signals which are integrated and processed by the cell-cycle regulatory machinery, and the cell makes a decision whether to divide or be quiescent.2 Cell division comprises four phases – the S phase of DNA synthesis, the M phase of mitosis, and two gap phases, G1 and G22 (Figure 1). There are four proliferative CDKs: CDK1 regulates the transition from G2 to M phase and CDK2, -4, and -6 regulate the transition from G1 to S phase.4 As the cell cycles, G1 and G2 act as checkpoints for progression, ensuring all necessary steps have been completed prior to entering the next phase.2 During G1, at the restriction or R point, the cell has two options, to continue cycling or to enter the non-dividing G0 state.5 Beyond the restriction point, progression through the cell cycle continues, independent of external stimuli.6 There is some redundancy within the system, as CDK2 knockdown models are viable, and therefore proliferation can bypass CDK2.7 However, CDK4 and -6 are critical drivers of oncogenesis in some tumors and therefore an attractive target for drug development.7
Transition from G1 to the S phase is controlled by the retinoblastoma tumor suppressor gene product (RB)2 (Figure 1). Retinoblastoma is a critical negative regulator of the cell cycle, preventing premature cell division by complexing with the E2F transcription factors, thereby inhibiting G1/S transition.6 Inactivation of RB releases the hold on the cell cycle, allowing division to proceed. This inactivation is achieved via sequential phosphorylation of retinoblastoma – as cells transition from M to G1, almost all phosphate groups are removed and RB is un-phosphorylated. During progression through G1, growth signals allow cyclin D to complex with either CDK4 or -6. This process facilitates the addition of phosphate groups to RB, resulting in hypophosphorylation. As the cell passes through the R point, cyclin E complexes with CDK2 and hyperphosphorylates RB, with release of bound E2F. This E2F activates S-phase genes allowing cell cycle progression.2 The CDK4 and -6 are functionally equivalent in their ability to phosphorylate RB, therefore when targeting this aspect of the cell cycle, inhibition of both CDK4 and CDK6 is required.7
Cell-cycle control in breast cancer
Loss of RB expression occurs in 20%–35% of breast cancers; thus, the majority of breast cancers maintain intact or functioning RB.8 The incidence of RB loss is dependent on the clinical subtype of breast cancer and is more common in triple-negative breast cancer (TNBC) compared with other subtypes.9 RB inactivation facilitates tumor progression through loss of proliferation control and conversion to an invasive phenotype.10 In general, it is associated with poorer differentiation and increased metastatic potential.10 In the absence of functional RB, cell–cell adhesion is disrupted, and epithelial-to-mesenchymal transition is facilitated, potentially underlying the increase in metastases observed.10 However, the impact of RB loss is dependent on the breast cancer subtype. RB dysregulation is associated with poor prognosis in estrogen receptor (ER)-positive breast cancer but better outcomes in ER-negative tumors.11 This difference in outcomes may be driven by differing responses to therapy,11 as RB loss increases sensitivity to DNA-damaging agents but results in continued proliferation in tamoxifen-treated xenografts.12
While the majority of tumors maintain functional RB, the CDK4/6–cyclin D pathway may be disrupted by a number of other mechanisms, with increased pathway activation facilitating G1 progression, providing a proliferative advantage to cancer cells.4 In tumors with intact RB, the process of RB phosphorylation and inactivation necessary to facilitate cell division is rate limited by cyclin D1 levels.13 Cyclin D1 is important for both breast development and breast cancer formation.13 Although cyclin D1 amplifications occur in 15% of breast cancer, protein overexpression occurs in >50%.14 In addition, signaling via the RAS/RAF/MAPK pathway increases cyclin D production.6 There are two isoforms of cyclin D1, cyclin D1a and cyclin D1b.13 High cyclin D1a levels have been shown to inversely correlate with Ki67 levels; however, they were not prognostic. In contrast, cyclin D1b elevation is associated with increased recurrence, distant metastases, and reduced survival. In particular, increased cyclin D1b is associated with poorer outcomes in ER-negative breast cancers.13
The formation of an activated cyclin D–CDK4/6 heterodimer also requires the cooperation of the CDK interacting protein/kinase inhibitory protein (Cip/Kip) family.4 Elevated cyclin E levels mediating RB pathway disruption15 and loss of p27kip1 occur frequently and are associated with poor outcome in breast cancer.16 Inactivation of the tumor suppressor and CDK inhibitor p16INKa occurs in 50% of invasive breast cancers. This can occur via multiple mechanisms, including gene deletion, point mutations, and methylation-mediated transcriptional silencing.17 Inactivation of p16INKa allows unchecked cell division and is associated with a loss of differentiation and high proliferation.17 However, the role of p16INKa in cancer is complex and depends on context.18 As noted, p16INKa is a tumor suppressor, and its loss is associated with tumor progression; however, its overproduction has also been associated with disease progression.18 Cells undergoing p16INKa-mediated senescence may acquire a secretory phenotype resulting in release of proinflammatory mediators that contribute to tumor growth via modulation of the microenvironment.18–20
As expected based on our understanding of the extensive but incomplete overlap between clinical and intrinsic subgroups of breast cancer, RB pathway alterations also differ by molecular subtype, as outlined by data from the Cancer Genome Atlas (Table 1).21 Luminal A tumors are more likely than other subtypes to retain intact RB. However, cyclin D1 amplification frequently occurs in the luminal subtypes, albeit most notably in luminal B tumors. In contrast to luminal subtypes, and in keeping with the overlap with clinically TNBC, basal like breast cancers have frequent RB loss. These tumors are also associated with cyclin E amplification. In HER2-enriched breast cancers, DNA amplification of CDK4 and cyclin D1 is found.21
 | Table 1 RB pathway alterations by breast cancer subtype |
Differences in outcomes associated with RB function may also be driven by the differences in underlying molecular biology. In an analysis of 201 ER-positive breast cancers of known luminal A or luminal B molecular subtype, the luminal B cancers were shown to be associated with a transcriptional signature of RB loss. These luminal B cancers were shown to have a poorer response to endocrine therapy compared with the luminal A cancers.22 The RB signature was also able to stratify tumors within the luminal B group based on low or high expression of RB loss. Those with low expression were more sensitive to endocrine therapy and had better survival.22
As outlined, dysregulation of cell-cycle control is a frequent event in breast cancer and occurs via a number of different mechanisms. The ubiquitous nature of these disruptions involving components of CDK4/6–cyclin D regulation implies that they confer a survival advantage to the cancer cell.4 Interestingly, in the setting of ductal carcinoma in situ, increased expression of p16ink4 and loss of RB function increases risk of recurrence and progression to invasive disease.23 In addition, elevated expression of p16ink4 in the surrounding non-proliferative stroma has been associated with disease recurrence, independent of histopathologic features, speaking to the importance of the cells in the surrounding microenvironment.23 Given the frequency of cell-cycle machinery alterations in breast cancer, this is an important arena for therapeutic targeting.
