")
Back to Journals » OncoTargets and Therapy » Volume 13
P55PIK Regulates P53-Dependent Apoptosis in Cancer Cells by Interacting with P53 DNA-Specific Domain
Authors Li C, Li W, Cheng X, Zhang D , Sun X, Zhou J, Zhou Y, Huang Y, Xia X , Ma Q, Su Z
Received 24 January 2020
Accepted for publication 6 May 2020
Published 8 June 2020 Volume 2020:13 Pages 5177—5190
DOI https://doi.org/10.2147/OTT.S247200
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Federico Perche
Chaoxing Li,1 Wenwen Li,2 Xiyao Cheng,1 Dapeng Zhang,2 Xiang Sun,1 Jingjing Zhou,1 Yin Zhou,1 Yongqi Huang,1 Xianmin Xia,1 Qi Ma,2,3 Zhengding Su1
1Key Laboratory of Industrial Fermentation (Ministry of Education), Hubei Key Laboratory of Industrial Microbiology, National “ 111” Center for Cellular Regulation and Molecular Pharmaceutics, Hubei University of Technology, Wuhan 430068, People’s Republic of China; 2State Key Laboratory of Membrane Biology, Beijing Key Laboratory of Cardiometabolic Molecular Medicine, Peking-Tsinghua Center for Life Sciences, Institute of Molecular Medicine, Peking University, Beijing 100871, People’s Republic of China; 3Department of Drug Discovery, PKU-Nanjing Joint Institute of Translational Medicine, Nanjing 211800, People’s Republic of China
Correspondence: Zhengding Su; Qi Ma
Email [email protected]; [email protected]
Purpose: Phosphatidylinositol 3-kinase (PI3K) plays an important role in tumorigenesis by cross-talking with several signaling pathways. p55PIK is a unique regulatory subunit of PI3K and contains an extra 24-residue N-terminal domain (N24). This study aimed to explore the interaction of p55PIK with p53 and the role of p55PIK in regulating p53-dependent apoptosis in cancer cells.
Materials and Methods: The expression of p55PIK was detected in cancer cells, and the interaction of p55PIK with p53 was examined by immunoprecipitation and pull-down assay. The expression of p53-dependent apoptosis-related genes was detected by PCR.
Results: N24 domain of p55PIK interacted with DNA-specific binding domain (DBD) of p53. The increase or decrease of p55PIK expression led to the change of the expression of p53 and p53-regulated genes in cancer cells. Moreover, N24 peptide led to the change of the expression of p53-regulated genes. Moreover, a membrane-permeable N24 peptide enhanced p53-dependent apoptosis induced by methyl methanesulfonate.
Conclusion: Our results reveal a novel mechanism that regulates p53-dependent apoptosis in cancer cells via p55PIK-p53 interaction.
Keywords: N24, p55PIK, p53, cancer, apoptosis
Introduction
Phosphatidylinositol 3-kinase (PI3K) plays important roles in the regulation of diverse cellular processes such as autophagy, proliferation, differentiation, migration, glucose and lipid metabolism, apoptosis, and is involved in inflammation, autoimmune disease and tumor through the activation of PI3K-related pathways.1,2 PI3K is composed of a 110-kDa catalytic subunit and a regulatory subunit.3 As a regulatory subunit of PI3K, p55PIK is composed of a unique 24-residue N-terminus (N24) followed by a proline-rich motif and two Src homology 2 (SH2) domains with significant sequence identify to those in p85.4,5 p55PIK is generated by alternative splicing of the p85α gene and the N24 domain has a specific role that p85 does not.5,6
In many cancers, the overexpression of p55PIK is highly relevant to several oncogenic processes such as cell growth, differentiation, metastasis and angiogenesis.5,7–11 p55PIK recruits downstream proteins to bind to the catalytic subunits of PI3K to catalyze the phosphorylation of the downstream proteins and activate cell signaling.12 The transcriptional activation of p55PIK by MZF1 increases the proliferation of colorectal cancer cells.13 Overexpression of p55PIK in colorectal cancer activates NF-κB pathway, decreases HIF-1α expression and increases VEGF expression.9 The downregulation of p55PIK inhibits the proliferation of ovarian cancer cells.8 In HT-29 cells, the downregulation of p55PIK expression inhibits colony growth.14 In gastrointestinal stromal tumors, the downregulation of p55PIK significantly decreases KIT expression and increases chemosensitivity in vitro and in vivo.11 On the other hand, small molecules can significantly affect p55PIK signaling. The treatment of MDA-MB-231 breast cancer cells by doxorubicin induced the expression of p55PIK.15 The treatment of SH-SY5Y cells and 293 cells by neurotoxin paraquat reduced p55PIK expression at both transcriptional and translational levels.16
