")
Back to Journals » OncoTargets and Therapy » Volume 13
p70S6K Promotes Acquired Resistance of Erlotinib Through Induction of Epithelial-Mesenchymal Transition in Non-Small Cell Lung Carcinoma
Authors Li M, Chen H, Sun T, Ma Z, Chen X, Wu D, Huang W, Wang X
Received 13 February 2020
Accepted for publication 11 May 2020
Published 9 June 2020 Volume 2020:13 Pages 5257—5270
DOI https://doi.org/10.2147/OTT.S249695
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Min Li,1,* Hongling Chen,1,* Tong Sun,2 Zhuo Ma,1 Xi Chen,1 Dandan Wu,1 Wenbin Huang,3 Xuerong Wang1
1Department of Pharmacology, Nanjing Medical University, Nanjing, Jiangsu Province, People’s Republic of China, 210029; 2Key Laboratory of Human Functional Genomics of Jiangsu Province, Nanjing Medical University, Nanjing, Jiangsu Province, People’s Republic of China, 210029; 3Department of Pathology, Nanjing First Hospital, Nanjing Medical University, Nanjing, Jiangsu Province, People’s Republic of China, 210006
*These authors contributed equally to this work
Correspondence: Wenbin Huang; Xuerong Wang Email [email protected]; [email protected]
Background: Lung cancer is the leading cause of cancer-related deaths. EGFR tyrosine kinase inhibitors, such as erlotinib, were approved for non-small cell lung carcinoma patients with EGFR mutations. However, the acquired resistance of these inhibitors has not been fully clarified. Therefore, clarifying the mechanism and developing new rationales to overcome the drug resistance are urgently needed.
Methods: A pair of erlotinib sensitive and resistant cells was used to identify the key molecules in mediating erlotinib resistance. Loss- or gain-of-function study was used to confirm the effects of the key molecules. Xenograft mouse model and human cancer tissue sample studies were conducted for further corroboration.
Results: HCC827 cells with acquired resistance to erlotinib underwent epithelial-mesenchymal transition and exhibited enhanced p70S6K signaling compared to parental sensitive cells. Moreover, in erlotinib resistant cells, downregulation of p70S6K expression using either siRNA or shRNA reversed EMT and partially overcame erlotinib resistance. Meanwhile, in erlotinib sensitive cells, overexpression of p70S6K promoted EMT and induced erlotinib resistance. Upregulation of p70S6K signaling in erlotinib resistant cells was caused by reduced GSK3β-mediated protein degradation of mTOR and raptor. Additionally, p70S6K silencing suppressed the growth of erlotinib resistant cells in a xenograft mouse model. Finally, we found a correlation between p70S6K and E-cadherin expression in human non-small-cell lung cancer (NSCLC) tissue samples.
Conclusion: Our findings suggest that p70S6K-induced EMT plays an important role in the acquired resistance of erlotinib and provides a novel therapeutic rationale of targeting p70S6K in NSCLC therapy.
Keywords: NSCLC, drug resistance, epithelial-mesenchymal transition, p70S6K, GSK3β
Background
Lung cancer is the leading cause of cancer-related deaths.1–3 It has one of the lowest survival rates among all cancers with a 5-year survival rate of around 19%.3 An estimated 135,720 deaths accounting for about 26% of all cancer deaths are expected to occur in 2020.3 Non-small-cell lung cancer (NSCLC), which is composed of adenocarcinomas, squamous cell carcinomas and large cell carcinomas, accounts for vast majority (around 85%) of all lung cancer.4 For the reason that most of the NSCLC patients are diagnosed at late stage, chemotherapy and targeted cancer therapy are more likely used for these patients, either solely or in combination with surgery and radiotherapy.5 Accumulating studies showed that targeting epidermal growth factor receptor (EGFR) using EGFR tyrosine kinase inhibitors (EGFR-TKIs), such as erlotinib or gefitinib, is a breakthrough in lung cancer treatment. However, its outcome is quite limited for the majority of lung cancer patients.6 Moreover, all the patients who are initially sensitive to EGFR inhibitors develop resistance after a 6–12 months therapy, resulting in reoccurring disease.7 Therefore, better understanding the effects of EGFR inhibitors behind inhibition of EGFR and clarifying the mechanism of drug resistance are urgently needed for improving the current therapies.
Epithelial-mesenchymal transition (EMT) promotes cell proliferation, migration, invasion, anti-apoptosis, and plays an important role in tumor malignancy, metastasis, and reoccurrence.8 Tumor cells undergoing EMT lose their epithelial markers, such as E-cadherin, and gain mesenchymal cell features, such as increased expression of vimentin and/or N-cadherin.9 Meanwhile, β-catenin is released from E-cadherin and translocates from membrane to nucleus, where it promotes robust gene transcription.10 Several findings support that EMT is involved in the resistance of chemotherapy.11 More and more studies suggest that EMT determines the response of tumors to EGFR inhibitors.12,13 Byers et al reported that EMT gene signature predicts resistance to EGFR inhibitors.14 Therefore, targeting EMT may overcome the resistance of EGFR inhibitors and subsequently enhance EGFR-targeted cancer therapy.
The 70-kD ribosomal S6 kinase (p70S6K) is a serine/threonine kinase. p70S6K belongs to the AGC-kinase family which includes Akt, the protein kinase C (PKC), and the 90-kD ribosomal S6 kinase (p90RSK).15,16 As a well-known downstream target of mTORC1 (mammalian target of rapamycin complex 1), p70S6K facilitates mRNA translation by increasing ribosomal production, which subsequently stimulates global protein synthesis and cell growth.17 However, the physiological function of p70S6K and its downstream targets are far from being elucidated. Even though many studies have confirmed the crucial role of mTORC1 in EMT and EGFR-TKIs resistance, the role and the mechanism of p70S6K in EMT and erlotinib resistance remain largely unknown.17,18
In this study, we investigated the role and mechanism of p70S6K-induced EMT in the erlotinib resistance in a pair of sensitive and resistant NSCLC cells. Furthermore, nude mice with erlotinib resistant cell xenografts were used to assess the inhibitory effects of p70S6K knockdown on tumor growth. Additionally, we analyzed the correlation between the expression of p70S6K and E-cadherin in human lung cancer tissue samples.
Methods
Reagents
Erlotinib was purchased from LC Laboratories, dissolved in DMSO and stored at −20°C. Lipofectamine 2000 transfection reagent was purchased from Life Technologies Co., Invitrogen. Rabbit polyclonal antibodies against p-p70S6K (Thr389) (70 kDa), p-GSK3β (Ser9) (46 kDa), p70S6K (85 and 70 kDa), mTOR (289 kDa), and rabbit monoclonal antibodies against p-S6 (Ser235/236) and S6 (32 kDa) were purchased from Cell Signaling Technology, Inc. Mouse monoclonal antibodies against vimentin (57 kDa) and actin (43 kDa) were purchased from Santa Cruz Biotechnology Inc. E-cadherin (130 kDa), N-cadherin (140 kDa), GSK3β (47 kDa), and GAPDH (36 kDa) antibodies were purchased from Bioworld Technology Inc. β-catenin antibody (92 kDa) was purchased from BD Transduction Laboratories. Rabbit polyclonal raptor antibody (149 kDa) was purchased from Bethyl Laboratories.
Cell Lines and Cell Culture
Erlotinib sensitive (HCC827-EP) and resistant (HCC827-ER) cell lines were kindly provided by Dr. Shi-Yong Sun (Emory University), which were established, confirmed, and cultured as previously described.19 The use of the gifted cells was approved by the research ethics committee of Nanjing Medical University.