ER-positive breast cancer, therapeutic targeting, and resistance
The majority (75%) of breast cancers are hormone receptor positive,24 depending on estrogen signaling for proliferation and survival. Estrogen can recruit cells from G0 and increase the rate of progression from G1 to S phase.3 Therapeutic withdrawal of estrogen (eg, via aromatase inhibition) and ER modulators (eg, tamoxifen) or antagonists (eg, fulvestrant) reduce CDK/cyclin complexing. This results in G0/G1 phase cell-cycle arrest and reduced tumor cell viability.22 However, de novo and acquired resistance to endocrine therapy are common, limiting the efficacy of these well-tolerated therapies.25 Efforts continue to try to better understand the mechanisms driving this resistance and to develop therapies and combinations that overcome this issue.25
The cell-cycle machinery is important for efficacy of hormonal therapy in breast cancer as ER-positive RB-negative xenograft models are resistant to tamoxifen.12 In a study of postmenopausal patients randomized to 2 years of adjuvant tamoxifen or placebo, in tumors with a functional RB pathway (N=204), there was significant benefit derived from adjuvant tamoxifen (hazard ratio [HR] =0.53, 95% confidence interval [CI] 0.34–0.81, P=0.003). However, in tumors with nonfunctioning RB (N=57), there was no benefit observed (HR =2.28, 95% CI 0.51–10.3, P=0.28).26
Cyclin D1 is essential for the regulation of breast epithelial cell division.27 Binding to ER-alpha drives cyclin D1 transcription, with activation of CDK4/6 and phosphorylation of RB and cell cycling.22 However, cyclin D1 can also activate ER-alpha, driving transcription and cell division independent of estrogen and CDK.27 This independent activity is not inhibited by anti-estrogen strategies,27 and cyclin D1 overexpression has been associated with tamoxifen resistance.28 This resistance is mediated via formation of cyclin D1–CDK4/6 complexes which sequester the inhibitory proteins p21Cip1 and p27kip1, with activation of cyclin E1–CDK2 complexes.29 In addition, overexpression specifically of the cyclin D1b isoform overcomes anti-estrogen therapy effect.30
In ER-positive cell lines, E2F transcription and cell-cycle progression can occur independent of estrogen following exposure to an aromatase inhibitor. Some of these cell lines drive E2F transcription via the ligand-free ER; these are resistant to aromatase inhibitors but respond to ER downregulation with fulvestrant. Other cells drive E2F signaling, independent of the ER. These cells function as if ER-negative and do not respond to fulvestrant; however, they rely on CDK4 to activate E2F. Both of these models, the first which is estrogen independent and ER dependent and the second which is estrogen and ER independent, respond to CDK4/6 inhibition.31 For this reason, it has been suggested that following tumor progression with aromatase inhibition, CDK4/6 inhibition may be more appropriate and effective than an ER downregulator.31
A separate study confirmed that in the setting of endocrine therapy resistance, cell cycling can occur independent of the activity and functional state of the ER.22 In this setting, cyclin D1 expression, with RB phosphorylation and inactivation, is maintained despite efficient ER antagonism.22 However, CDK4/6 inhibition can suppress cyclin D1 activity, restoring RB transcriptional repression, and inhibit proliferation in these tumors that are resistant to endocrine therapy.22 This acquired resistance to endocrine therapy driven by dysregulated RB/E2F transcriptional control can be negated by CDK4/6 inhibition, with the potential to re-sensitize these tumors to hormonal therapy.22
The cell-cycle regulatory machinery has been implicated in the development of resistance to endocrine therapy via multiple mechanisms. CDK4/6 inhibition alone or in combination with hormonal therapy may be able to overcome this resistance and provide durable tumor responses.