Western Blot Analysis
The procedures for preparation of whole-cell lysates and for Western blotting were described previously. Enhanced Chemiluminescence Substrate from PerkinElmer was used before exposure.20,21 Quantification of Western blots was conducted. First, the chemiluminescent signal was collected and analyzed by ImageJ. Index of Density (IOD) of E-cadherin and actin were obtained (IOD = density × area). Then the value of IOD ratio (IOD ratio = IOD of E-cadherin/IOD of actin) was calculated. And then, the fold-change (Fold change = IOD ratio of treatment/IOD ratio of control) was presented under the blot of E-cadherin.
RNA Isolation and Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA extraction and quantitative reverse transcription PCR were conducted according to manufacturer’s instructions as we previously described.22 Forward (F) and reverse (R) primers were used as follows: p70S6K, F: 5ʹ-TACTTCGGGTACTTGGTAA-3ʹ and R: 5ʹ-GATGAAGGGATGCTTTACT-3ʹ;23 mTOR, F: 5ʹ-TCCGAGAGATGAGTCAAGAGG-3ʹ, R: 5ʹ-CACCTTCCACTCCTATGAGGC-3ʹ; raptor, F: 5ʹ-ACTGGAACCTACCTTTGGCTT-3ʹ, R: 5ʹ-ACTGTCTTCATCCGATCCTTCA-3ʹ; GAPDH, F: 5ʹ-ATGGGGAAGGTGAAGGTCG-3ʹ, R: 5ʹ-GGGGTCATTGATGGCAACAATA-3ʹ,24 and synthesized by Invitrogen. GAPDH was amplified in parallel with target gene in each sample and used for normalization. The fold-change of mRNA was calculated using the 2−ΔΔCT method.
Growth Inhibition Assay
Cell number was examined by the sulforhodamine B (SRB) assay. The growth rate of the cells and the inhibition rate of erlotinib were calculated as previously described.20,22 In brief, cells were transfected with siRNAs or plasmids in 6-well plates using Lipofectamine 2000 for 24 h, and reseeded to 96-well plates at a density of 2000 cells/well for subsequent 5-d SRB assay. Cells after transfection were reseeded to 96-well plates at a density of 3000 cells/well, then treated with or without erlotinib on the second day for another 3 days, and then subjected to SRB assay.
Colony Formation Assay
Cells were seeded in 12-well plates at a density of 300 cells/well and treated with erlotinib diluted in media on the second day. Every 3 days, the media were replaced with fresh media containing erlotinib. After 21 days, cell colonies were stained with crystal violet. Pictures were taken using a digital camera.
Migration Assay
This migration assay was performed using Transwell Chambers (8.0-μm pore size) from BD Transduction Laboratories. 5×104 Cells in 0.2 mL serum-free medium were seeded in the upper chamber and 0.6 mL medium containing 10% FBS was added to the lower chamber. After 24 h, the non-migratory cells on the top side of the membrane were removed with a wipe cotton swab, and the migratory cells on the bottom side of the membrane were fixed with methanol and stained with 0.1% crystal violet. The migrated cells were counted and pictures were taken under a microscope.
Gene Silencing by Small Interfering RNA (siRNA)
Control (non-target) small interfering RNA (siRNA) was purchased from Invitrogen. p70S6K siRNA pools that target 5ʹ-CCAAGGUCAUGUGAAACUA-3ʹ; 5ʹ-CAUGGAACAUUGUGAGAAA-3ʹ; 5ʹ-GACAAAAUCCUCAAAUGUA-3ʹ; 5ʹ-GCAGGAGUGUUUGACAUAG-3ʹ25 and β-catenin siRNA pools that target 5ʹ-AGCUGAUAUUGAUGGACAG-3ʹ; 5ʹ-CAGUUGUGGUUAAGCUCUU-3ʹ26 were described previously and synthesized by Invitrogen. Cells were transfected with 100 nmol/L siRNAs in 6-well plates using Lipofectamine 2000 for 48 h, and subjected to Western blot analysis. Cells cultured 24 h after transfection were reseeded to 96-well plates, and subjected to growth inhibition assay. 100 nmol/L siRNA pools meant total siRNA of different target sequences was 100 nmol/L.
Gene Silencing by Lentivirus-Mediated shRNA
HCC827-ER cells were infected once with lentivirus carrying control, p70S6K shRNA (LV-p70S6K-RNAi, Genechem,Inc. China) or β-catenin shRNA (LV-β-catenin-RNAi, Genechem, Inc. China) according to the manufacturer’s protocol. The target sequence for p70S6K (5ʹ-CAUGGAACAUUGUGAGAAA-3ʹ; 5ʹ-GACGGGGUCCUCAAAUGUA-3ʹ) and β-catenin (5ʹ-CAGUUGUGGUUAAGCUCUU-3ʹ) were selected from the target sequences in the pool siRNA aforementioned. The cells were then subjected to selection with 1 μg/mL puromycin for 15 days and further cultured in puromycin-free medium for another 10 days. The expression of p70S6K and β-catenin in these cell lines were confirmed by Western blotting.
Gene Overexpression by Plasmids
The pRK7-S6K1-WT plasmid that encodes the wild-type form of p70S6K and its control vector pRK7 were described previously and kindly provided by Dr. John Blenis (Harvard Medical School).27 The HA-GSK3β-WT and HA- GSK3β-S9A plasmids that encode the wild-type and constitutively active form of GSK3β were described previously and purchased from Addgene, Inc. pcDNA plasmids were used as control. Transfection was conducted as described previously.20
Lung Cancer Xenografts in Nude Mice
Animal experiments were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University. Guides for the Care and Use of Laboratory Animals (8th edition) was followed for the welfare of the laboratory animals. Four- to six-weeks old female anthymic (nu/nu) mice were housed under pathogen-free conditions with laboratory chow and water ad libitum. HCC827 ER-Ctrl- shRNA and HCC827 ER-p70S6K-shRNA cells at 3×106 cells in serum-free medium were injected s.c. into the left and right flank region of each nude mouse (mouse number, n=7) respectively. Tumor volumes were measured using caliper measurement once every two days and calculated with the formula V = π (length × width2)/6. After 47 days, the tumors were removed and weighed.
Human Tissue Samples
The study was approved by the ethics committee of Nanjing Medical University in accordance with the Declaration of Helsinki. All patients involved in this study provided written informed consent for the use of their tissue in research. Continuous sections of formalin-fixed paraffin-embedded (FFPE) tumor tissues were collected from 96 lung cancer patients with NSCLC, who visited Nanjing First Hospital during 2010 to 2013. The mean age of patients was 66 years and ranged from 50 to 90 years. The NSCLC histological types, pathological T (pT) stage, and pathological tumor nodal metastasis (pTNM) stage were determined according to WHO criteria of lung cancer and AJCC stage manual (2010 version). No patient underwent radiation and chemotherapy before surgery.
Immunohistochemistry
Immunohistochemical staining was done using Dako EnVision system (Dako, USA) as described previously.22 Anti-p70S6K (Cell Signaling Technology) and anti-E-cadherin (Abcam) antibody were used. Expression of p70S6K was assessed semi-quantitatively according to criteria that evaluated the staining intensity and the proportion of positive tumor cells. The staining intensity was defined as follows: 0, no staining; 1, light yellow; 2, yellow; and 3, dark yellow. The proportion of positive tumor cells was scored as 0, negative; 1, <10%; 2, 10–50%; and 3, >50%. The total staining score was calculated by staining intensity score plus frequency of positive tumor cells. For statistical analysis, total scores of 0 to 4 were considered negative expression, and 5 to 6 were positive expression. The E-cadherin expression in NSCLC was leveled depending on the positive cells proportion: +, >90% out of tumor cells were membrane staining; ±, 10–90% of the tumor cells were membranous and cytoplasmic staining; -, negative or <10% of the tumor cell were membrane staining. + was considered as being normal, ± or – was defined as aberrant expression of E-cadherin.
Statistical Analysis
All data were presented as the mean ± SD and were representatives of three independent experiments. The statistical significance of different treatments were analyzed using the two-sided unpaired Student’s t-tests and P<0.05 was considered statistically significant, except that tumor size and weight was analyzed using the two-side paired Student’s t-tests. The correlation of p70S6K expression, aberrant E-cadherin expression, and clinicopathologic parameters was analyzed using Chi-square test. GraphPad InStat 3 software was used for Statistical analysis.
Results
Epithelial-Mesenchymal Transition Promotes the Acquired Resistance of Erlotinib in NSCLCs
EMT has been suggested to promote cell proliferation and the resistance of EGFR inhibitors.14 HCC827 cell line, which harbors the erlotinib sensitive mutation (Del E746-A750) of EGFR gene, is very sensitive to erlotinib treatment. Li et al developed erlotinib resistant HCC827 cells (HCC827-ER) by chronically exposure HCC827 cells (HCC827-EP) to increased concentrations of erlotinib.19 Subsequent DNA sequencing has proved no secondary T790M mutation of EGFR genes in these cells.7 Thereby, it provides an ideal model for studying the acquired resistance of erlotinib.19 Using this pair of cells, we found that the expression levels of epithelial marker E-cadherin decreased, and mesenchymal marker vimentin and N-cadherin increased in HCC827-ER cells compared to HCC827-EP cells (Figure 1A). Moreover, the migratory potency of HCC827-ER cells was around 1.8-fold stronger than HCC827-EP cells by migration assay, and quantitative analysis showed a significant difference (p<0.05) (Figure 1B and C). These findings suggest that HCC827-ER cells undergo the process of epithelial-mesenchymal transition (EMT).
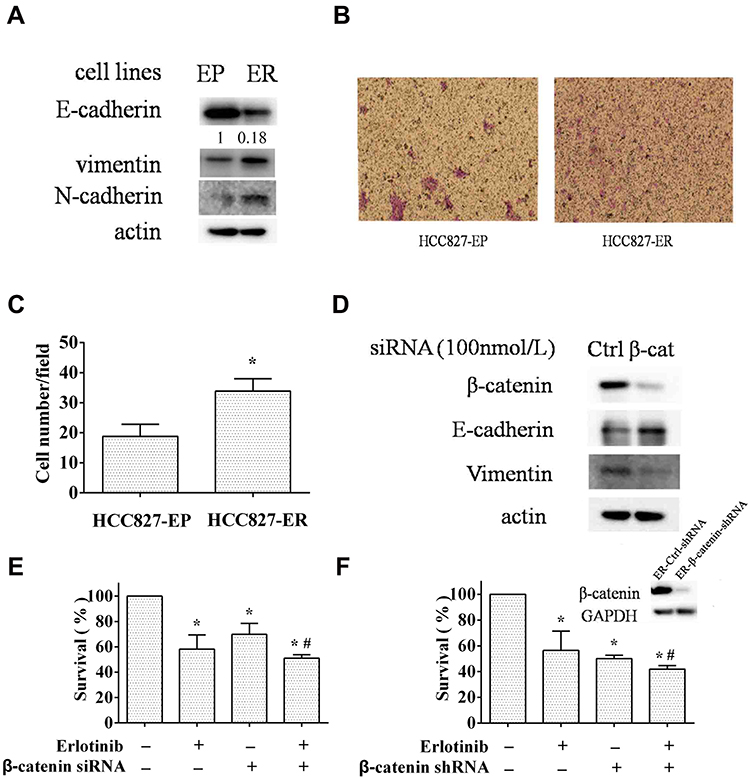 |
Figure 1 EMT promotes acquired resistance of erlotinib. (A and B) the erlotinib sensitive (EP) and resistant (ER) HCC827 cells were subjected to Western blot analysis for EMT markers (A), transwell migration assay (B). (C) migrated cell numbers/field was counted under microscope for five randomly selected fields and average cell numbers/field was presented. Columns, means of three replicate determinations; bars, SD; *p<0.05. (D and E) HCC827-ER cells transfected with β-catenin or control siRNA were subjected to Western blot analysis (D) and SRB assay with or without erlotinib treatment (E). Ctrl, control; β-cat, β-catenin. (F) HCC827 ER-β-catenin-shRNA cells (right) and HCC827 ER-Ctrl-shRNA cells (left) were subjected to Western blot analysis and SRB assay with or without erlotinib treatment. Columns, means of four replicate determinations; bars, SD; *p<0.05 vs control; #p<0.05 vs erlotinib. |
Since β-catenin plays an important role in EMT through translocation from cytoplasm to nucleus to facilitate transcription of a number of genes that are upregulated in EMT, we evaluated the efficacy of erlotinib on HCC827-ER cells when EMT was reversed by β-catenin silencing. First, knockdown of β-catenin by siRNA significantly decreased β-catenin protein levels in parallel with an increase of E-cadherin and a decrease of vimentin, compared to that in control, suggesting the reverse of EMT (Figure 1D). Moreover, knockdown of β-catenin enhanced the inhibitory effect of erlotinib on HCC827-ER cells, indicating that inhibition of EMT sensitized the erlotinib resistant cells (Figure 1E). We established a stable β-catenin silencing cell line and a control cell line by using β-catenin shRNA (HCC827 ER-β-catenin-shRNA) or control shRNA (HCC827 ER-control-shRNA) in HCC827-ER cells, respectively. In consistence, β-catenin knockdown sensitized HCC827-ER cells to erlotinib treatment with the cell survival rate decreased (Figure 1F). Successful downregulation of β-catenin expression were confirmed by Western blot (Figure 1F). These results suggest that EMT promotes the acquired resistance of erlotinib in NSCLC cells.
p70S6K Is Upregulated in Erlotinib Resistant Cells
It has been reported that p70S6K induces EMT in ovarian cancer, and mTOR signaling pathway is involved in erlotinib resistance, so we propose that p70S6K may promote acquired resistance of erlotinib through inducing EMT.5,25 We found that both mRNA and protein levels of p70S6K were increased in HCC827-ER cells compared to HCC827-EP cells (Figure 2A and B). Moreover, phosphorylation levels of both p70S6K and its downstream target S6 were higher in HCC827-ER cells than in HCC827-EP cells, suggesting an elevated activation of p70S6K (Figure 2B). The upregulation of p70S6K signaling in HCC827-ER cells indicates the involvement of p70S6K in the acquired resistance of erlotinib.
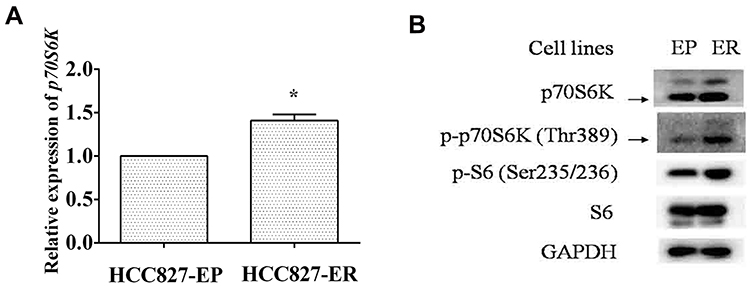 |
Figure 2 p70S6K is upregulated in erlotinib resistant cells. HCC827-EP and HCC827-ER cells were subjected to qRT-PCR assay (A) and Western blot analysis (B). Columns, means of three replicate determinations; bars, SD; *p<0.05. |
Downregulation of p70S6K Reverses EMT and Increases the Growth Inhibitory Effect of Erlotinib in Erlotinib Resistant Cells
To identify the role of p70S6K in EMT-mediated erlotinib resistance, we firstly examined the effects of p70S6K silencing on EMT and erlotinib resistance in HCC827-ER cells. A pool of 4 sequences of p70S6K siRNA was used to knockdown of p70S6K protein expression. We observed that total p70S6K, p-p70S6K, and p-S6 levels were significantly decreased, suggesting the downregulation of p70S6K signaling pathway. In parallel, E-cadherin was increased and N-cadherin was decreased when p70S6K was knocked down, suggesting a reverse of EMT in erlotinib resistant cells (Figure 3A). We also noted that the expression of E-cadherin in HCC827-EP cells decreased by 5.6-fold when compared to HCC827-ER cells after quantification of Western blots (Figure 1A). However, p70S6K knockdown increased the expression of E-cadherin by 3.1-fold in HCC827-ER cells, suggesting p70S6K partially reversed the EMT in erlotinib resistance (Figure 3A). SRB assay showed that p70S6K siRNA inhibited the growth of HCC827-ER cells compared to control siRNA (Figure 3B). Moreover, p70S6K knockdown enhanced the inhibitory effects of 0.5 μmol/L erlotinib on HCC827-ER cells, compared to erlotinib or p70S6K siRNA single treatment (Figure 3C).