Palbociclib preclinical data
Palbociclib, or PD-0332991 (Pfizer, Inc., New York, NY, USA), is an orally available inhibitor of CDK4 and -67 (Figure 2). PD-0332991 was developed from a group of pyridopyrimidine compounds due to its favorable physical and pharmaceutical properties.7 PD-0332991 potently and equally inhibits both CDK4– and CDK6–cyclin D1 kinase activity.7 In addition, it is highly selective for CDK4/6, with little activity against a panel of multiple other protein kinases. PD-0332991 inhibits cell growth and suppresses DNA replication at low nanomolar concentrations in a number of RB proficient human cancer cells, including breast cancer. As expected, no activity is seen in RB-deficient cells.7 Consistent with the anticipated impact of a CDK4/6 inhibitor on cell-cycle progression, these cells show significant increase in the proportion of cells in G1. In vivo, significant antitumor activity is demonstrated in breast cancer xenografts, with near to complete suppression of tumor growth. In addition, sustained inhibition of tumor RB phosphorylation is possible.7 The specificity of PD-0332991 in targeting CDK4/6–cyclin D is important, allowing inhibition of oncogenic events while sparing normal tissue which is in a relatively inactive, non-cycling state.16
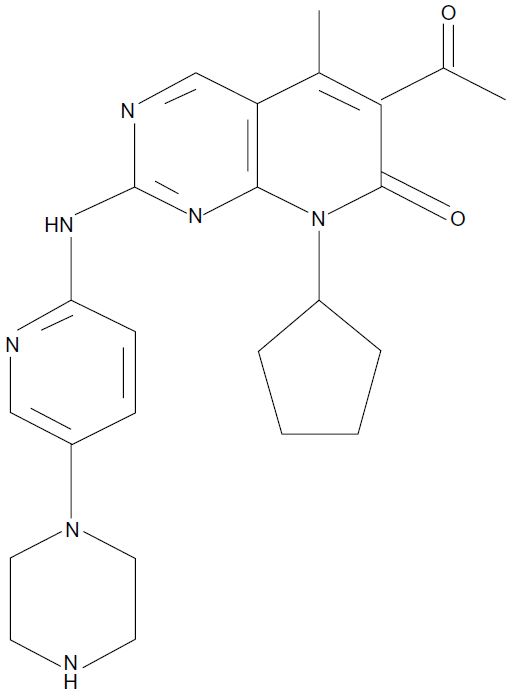 | Figure 2 Palbociclib, or PD-0332991, a CDK4/6 inhibitor from Pfizer, Inc. (New York, NY, USA). |
It is not surprising given our understanding of the differing drivers of the intrinsic subtypes of breast cancer32 and their corresponding differences in RB pathway alterations,21 that sensitivity to CDK4/6 inhibition depends on molecular phenotype. Finn et al33 investigated the activity of PD-0332991 in a number of cell lines representing the molecular subtypes of breast cancer and found a significant correlation between subtype and sensitivity to the drug. The authors found that ER-positive cell lines with luminal features are most sensitive. In addition, HER2-amplified cell lines that respond to PD-0332991 primarily have luminal features. In contrast, basal cell lines are resistant (Figure 3).
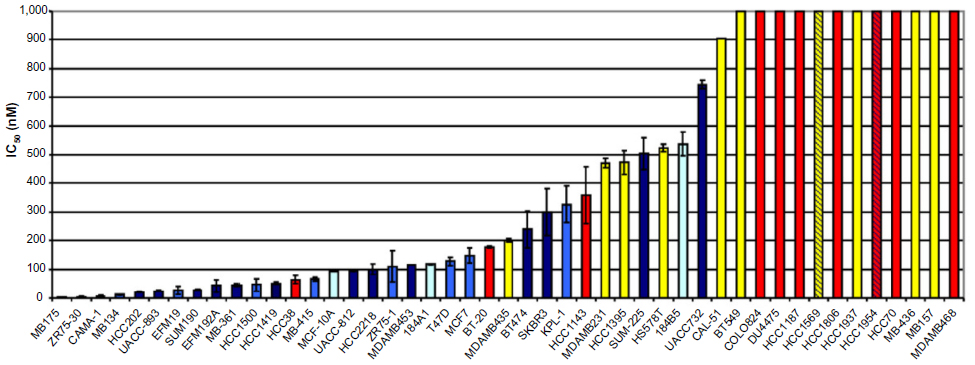 | Figure 3 Inhibitory concentration and cell type. |
PD-0332991 effectively dephosphorylates RB and inhibits the cell cycle in tamoxifen-resistant breast cancer models.22,33 In cell lines resistant to tamoxifen, there is activity with PD-0332991 monotherapy, and cells are re-sensitized to tamoxifen, with increased activity when the drugs are combined.31 In breast cancer models refractory to hormonal therapy, PD-0332991 can effectively halt proliferation and induce cellular senescence, resulting in a stable cell-cycle arrest that is distinct from the effect of ER antagonists.22 For tumors with acquired resistance to endocrine therapy, CDK4/6 inhibition represents an important and viable therapeutic strategy.22
Further preclinical data have confirmed the activity and specificity of PD-0332991 in a number of breast cancer models but have also elucidated a number of mechanisms through which resistance to this agent may arise.16 Increased RB and cyclin D1 and decreased p16 are associated with sensitivity to PD-0332991.33 Tumor cells without endogenous RB are resistant to PD-03329917,16 and in fact show a significant growth advantage when exposed to the drug.16 Tumor cells without endogenous RB also have abundant expression of p16ink4a, which separately inhibits CDK4/6 activity. These tumors have acquired independence from CDK4/6 signaling and therefore do not appear responsive to CDK4/6 inhibition.19
As expected by its mechanism of action, cell lines sensitive to PD-0332991 decrease RB phosphorylation, while those that are resistant to the drug exhibit little effect on RB phosphorylation. In addition, phosphorylated RB is seen in some non-luminal cell lines that are resistant to the anti-proliferative effects of PD-0332991; the mechanism of this resistance is not clear but may reflect CDK4/6 mutations, greater dependence on CDK1/2–cyclin E, or loss of negative regulators of CDK. In terms of clinical translation, the implication is that intact RB is required for but does not predict response to this agent.33
PD-0332991 stabilizes cyclin D1 putatively through proximal effects on the CDK4/6–cyclin D1 complex. Elevated cyclin D1, as is frequently seen in breast cancers, has the potential to activate CDK4/6 if the levels of the drug are limited, with resultant therapeutic resistance and tumor growth.16