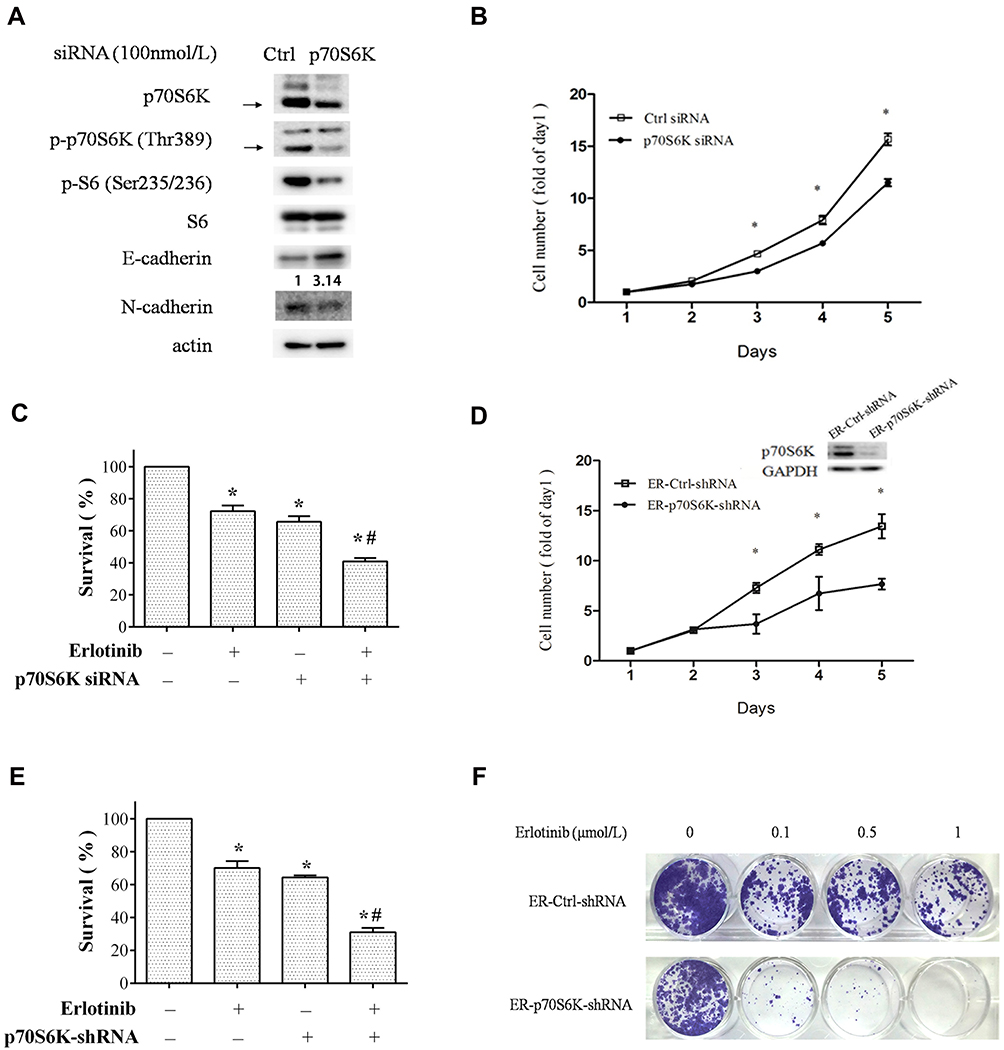 |
Figure 3 Downregulation of p70S6K expression in HCC827-ER cells partially reverses EMT and erlotinib resistance. (A–D) HCC827-ER cells transfected with p70S6K siRNA or control siRNA were subjected to Western blot analysis for EMT markers (A), SRB assay for cell growth (B and C). Ctrl, control. (D–F) HCC827 ER-p70S6K-shRNA and ER-Ctrl-shRNA cells were subjected to SRB assay (D and E) and colony formation assay for cell growth. Representative images of colony formation assay (E). (F) Points and columns, means of four replicate determinations; bars, SD; *p<0.05 vs control; #p<0.05 vs erlotinib. HCC827 ER-p70S6K-shRNA (right) and its control (left) cells were subjected to Western blot analysis (D). |
We then established a stable p70S6K silencing cell line (HCC827 ER-S6K-shRNA cells) and a control cell line (HCC827 ER-Ctrl-shRNA cells) in HCC827-ER cells by infection of lentivirus carrying p70S6K shRNA or scramble shRNA. We observed that knockdown of p70S6K inhibited cell growth by SRB assay (Figure 3D). The silencing of p70S6K was confirmed by Western blot assay (Figure 3D). Moreover, p70S6K stable silencing enhanced the inhibitory effect of 0.5 μmol/L erlotinib (Figure 3E). Additionally, colony formation assay confirmed that erlotinib inhibited cell growth more dramatically in p70S6K stable silencing cells than that in control cells (Figure 3F). These results suggest that downregulation of p70S6K expression could reverse EMT and sensitize erlotinib resistant cells.
Overexpression of p70S6K Induces EMT and Decreases the Inhibitory Effect of Erlotinib on Growth in Erlotinib Sensitive Cells
We then investigated the role of p70S6K-induced EMT in erlotinib resistance by overexpression of p70S6K in erlotinib sensitive cells (HCC827-EP cells). We observed that transfection of HCC827-EP cells with wild-type p70S6K constructs decreased E-cadherin expression and increased vimentin levels compared to that in control, suggesting that p70S6K induces EMT in erlotinib sensitive cells (Figure 4A). We observed that p70S6K overexpression in HCC827-EP cells decreased E-cadherin expression by 3.6-fold, while there was a 5.6-fold decrease of E-cadherin expression in HCC827-ER cells compared with that in HCC827-EP cells (Figure 1A). These results suggested that p70S6K partially reversed the EMT in erlotinib resistance. Meanwhile, erlotinib inhibited cell growth while p70S6K overexpression promoted cell growth. The inhibitory effect of 0.5 μmol/L erlotinib was attenuated by over 5-fold (p<0.05) when p70S6K was overexpressed, suggesting that p70S6K is involved in erlotinib resistance (Figure 4B). It should be noted that the survival rate of erlotinib in HCC827-ER cells with p70S6K knockdown was around 40.8±2.2% (Figure 3C), albeit in HCC827-EP cells with p70S6K overexpression was 56.3±3.2% under 0.5 μmol/L erlotinib treatment. Therefore, increased expression p70S6K-induced a dramatic resistance to erlotinib treatment when we considering that the survival rate of 0.5 μmol/L erlotinib in HCC827-EP cells was only 11.0±3.5% (Figure 4B). Moreover, EMT features induced by p70S6K, such as increased E-cadherin expression and decreased vimentin expression, was partially reversed by β-catenin knockdown using siRNA (Figure 4C). In addition, β-catenin silencing suppressed p70S6K-induced cell growth (Figure 4D). These findings suggest that p70S6K-induced EMT could contribute to the erlotinib resistance.