Dean et al16 determined that a crucial downstream effect of CDK4/6 inhibition is modulation of E2F targets, and their deregulation can overcome PD-0332991 efficacy. While RB status is critical for acute response to PD-0332991, compensatory mechanisms controlling E2F have significant implications in the development of resistance with longer-term use.16 Resistance to extended CDK4/6 inhibition may be due to loss of RB function, with increase in the E2F-target genes cyclins A and E. These are required for CDK2 function, potentially providing a mechanism to acquire independence from CDK4/6. Loss of functioning RB may facilitate this independence from CDK4/6, allowing CDK2 to drive proliferation.16 Cell-cycle arrest in the absence of RB is associated with suppression of downstream targets – for example, due to accumulation of p107.16 In the setting of p107 accumulation and RB deficiency, PD-0332991 generates moderate but incomplete repression of E2F-regulated proteins. Dean et al16 showed that cell populations initially sensitive to CDK4/6 inhibition that subsequently develop resistance with extended exposure demonstrate high p107, CDK2 protein, and loss of the inhibitory proteins p21Cip1 and p27kip1. Loss of p27kip1 has been associated with poor prognosis in breast cancer, and p27kip1 protein levels may be predictive of poor response to CDK4/6 inhibition in addition to this prognostic utility.
Thus, the cell-cycle regulatory network is complex, influenced by RB, E2F, and p16ink4a in tumors, with implications for tumor response that may be important in patient selection.16 Redundancies in the CDK/cyclin mechanism and the potential for bypass of this mechanism in cell-cycle control suggest that CDK4/6 inhibition may be better considered in combination therapy.
Preclinical data PD-0332991 combination therapy
Anti-estrogen strategies repress transcription of ER-alpha target genes in S phase. In cells that are therapeutically arrested (eg, with thymidine or hydroxyurea), an increase in apoptosis is possible over that seen with anti-estrogen therapy alone. This synergy of anti-estrogen therapy with cell-cycle effect underpins the possibility of enhanced efficacy when combined with cell-cycle-targeting drugs.34 This has been borne out preclinically, as PD-0332991 is synergistic with tamoxifen in ER-positive cell lines, with a mean combination index <1 across clinically relevant drug concentrations.33 PD-0332991 is similarly synergistic with trastuzumab in HER2-amplified cell lines. PD-0332991 can sensitize resistant cells to tamoxifen again33 and inhibit growth in models that have acquired resistance to aromatase inhibition and ER antagonists.31
However, combining CDK4/6 inhibition with DNA-damaging techniques may be more complex. Radiation-induced cytotoxicity is dependent on the phase of the cell cycle such that early G1 and late S phases are relatively resistant compared with G1/S and G2/M transition phases which are more radiosensitive.35 Johnson et al35 demonstrated in cell lines dependent on CDK 4/6, that its inhibition lengthens G1 and increases radio-resistance. As expected, there is little effect on tumor models proliferating independently of CDK4/6. They also showed that CDK4/6 inhibition can alleviate the myelosuppression of lethal doses of total body irradiation, thereby increasing survival. This is achieved by reversible quiescence of early hematopoietic stem/progenitor cells. They postulated that for patients who have tumors that are RB deficient, the addition of CDK4/6 inhibition could aid bone marrow tolerance of cytotoxic agents without compromising efficacy. Furthering this theory, they explored the role of PD-0332991 with carboplatin in mouse models of breast cancer with intact and absent RB.4 As expected, single agent PD-0332991 inhibited tumor growth in RB-intact but not RB-deficient tumors. Combination therapy was shown to be myeloprotective but compromised efficacy in RB-intact mice, compared with carboplatin alone. In RB-deficient mice there was no deleterious effect on tumor growth with the combination, but statistically significant platelet protection was demonstrated. This study suggested two distinct potential roles for PD-0332991, as an anti-proliferative agent in RB-dependent tumors and as a myeloprotective agent that can facilitate delivery of cytotoxic therapy in RB-independent tumors.
RB-deficient TNBC cell lines are more sensitive to the effects of doxorubicin and methotrexate than those with intact RB; however response to multiple other drugs (including cisplatin) does not vary by RB status.36 Like radiation therapy, cytotoxic agents such as doxorubicin rely on active cell cycling for effect, raising questions about the role of CDK4/6 inhibition in combination with anti-proliferative agents.37 Continuous CDK4/6 inhibition facilitates prolonged cell-cycle arrest, thereby preventing cytotoxic-mediated cell death.38 In RB-intact TNBC models, PD-0332991 protects cells from doxorubicin cytotoxicity.37 Furthermore when the same cell line (MDA-MB-231) has continuous PD-0332991 exposure before or with paclitaxel administration, there is an increase in cancer cell number.38
While anthracyclines and taxanes have different mechanisms of action, both drug classes ultimately result in DNA damage, and PD-0332991-mediated G1 cell-cycle arrest protects RB-intact cells from this cytotoxic driven apoptosis. CDK4/6 inhibition does not alter the rate of DNA repair but alters the repair mechanism with a shift from predominantly homologous recombination to non-homologous end joining.38 Therefore in addition to antagonizing the apoptotic effect of cytotoxic therapy, CDK4/6 inhibition can also potentiate disease progression via an increase in error-prone DNA-repair mechanisms.38 In contrast to the result with continuous exposure, when PD-0332991 is used to synchronize cells for 24 hours before paclitaxel administration, there is an increase in cytotoxicity, suggesting a potential role in a metronomic cytotoxic regimen.37 It is possible that pretreatment with a CDK4/6 inhibitor may facilitate cells synchronously entering S phase, the point at which they are most susceptible to cytotoxic DNA damage;39 however, further validation is required.
PD-0332991 (palbociclib) clinical development
Schwartz et al40 reported the first in-human, Phase I dose-finding study of PD-0332991 (palbociclib) in 33 patients with RB-intact advanced solid tumors and non-Hodgkin lymphoma. Palbociclib was administered 2 weeks on, 1 week off per cycle, with cohort-based dose escalation. The maximum dose administered was 225 mg, with two of six (33%) patients treated at that dose level experiencing dose-limiting toxicities (DLTs) of neutropenia and thrombocytopenia. A 200 mg dose was selected for further evaluation initially with six patients and subsequently expanded to include 20 patients. There were four (20%) DLTs of neutropenia and thrombocytopenia at this dose, falling below the 33% pre-specified definition of maximum tolerated dose. Therefore, 200 mg became the recommended Phase II dose. Palbociclib was well tolerated, with no patients discontinuing therapy for a treatment-related adverse event (AE). Hematological toxicity was common, with grade 3/4 lymphopenia (36%), neutropenia (24%), leucopenia (21%), thrombocytopenia (9%), and anemia (3%). Non-hematologic toxicity was mild, with no grade 4 and rare grade 3 AEs including fatigue, hyperglycemia, hyponatremia, nausea, and vomiting. The most common non-hematological AEs of any grade were fatigue, nausea, diarrhea, constipation, rash, and epistaxis. Thirty-one patients were evaluable for response. There was one partial response (PR) in a patient with a non-seminomatous germ cell tumor. Nine patients with mixed tumor subtypes had stable disease (SD).
A second Phase I study examined palbociclib in a 3 weeks on, 1 week off schedule, in patients with advanced malignancy refractory to standard therapy.41 Five of 41 patients experienced a DLT, all of which were neutropenia, at varied dose levels. The recommended Phase II dose was deemed to be 125 mg once daily on this schedule. Hematologic toxicities again predominated with grade 3 neutropenia (12%), anemia (7%), and leucopenia (2%). Non-hematologic toxicity was mild, including fatigue, nausea, and diarrhea. Thirty-seven patients were evaluable for response. Of these, 13 (35%) had stable disease as best response, including six patients (16.2%) who received ≥10 cycles of therapy.
Pharmacokinetic data from the two schedules40,41 determined that palbociclib is slowly absorbed (mean Tmax 4.2–5.5 hours, respectively), with a large volume of distribution and tissue penetration. Palbociclib is also eliminated slowly, with mean terminal half-life of 26 hours and mean apparent plasma clearance of 80–89 hours. There is dose proportional exposure over the ranges explored, with neutropenia proportional to exposure. However, decreases in neutrophil and platelet numbers are not cumulative and it has been shown that the effect of palbociclib on these cells is saturable.40
Palbociclib of 125 mg daily, 3 weeks on, 1 week off was selected for further study. A Phase II study of multiple solid tumors noted activity in patients with breast cancer and was expanded to include a total of 37 patients with breast cancer.42 Patients were heavily pretreated with a median 3 prior lines of chemotherapy for advanced disease. The majority of patients enrolled (31/37, two of whom had HER2-positive disease) had hormone receptor-positive breast cancer. Two of 30 patients had PR, and 17 had disease stabilization as best response. Six patients enrolled in this study had TNBC. In this small subgroup, there were no responses; however, one patient had disease stabilization. The progression-free survival (PFS) was 3.8 months (95% CI 2.0–7.1) in patients with hormone receptor-positive breast cancer, 1.9 months (95% CI 1.8–infinity) for those with TNBC. The drug was well tolerated. Grade 3/4 toxicities were hematologic (neutropenia, leucopenia, lymphopenia, and thrombocytopenia) and most were uncomplicated (only one episode of febrile neutropenia). Sixteen (43%) patients in this heavily pretreated cohort required dose modification for hematologic toxicity. This study underlines the potential for CDK 4/6 inhibition in luminal breast cancers, and further expansion of this study with specific breast cancer subtypes is ongoing.