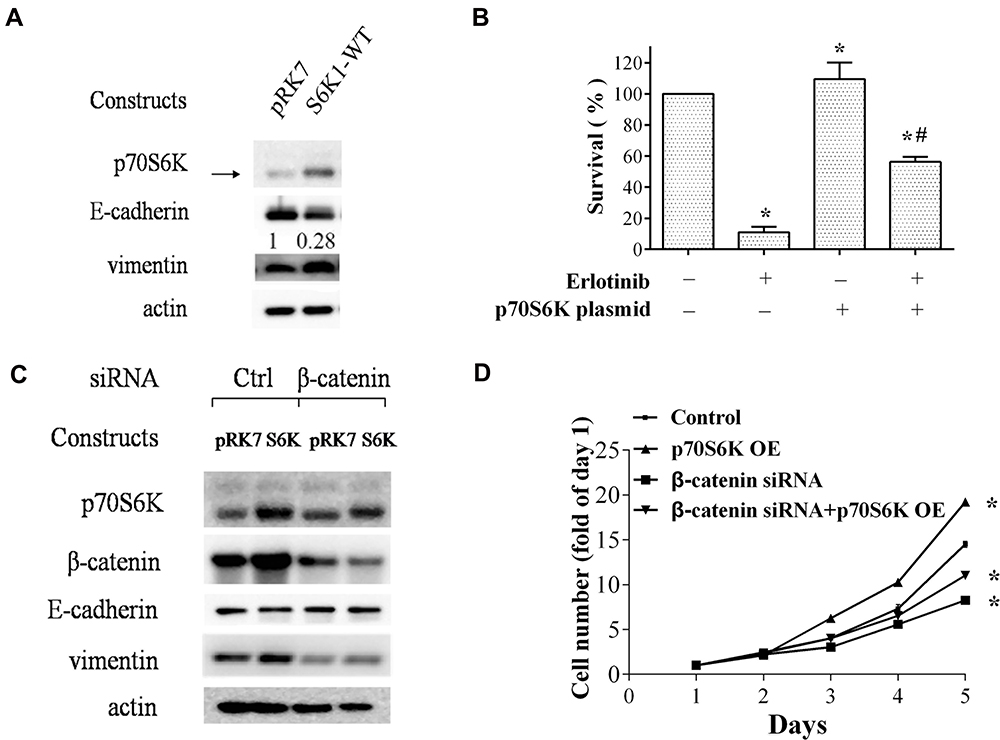 |
Figure 4 Overexpression of p70S6K in HCC827-EP cells induces EMT and erlotinib resistance. (A and B) HCC827-EP cells transfected with the constructs encoding the wild-type p70S6K (pRK7-S6K1-WT) and its control vector pRK7 were subjected to Western blot analysis for EMT markers (A) and SRB assay for cell numbers (B). (C and D) HCC827-EP cells were co-transfected with p70S6K constructs and β–catenin siRNA as indicated, and subjected to Western blot analysis for EMT markers (C) and SRB assay for cell numbers (D). Points and Columns, means of four replicate determinations; bars, SD; *p<0.05 vs control; #p<0.05 vs erlotinib. |
GSK3β Inhibited p70S6K Expression Through Promoting p70S6K Degradation
We then explored how p70S6K signaling pathway was upregulated in erlotinib resistant cells (Figure 2B). First, we checked its upstream signal - mTORC1 expression in both HCC827-EP and HCC827-ER cells. We found that the expression levels of mTOR and raptor, the two main components of mTOR complex 1 (mTORC1), was higher in HCC827-ER cells than that in HCC827-EP cells, suggesting that the hyper-active mTORC1 signaling might contribute to the upregulation of p70S6K signaling in HCC827-ER cells (Figure 5A). However, the mRNA levels of mTOR and raptor were decreased by 18% and 27% respectively in HCC827-ER cells, suggesting that it was not a transcriptional regulation (Figure 5B). We then detected the protein degradation of these proteins. Western blotting showed that MG132, a proteasome inhibitor, increased mTOR, raptor, and p70S6K protein levels significantly in HCC827-EP cells, but not in HCC827-ER cells (Figure 5C). Moreover, the expression of GSK3β, a kinase that promoted mTOR and raptor degradation, was decreased in HCC827-ER cells compared to that in HCC827-EP cells (Figure 5A). Transfection of wild-type (GSK3β-WT) or active form of GSK3β constructs (GSK3β-S9A) in HCC827-ER cells decreased p70S6K, mTOR, and raptor protein levels significantly compared to that in control (pcDNA) (Figure 5D). These results suggest that the upregulated p70S6K pathway in HCC827-ER cells, at least partially, results from the decrease of GSK3β-mediated protein degradation of p70S6K, mTOR, and raptor.
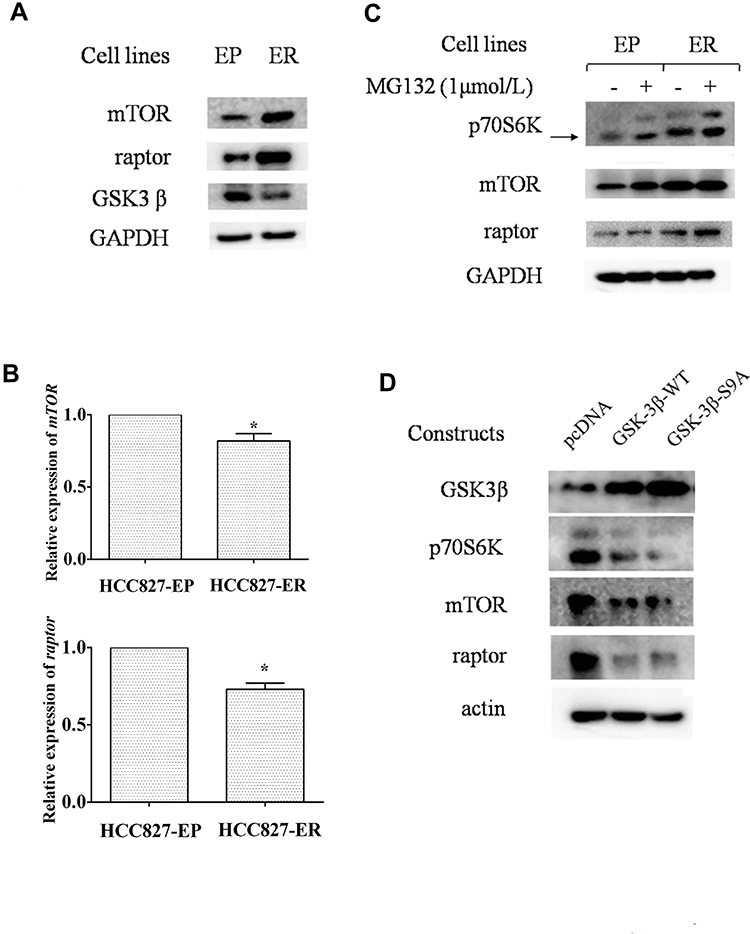 |
Figure 5 Upregulation of p70S6K is a consequence of decreased protein degradation regulated by GSK3β. (A and B) HCC827-EP and ER cells were subjected to Western blot analysis (A) and qRT-PCR assay (B). Columns, means of three replicate determinations; bars, SD; *p<0.05. (C) cell lines aforementioned were treated with or without MG132 for 6h and subjected to Western blot analysis. (D) HCC827-ER cells transfected with control vectors (pcDNA), constructs encoding the wild-type GSK3β (GSK3β-WT), and the constitutive active form of GSK3β (GSK3β-S9A) were subjected to Western blot analysis. |
Downregulation of p70S6K Expression Inhibits Tumor Growth in a Xenograft Mouse Model
To determine the inhibitory effects of targeting p70S6K in erlotinib resistance cells in vivo, we inoculated HCC827 ER-p70S6K-shRNA cells and HCC827 ER-Ctrl-shRNA cells to the nude mice subcutaneously and evaluated the growth of the xenografts. The average size of HCC827 ER-p70S6K-shRNA cell-tumors was smaller than that of control tumors (Figure 6A). Accordingly, the weight of HCC827 ER-p70S6K-shRNA cell-tumors was much lighter than that of control tumors (Figure 6B). These results suggest that p70S6K knockdown significantly inhibited the growth of erlotinib resistant cell xenografts in vivo.
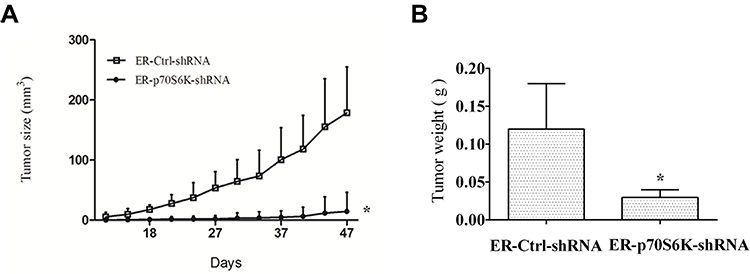 |
Figure 6 Downregulation of p70S6K expression inhibits the growth of lung cancer xenografts in nude mice. HCC827 ER-p70S6K-shRNA and ER-Ctrl-shRNA cells were injected s.c. into the left or right flank region of each nude mouse respectively. After 47 days, the mice were sacrificed and the tumors were removed. (A) tumor size was measured once every 2 days. (B) separated tumors were weighed. Means ± SD (n=7); *p<0.05 compare to control xenografts. |
p70S6K Expression Correlates with the Aberrant E-Cadherin Expression in Human NSCLC Tissue Samples
We performed the immunohistochemical staining of p70S6K and E-cadherin in 96 cases of human NSCLC to evaluate the correlation between p70S6K and EMT. Theoretically, cancer cells undergoing EMT show decreased expression of E-cadherin; however, we detected high expression levels of E-cadherin in human lung cancer tissues. Thus, we defined weak cytoplasmic staining or incomplete membrane staining of E-cadherin as aberrant expression. In adenocarcinoma, there displayed a strong nuclear staining of p70S6K together with an incomplete and weak cytoplasmic staining of E-cadherin (Figure 7, upper panel). In squamous cell carcinoma, there showed a strong nuclear staining of p70S6K in company with cytoplasmic and incomplete membrane staining of E-cadherin, which we considered as aberrant expression (Figure 7, lower panel). Moreover, 72/96 (75%) cases showed positive signal for p70S6K and 24/96 (25%) cases were negative, whereas E-cadherin was aberrantly expressed in 50/96 (52.1%) cases and normally expressed in 46/96 (47.9%) cases. Statistical analysis revealed a significant correlation between p70S6K expression and aberrant E-cadherin expression (p<0.05) (Table 1). We further analyzed the correlation between their expression and clinicopathologic parameters in NSCLC. The aberrant expression of E-cadherin was positively correlated with lymph node metastasis, high pT stage, and high pTNM stage with statistical significance (p<0.05) (Table 2). However, we did not observe any correlation between p70S6K expression and clinicopathologic parameters (Table 2). These results suggest that p70S6K overexpression is correlated with EMT in human NSCLC tissue samples.
 |
Table 1 Correlation Between p70S6K and E-Cadherin Expression in NSCLC |
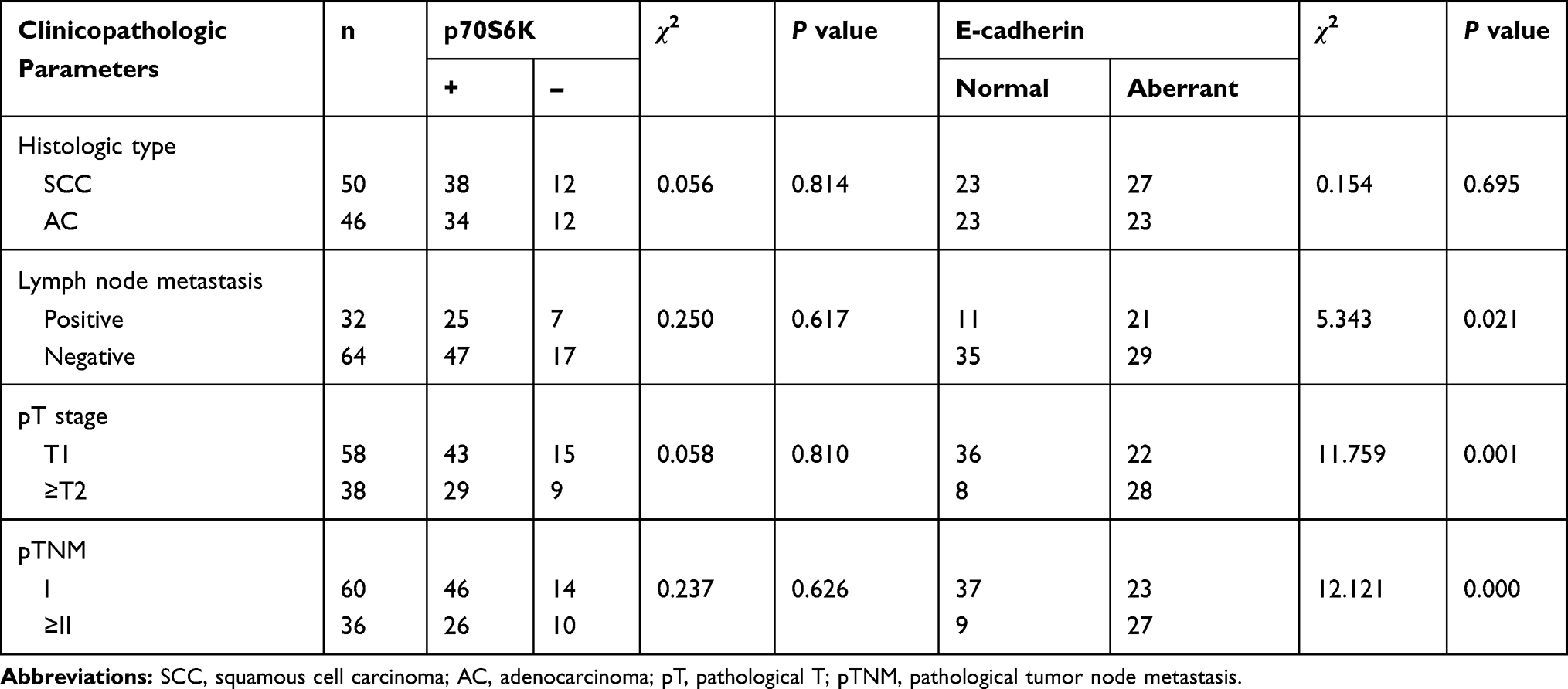 |
Table 2 Correlation Between p70S6K or E-Cadherin Expression and Clinicopathologic Parameters in NSCLC |
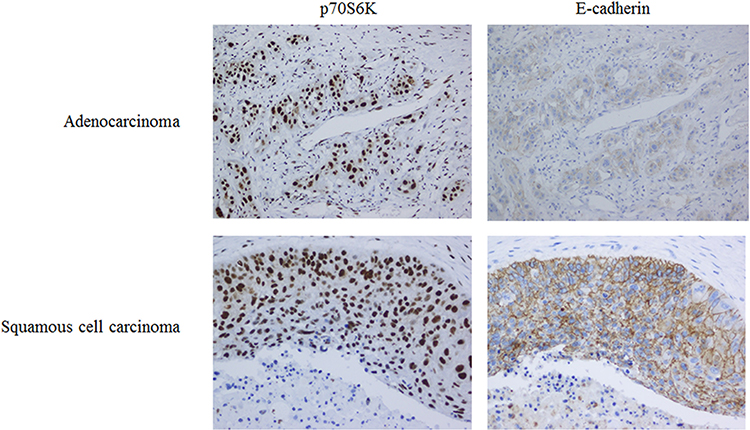 |
Figure 7 p70S6K and E-cadherin expression in the specimens of human NSCLC by immunohistochemistry staining. Serial sections of lung adenocarcinoma and squamous cell carcinoma specimens were subjected to IHC staining of p70S6K and E-cadherin. Up, strong nuclear positive expression of p70S6K, and weak and discontinuous membrane expression of E-cadherin in adenocarcinoma. Low, strong nuclear positive expression of p70S6K and discontinuous membrane expression of E-cadherin in squamous cell carcinoma. Representatives of IHC staining as indicated. Magnification, ×200. |
Discussion
Until now, several mechanisms of EGFR-TKI (tyrosine kinase inhibitor) have been identified.28 EGFR T790M mutant accounts for approximately 50% of the patients that lose the response to EGFR-TKIs.29 Amplification of met gene accounts for about 30% of the patients with acquired resistance.30 However, there are still 15–20% of patients whose EGFR-TKI resistance is unknown.31 In this study, we showed that the mRNA, protein, and phosphorylation levels of p70S6K were all increased in erlotinib resistant cells compare to their parental erlotinib sensitive cells, suggesting an upregulation and activation of p70S6K in erlotinib resistant cells. Moreover, downregulation of p70S6K expression enhanced the growth inhibitory effects of erlotinib in erlotinib resistant cells, whereas overexpression of p70S6K decreased the inhibitory effects of erlotinib in erlotinib sensitive cells. Additionally, silencing of p70S6K inhibited the growth of erlotinib resistant xenografts in a nude mouse model. Our findings suggest p70S6K plays an important role in erlotinib resistance.