Given preclinical data suggesting greater sensitivity of luminal breast cancer to CDK4/6 inhibition and synergy with endocrine therapy,33 a Phase IB/II study was initiated to explore combination therapy with letrozole in the context of ER-positive HER2-negative MBC.43 Twelve patients received palbociclib (125 mg daily, 3 weeks on, 1 week off) with letrozole (2.5 mg daily). There were three DLTs due to neutropenia. The most common AEs were neutropenia, leucopenia, and fatigue. There was no drug–drug interaction with letrozole observed. Three patients experienced PR, and nine had disease stabilization. This established the recommended Phase II dose of palbociclib as 125 mg once daily, 3 weeks on, 1 week off when combined with letrozole 2.5 mg daily.
This encouraging signal of activity led to the Phase II study expansion where patients were randomized to combination therapy compared with letrozole alone as first-line treatment for MBC.44,45 The Phase II study was designed in two parts. In part one, patients were clinically selected based on ER-positive HER2-negative MBC. In the second part, additional eligibility criteria included tumor cyclin D1 gene (CCND1) amplification and/or p16 loss. Sixty-six patients were randomized in part one. The primary endpoint of improved PFS was met at 18.2 months for the combination compared with 5.7 months for letrozole alone (HR 0.35, 95% CI 0.17–0.72, P=0.006). PR rate and disease stabilization were higher for the combination (52% versus 32% and 35% versus 25%, respectively). There were no grade 3/4 AEs with letrozole alone. For combination therapy, the most common drug-related grade 3/4 AEs were neutropenia, leucopenia, and fatigue. Three patients discontinued study therapy secondary to an AE. The median duration of therapy was 14 months (<1–24). A second interim analysis of this study combining parts 1 and 2 with a total of 165 patients was presented at the annual San Antonio Breast Cancer posium in 2012.45 The significant improvement in PFS with the combination was maintained (26.1 versus 7.5 months, HR 0.37, 95% CI 0.21–0.63, P<0.001). No new safety signals emerged. This exciting improvement in PFS led to the US Food and Drug Administration breakthrough therapy designation in April 2013.
The final PFS analysis for this study was presented at the American Association for Cancer Research Annual Meeting 2014 and the statistically significant difference in PFS persisted (20.2 months for combination versus 10.2 months for letrozole alone).46 Patients enrolled in part 2 were selected for eligibility based on tumor CCND1 amplification and/or loss of p16, as possible biomarkers of sensitivity to palbociclib. However, there was no correlation found between these markers and outcomes. An interim analysis of overall survival, based on 61 patients, showed a nonsignificant trend in favor of the combination (37.5 months versus 33.3 months, respectively; HR 0.813; P=0.2105), these data were immature, and final analysis is outstanding.
These studies of palbociclib in breast cancer42–46 (Table 2) have been reported in abstract form only and full published reports are eagerly awaited. In addition further data from exploratory correlatives regarding the role of selected biomarkers (Ki67, CCND1 amplification, and p16 loss) to guide patient selection are outstanding.42–46 From a clinical perspective CDK 4/6 inhibition is most likely to be effective in hormone receptor-positive and HER2-positive breast cancers. As noted, luminal breast cancers frequently have intact RB,21 and preclinical data have suggested these tumors are sensitive to CDK4/6 inhibition.33 In addition, synergy with endocrine therapy has been demonstrated.33,43–46 Luminal HER2-positive breast cancers are also sensitive to CDK inhibition,33 and synergy with trastuzumab has been demonstrated.7 In contrast, this strategy is ineffective in the setting of RB loss,7 an event which is common in TNBC.9
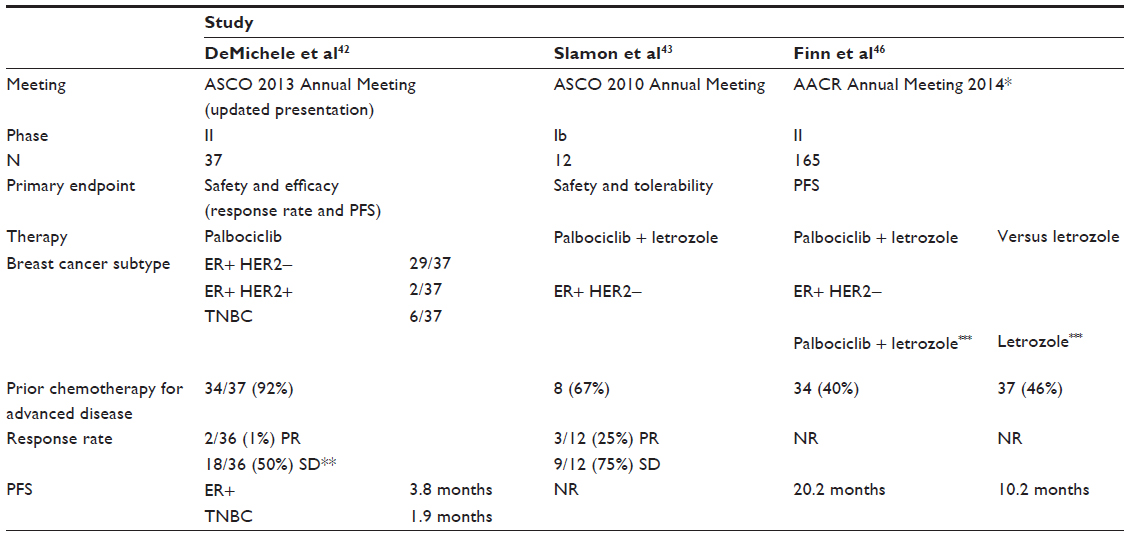 | Table 2 Clinical studies of Palbociclib in breast cancer |
Given the statistically and clinically significant PFS benefit seen in the Phase II combination study,44–46 a Phase III randomized, double-blind study is underway comparing palbociclib and letrozole with letrozole alone as first-line therapy for postmenopausal women with ER-positive HER2-negative MBC.47 There are no biomarker-driven eligibility specifications for this study. There are a number of additional studies, open or planned, exploring the role of palbociclib in MBC. For hormone receptor-positive disease, these include combination therapy with fulvestrant (NCT01942135) or with letrozole versus capecitabine (NCT02028507) following failure of prior endocrine therapy. Combinations of palbociclib with hormonal therapy in the adjuvant setting (NCT02040857) and with residual disease following neoadjuvant chemotherapy (NCT01864746) are being investigated (NCT01864746). In addition, palbociclib is being combined with neoadjuvant endocrine therapy (NCT01723774 and NCT01709370). In HER2-positive MBC, palbociclib combined with trastuzumab emtansine (TDM1) is being explored (NCT01976169), and palbociclib is being combined with paclitaxel in a Phase Ib feasibility study accruing patients with any subtype of breast cancer (NCT01320592).