Epithelial-mesenchymal transition (EMT), which is correlated with cancer metastasis, stem cells, and drug resistance, has attracted great research interests.32,33 Witta et al identified an inverse correlation between sensitivity to gefitinib and expression of E-cadherin. Forced expression of E-cadherin in gefitinib-resistant cell increased its sensitivity to gefitinib, suggesting that EMT promotes EGFR-TKI resistance.34 Zhang et al reported that activation of AXL kinase causes resistance to EGFR-TKIs in lung cancer, and evidenced EMT in resistant cells and tumor tissues.35 In agreement with the studies above, we found, in this study, that erlotinib resistant cells showed lower levels of E-cadherin, higher levels of vimentin and N-cadherin, than that in erlotinib sensitive cells. β-catenin silencing sensitized erlotinib resistant cells and, in the mean time, reversed EMT. These findings suggest that erlotinib resistant cells display mesenchymal cell properties. Manipulating EMT could, at least partially, overcome erlotinib resistance.
The ribosomal protein S6 kinase (p70S6K) is one of the best characterized downstream targets of mTORC1. Recent studies revealed that activation of mTORC1-p70S6K signaling promotes, while blocking this pathway inhibits, EMT, cell migration, invasion, and cancer metastasis.17,18,36,37 Pon et al reported that p70S6K induced EMT in ovarian cancer through increasing snail transcription.25,38 However, to date, there are limited intensive studies focusing on the role and the mechanism of p70S6K in EMT. In the present study, we found that p70S6K overexpression induced EMT in erlotinib sensitive cells, while knockdown of p70S6K reversed EMT in erlotinib resistant cells (mesenchymal-like cells). Moreover, in human lung cancer tissue sections, we found that p70S6K expression is correlated with the aberrant E-cadherin expression. To our surprise, we detected enriched p70S6K expression in the nuclear of cancer cells, which is inconsistent with the fact that p70S6K is known as a translational regulator. Thus, clarifying the role of p70S6K in the nuclear as well as the shuttling mechanism of p70S6K between the nuclear and cytosol would be interesting research directions in the future. This study, for the first time, suggests that p70S6K-induced EMT plays an important role in the acquired resistance of EGFR-TKIs in NSCLC. Notably, identifying downstream proteins in mTOR signaling pathway involved in EMT would provide novel targets for treatment with less toxicity.
We and others have reported that mTOR, raptor, rictor are all substrates of an E3-ligase named FBWX7, and their degradation is regulated by GSK3β through ubiquitination.39,40 In this study, we found that the increase of mTOR and raptor expression levels was not resulted from transcriptional regulation, since their mRNA levels was not elevated. Moreover, in erlotinib sensitive cells, MG132 treatment increased protein levels of p70S6K, mTOR, and raptor. Introducing wild-type and active form of GSK3β to erlotinib resistant cells decreased p70S6K, mTOR, and raptor protein levels significantly. Therefore, the upregulation of p70S6K in erlotinib resistant cells results from the suppression of GSK3β-mediated degradation of mTOR and raptor. Currently, we do not know how GSK3β was downregulated in erlotinib resistant cells. Further studies are needed to clarify this mechanism.
In conclusion, our findings suggest that p70S6K induced EMT play an important role in the acquired resistance of erlotinib (Figure 8). Targeting p70S6K might provide a novel therapeutic strategy to increase the survival rate and decrease the reoccurring chance of NSCLC patients.
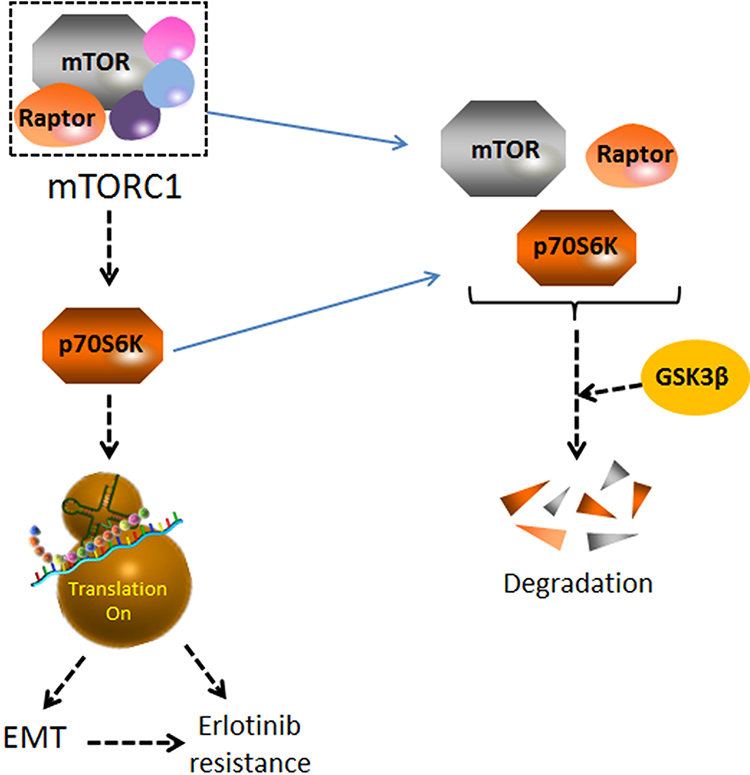 |
Figure 8 A schematic diagram of p70S6K-induced EMT in the acquired resistance of erlotinib in NSCLC. p70S6K, a downstream molecule of mTORC1 signaling, facilitates global protein synthesis. p70S6K played a simulative role in EMT as well as the acquired resistance of erlotinib in NSCLCs. GSK3β promoted the degradation of mTOR, raptor and p70S6K. The decreased expression of GSK3β in HCC827-ER cells could lead to the enhancement of mTORC1-p70S6K signaling pathway, which further stimulated EMT and erlotinib resistance. |
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval
The study was approved by the ethics committee of Nanjing Medical University. Animal experiments were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University. Guides for the Care and Use of Laboratory Animals (8th edition) was followed for the welfare of the laboratory animals. All patients involved in this study provided written informed consent for the use of their tissue in research.