Conclusion
The cell-cycle regulatory machinery, critical to cellular division, is frequently disrupted in cancer contributing to sustained tumor growth.2 In tumors with functional RB, CDK4/6 inhibition can inhibit cell growth and suppress DNA replication.7 Palbociclib is a potent and selective CDK4/6 inhibitor that is active in breast cancer preclinical models.7 As expected due to its mechanism of action, it has no antitumor activity in RB-deficient tumor models.16 Palbociclib is synergistic with tamoxifen and trastuzumab in ER-positive and HER2-positive cell lines respectively.33 In addition, it can effectively inhibit the cell cycle in tamoxifen-resistant breast cancer cell lines.33 Palbociclib can protect rapidly dividing cells from DNA damage; in tumors with functional RB, this may protect tumor cells from cytotoxicity; however, in tumors that do not have functional RB, there is the potential to protect bone marrow without compromising efficacy.4 This provides the possibility for a supportive role for this therapy, separate from its efficacy. Phase I studies have demonstrated that palbociclib is well tolerated, with neutropenia being the most significant and dose-limiting toxicity.40,41 A Phase II monotherapy study provided a signal for efficacy in breast cancer;42 however it is in combination with letrozole that a striking improvement in PFS was demonstrated in a randomized Phase II trial.45 This combination and a number of other therapeutic strategies are currently being explored.47 In addition, there are a number of other CDK4/6 inhibitors, including LEE001 from Novartis and LY2835210 from Lily, being explored in breast cancer. These drugs are exciting but are not as far in development as palbociclib. Palbociclib in combination with hormonal therapy offers exciting promise in ER-positive breast cancer.
Disclosure
The authors have no conflicts of interest to report.
References
Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–529. | |
Weinberg RA. The Biology of Cancer. 2nd ed. New York: Garland Science; 2014. | |
Nair BC, Vadlamudi RK. Regulation of hormonal therapy resistance by cell cycle machinery. Gene Ther Mol Biol. 2008;12:395. | |
Roberts PJ, Bisi JE, Strum JC, et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104(6):476–487. | |
Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81(3):323–330. | |
Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol. 2005;23(36):9408–9421. | |
Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–1438. | |
Bosco EE, Knudsen ES. RB in breast cancer: at the crossroads of tumorigenesis and treatment. Cell Cycle. 2007;6(6):667–671. | |
Trere D, Brighenti E, Donati G, et al. High prevalence of retinoblastoma protein loss in triple-negative breast cancers and its association with a good prognosis in patients treated with adjuvant chemotherapy. Ann Oncol. 2009;20(11):1818–1823. | |
Arima Y, Inoue Y, Shibata T, et al. Rb depletion results in deregulation of E-cadherin and induction of cellular phenotypic changes that are characteristic of the epithelial-to-mesenchymal transition. Cancer Res. 2008;68(13):5104–5112. | |
Musgrove EA, Sutherland RL. RB in breast cancer: differential effects in estrogen receptor-positive and estrogen receptor-negative disease. Cell Cycle. 2010;9(23):4607. | |
Bosco EE, Wang Y, Xu H, et al. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Invest. 2007;117(1):218–228. | |
Millar EK, Dean JL, McNeil CM, et al. Cyclin D1b protein expression in breast cancer is independent of cyclin D1a and associated with poor disease outcome. Oncogene. 2009;28(15):1812–1820. | |
Arnold A, Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. J Clin Oncol. 2005;23(18):4215–4224. | |
Loden M, Stighall M, Nielsen NH, et al. The cyclin D1 high and cyclin E high subgroups of breast cancer: separate pathways in tumorogenesis based on pattern of genetic aberrations and inactivation of the pRb node. Oncogene. 2002;21(30):4680–4690. | |
Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29(28):4018–4032. | |
Lee JJ, Ko E, Cho J, et al. Methylation and immunoexpression of p16(INK4a) tumor suppressor gene in primary breast cancer tissue and their quantitative p16(INK4a) hypermethylation in plasma by real-time PCR. Korean J Pathol. 2012;46(6):554–561. | |
Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29(2):273–283. | |
Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. | |
Laberge RM, Awad P, Campisi J, Desprez PY. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. 2012;5(1):39–44. | |
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. | |
Thangavel C, Dean JL, Ertel A, et al. Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr Relat Cancer. 2011;18(3):333–345. | |