Acknowledgments
We thank Dr. Shi-Yong Sun (Emory University) for providing us with erlotinib sensitive (HCC827-EP) and resistant (HCC827-ER) cell lines. We thank Dr. John Blenis (Harvard Medical School) for providing us with the pRK7-S6K1-WT plasmids.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Muller DC, Johansson M, Brennan P. Lung cancer risk prediction model incorporating lung function: development and validation in the UK biobank prospective cohort study. J Clin Oncol. 2017;35(8):861–869. doi:10.1200/JCO.2016.69.2467
2. Cai Z, Liu Q. Understanding the global cancer statistics 2018: implications for cancer control. Sci China Life Sci. 2019. doi:10.1007/s11427-019-9816-1
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
4. Jumeau R, Vilotte F, Durham AD, Ozsahin EM. Current landscape of palliative radiotherapy for non-small-cell lung cancer. Transl Lung Cancer Res. 2019;8(Suppl 2):S192–S201. doi:10.21037/tlcr.2019.08.10
5. Tang J, Salama R, Gadgeel SM, Sarkar FH, Ahmad A. Erlotinib resistance in lung cancer: current progress and future perspectives. Front Pharmacol. 2013;4:15. doi:10.3389/fphar.2013.00015
6. Doebele RC, Oton AB, Peled N, Camidge DR, Bunn PA
7. Li R, Hu Z, Sun SY, et al. Niclosamide overcomes acquired resistance to erlotinib through suppression of STAT3 in non-small cell lung cancer. Mol Cancer Ther. 2013;12(10):2200–2212. doi:10.1158/1535-7163.MCT-13-0095
8. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi:10.1016/j.cell.2009.11.007
9. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–454. doi:10.1038/nrc822
10. Hsu JM, Xia W, Hsu YH, et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun. 2018;9(1):1908. doi:10.1038/s41467-018-04313-6
11. Arumugam T, Ramachandran V, Fournier KF, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69(14):5820–5828. doi:10.1158/0008-5472.CAN-08-2819
12. Yauch RL, Januario T, Eberhard DA et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11(24):8686–8698. doi:10.1158/1078-0432.CCR-05-1492
13. Thomson S, Buck E, Petti F, et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65(20):9455–9462. doi:10.1158/0008-5472.CAN-05-1058
14. Byers LA, Diao L, Wang J, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19(1):279–290. doi:10.1158/1078-0432.CCR-12-1558
15. Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J. 2012;441(1):1–21. doi:10.1042/BJ20110892
16. Abe Y, Yoon SO, Kubota K, Mendoza MC, Gygi SP, Blenis J. p90 ribosomal S6 kinase and p70 ribosomal S6 kinase link phosphorylation of the eukaryotic chaperonin containing TCP-1 to growth factor, insulin, and nutrient signaling. J Biol Chem. 2009;284(22):14939–14948. doi:10.1074/jbc.M900097200
17. Zhou H, Huang S. Role of mTOR signaling in tumor cell motility, invasion and metastasis. Curr Protein Pept Sci. 2011;12(1):30–42. doi:10.2174/138920311795659407
18. Gulhati P, Bowen KA, Liu J, et al. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res. 2011;71(9):3246–3256. doi:10.1158/0008-5472.CAN-10-4058
19. Li Y, Fan S, Koo J, et al. Elevated expression of eukaryotic translation initiation factor 4E is associated with proliferation, invasion and acquired resistance to erlotinib in lung cancer. Cancer Biol Ther. 2012;13(5):272–280. doi:10.4161/cbt.18923
20. Ma Z, Zhu L, Luo X, Zhai S, Li P, Wang X. Perifosine enhances mTORC1-targeted cancer therapy by activation of GSK3beta in NSCLC cells. Cancer Biol Ther. 2012;13(11):1009–1017. doi:10.4161/cbt.20989
21. Huang W, Yang L, Liang S, et al. AEG-1 is a target of perifosine and is over-expressed in gastric dysplasia and cancers. Dig Dis Sci. 2013;58(10):2873–2880. doi:10.1007/s10620-013-2735-5
22. Luo X, Fan S, Huang W, et al. Downregulation of IRS-1 promotes metastasis of head and neck squamous cell carcinoma. Oncol Rep. 2012;28(2):659–667. doi:10.3892/or.2012.1846
23. Zhan HX, Cong L, Zhao YP, et al. Activated mTOR/P70S6K signaling pathway is involved in insulinoma tumorigenesis. J Surg Oncol. 2012;106(8):972–980. doi:10.1002/jso.23176
24. Dubrovska A, Kim S, Salamone RJ, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci U S A. 2009;106(1):268–273. doi:10.1073/pnas.0810956106
25. Pon YL, Zhou HY, Cheung AN, Ngan HY, Wong AS. p70 S6 kinase promotes epithelial to mesenchymal transition through snail induction in ovarian cancer cells. Cancer Res. 2008;68(16):6524–6532. doi:10.1158/0008-5472.CAN-07-6302
26. Verma UN, Surabhi RM, Schmaltieg A, Becerra C, Gaynor RB. Small interfering RNAs directed against beta-catenin inhibit the in vitro and in vivo growth of colon cancer cells. Clin Cancer Res. 2003;9(4):1291–1300.
27. Martin KA, Schalm SS, Richardson C, Romanelli A, Keon KL, Blenis J. Regulation of ribosomal S6 kinase 2 by effectors of the phosphoinositide 3-kinase pathway. J Biol Chem. 2001;276(11):7884–7891. doi:10.1074/jbc.M006969200
28. Nguyen KS, Kobayashi S, Costa DB. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer. 2009;10(4):281–289. doi:10.3816/CLC.2009.n.039
29. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. doi:10.1056/NEJMoa044238
30. Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14(10):2895–2899. doi:10.1158/1078-0432.CCR-07-2248
31. Pallis A, Briasoulis E, Linardou H, et al. Mechanisms of resistance to epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small-cell lung cancer: clinical and molecular considerations. Curr Med Chem. 2011;18(11):1613–1628. doi:10.2174/092986711795471383
32. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–4751. doi:10.1038/onc.2010.215
33. Voulgari A, Pintzas A. Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta. 2009;1796(2):75–90. doi:10.1016/j.bbcan.2009.03.002
34. Witta SE, Gemmill RM, Hirsch FR, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66(2):944–950. doi:10.1158/0008-5472.CAN-05-1988
35. Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44(8):852–860. doi:10.1038/ng.2330
36. Zhou HY, Wong AS. Activation of p70S6K induces expression of matrix metalloproteinase 9 associated with hepatocyte growth factor-mediated invasion in human ovarian cancer cells. Endocrinology. 2006;147(5):2557–2566. doi:10.1210/en.2005-1404
37. Liu L, Li F, Cardelli JA, Martin KA, Blenis J, Huang S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene. 2006;25(53):7029–7040. doi:10.1038/sj.onc.1209691
38. Ip CK, Wong AS. Exploiting p70 S6 kinase as a target for ovarian cancer. Expert Opin Ther Targets. 2012;16(6):619–630. doi:10.1517/14728222.2012.684680
39. Fu L, Kim YA, Wang X, et al. Perifosine inhibits mammalian target of rapamycin signaling through facilitating degradation of major components in the mTOR axis and induces autophagy. Cancer Res. 2009;69(23):8967–8976. doi:10.1158/0008-5472.CAN-09-2190
40. Mao JH, Kim IJ, Wu D, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321(5895):1499–1502. doi:10.1126/science.1162981
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.