Witkiewicz AK, Rivadeneira DB, Ertel A, et al. Association of RB/p16-pathway perturbations with DCIS recurrence: dependence on tumor versus tissue microenvironment. Am J Pathol. 2011;179(3):1171–1178. | |
Nadji M, Gomez-Fernandez C, Ganjei-Azar P, Morales AR. Immunohistochemistry of estrogen and progesterone receptors reconsidered: experience with 5,993 breast cancers. Am J Clin Pathol. 2005;123(1):21–27. | |
Gnant M. Overcoming endocrine resistance in breast cancer: importance of mTOR inhibition. Expert Rev Anticancer Ther. 2012;12(12):1579–1589. | |
Lehn S, Ferno M, Jirstrom K, Ryden L, Landberg G. A non-functional retinoblastoma tumor suppressor (RB) pathway in premenopausal breast cancer is associated with resistance to tamoxifen. Cell Cycle. 2011;10(6):956–962. | |
Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88(3):405–415. | |
Stendahl M, Kronblad A, Ryden L, Emdin S, Bengtsson NO, Landberg G. Cyclin D1 overexpression is a negative predictive factor for tamoxifen response in postmenopausal breast cancer patients. Br J Cancer. 2004;90(10):1942–1948. | |
Hui R, Finney GL, Carroll JS, Lee CS, Musgrove EA, Sutherland RL. Constitutive overexpression of cyclin D1 but not cyclin E confers acute resistance to antiestrogens in T-47D breast cancer cells. Cancer Res. 2002;62(23):6916–6923. | |
Wang Y, Dean JL, Millar EK, et al. Cyclin D1b is aberrantly regulated in response to therapeutic challenge and promotes resistance to estrogen antagonists. Cancer Res. 2008;68(14):5628–5638. | |
Miller TW, Balko JM, Fox EM, et al. ERα-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov. 2011;1(4):338–351. | |
Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. | |
Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. | |
Dalvai M, Bystricky K. Cell cycle and anti-estrogen effects synergize to regulate cell proliferation and ER target gene expression. PloS One. 2010;5(6):e11011. | |
Johnson SM, Torrice CD, Bell JF, et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J Clin Invest. 2010;120(7):2528–2536. | |
Robinson TJ, Liu JC, Vizeacoumar F, et al. RB1 status in triple negative breast cancer cells dictates response to radiation treatment and selective therapeutic drugs. PloS One. 2013;8(11):e78641. | |
McClendon AK, Dean JL, Rivadeneira DB, et al. CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle. 2012;11(14):2747–2755. | |
Dean JL, McClendon AK, Knudsen ES. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J Biol Chem. 2012;287(34):29075–29087. | |
Johnson N, Shapiro GI. Cyclin-dependent kinase 4/6 inhibition in cancer therapy. Cell Cycle. 2012;11(21):3913. | |
Schwartz GK, LoRusso PM, Dickson MA, et al. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011;104(12):1862–1868. | |
Flaherty KT, Lorusso PM, Demichele A, et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012;18(2):568–576. | |
DeMichele A, Sanders Clark A, Heitjan D, et al. A Phase II trial of an oral CDK 4/6 inhibitor, PD0332991, in advanced breast cancer. Paper presented at:2013 ASCO Annual Meeting; 2013. | |
Slamon DJ, Hurvitz SA, Applebaum S, et al. Phase I study of PD 0332991, cyclin-D kinase (CDK) 4/6 inhibitor in combination with letrozole for first-line treatment of patients with ER-positive, HER2-negative breast cancer. Paper presented at:2010 ASCO Annual Meeting; 2010; Chicago, IL. | |
Finn RS, Crown JP, Lang I, et al. Results of a randomized Phase 2 study of PD 0332991, a cyclin-dependent kinase (CDK) 4/6 inhibitor, in combination with letrozole vs letrozole alone for first-line treatment of ER+/HER2− advanced breast cancer (BC). Paper presented at: IMPAKT Breast Cancer Conference 2012; May 5; 2012; Belgium. | |
Finn RS, Crown JP, Lang I, et al. Results of a randomized phase 2 study of PD 0332991, a cyclin-dependent kinase (CDK) 4/6 inhibitor, in combination with letrozole vs letrozole alone for first-line treatment of ER+/HER2− advanced breast cancer (BC). Paper presented at: Thirty-Fifth Annual CTRC-AACR San Antonio Breast Cancer posium; December 4–8; 2012; San Antonio, TX. | |
Finn RS, Crown JP, Lang I, et al. Final results of a randomized Phase II study of PD 0332991, a cyclin-dependent kinase (CDK)-4/6 inhibitor, in combination with letrozole vs letrozole alone for first-line treatment of ER+/HER2− advanced breast cancer (PALOMA-1; TRIO-18) Paper presented at: AACR Annual Meeting 2014; April 6, 2014; San Diego, CA. | |
Finn RS, Dieras V, Gelmon KA, et al. A randomized, multicenter, double-blind phase III study of palbociclib (PD-0332991), an oral CDK 4/6 inhibitor, plus letrozole versus placebo plus letrozole for the treatment of postmenopausal women with ER(+), HER2(−) breast cancer who have not received any prior systemic anticancer treatment for advanced disease. ASCO Meeting Abstracts. 2013; 31(Suppl 15):TPS652. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.