PI3K pathway can crosstalk with other tumorigenic signaling pathways including Ras, p53, TOR, DJ1 and PTEN.17,18 PI3K can regulate the function of PTEN depending on its N-terminal specific structure.19–21 Activation of the RTK/Ras/PI3K signaling and inactivation of the p53 and the Rb pathways concurrently occur in cancer cells to promote cell proliferation and inhibit apoptosis.22,23 The p53/miR148b/p55PIK axis plays an important role in cell proliferation and tumor growth.24 The miRNA432 inhibits myogenesis in muscle cells by targeting E2F3 and P55PIK.25 Moreover, p53 and MDM2 are regulated by PI3K under stress induced by UV radiation and proteasome dysfunction.26 However, the association of p55PIK signaling with p53-dependent apoptosis is rarely explored.
N24 domain is crucial for p55PIK subcellular localization.16 Moreover, N24 domain can bind to different signaling proteins to regulate cell cycle and cytokine secretion.27 N24 domain significantly stimulated the expression of AFP by activating NF-κB signaling pathway.28 p55PIK can specifically interact with Rb through the N24 domain and induce cell cycle arrest at G0-G1 phase.7,29 Therefore, overexpression of N24 can competitively bind Rb protein to inhibit cell cycle process by blocking the p55PIK-Rb interaction.30 N24 domain is efficiently translocated into the nucleus after fusion with a cell-permeable peptide TAT.31 Systemically administered TAT-N24 significantly inhibits the growth of leukemia in animal models.10 TAT-N24 can inhibit tumor cell growth both in vitro and in vivo through decreasing the expression of Cyclin D1.32 TAT-N24 can effectively inhibit LPS-stimulated expression of TNF-alpha, IL-6, and IL-8 to block nuclear translocation of NF-κB p65.33 TAT-N24 disrupted the p55PIK-PCNA interaction in cancer cells and inhibited DNA synthesis and tumor growth in vitro and in vivo.30 However, it is unclear whether p55PIK can interact with p53 via N24 domain.
In this study, we aimed to explore the interaction of p55PIK with p53 and the role of p55PIK in regulating p53-dependent apoptosis in cancer cells. We found that p55PIK directly binds to the DBD domain of p53 via N24 domain. Moreover, the upregulation of p55PIK expression increases transcriptional levels of p53-dependent apoptosis-related genes including GADD45α, S100A9, MDM2 and AIP1. Furthermore, synthetic N24 translocated to nucleus can significantly inhibit cancer cell growth.
Materials and Methods
Materials
HeLa (ATCC CCL-2), HepG2 (ATCC HB-8065), HEK293A (ATCC CRL-1573), H1299 (ATCC CRL-5803) cell lines were purchased from ATCC (VA, USA) and routinely maintained in our laboratories. Colon cancer samples, SW480 (ATCC CCL-228), HCT116, P6C (CVCL-T056) cell lines were gifts from Professor Quan Chen (Institute of Zoology, CAS, China). DMEM and RPMI-1640 were obtained from MACGENE. FBS (Fetal Bovine Serum) (Gibco, USA), Lipofectamine® 2000 and lipofectamine-RNAiMAX were purchased from Invitrogen. Antibodies for p53 and P55PIK were purchased from CST. Annexin V-HITC Apoptosis Detection Kit was from Thermo Fisher. Colon tumor samples were collected immediately after surgery from 90 patients (age 26–78, median age 59.6 years) in Beijing Cancer Hospital Tumor Bank (Beijing, China) according to the approval of the Ethics Committee of Beijing Cancer Institute.34 All patients provided written informed consent. Six samples were randomly selected from this experiment.
Cell Culture
SW480, HeLa, HepG2, HEK293A, and P6C cells were inoculated in DMEM medium supplemented with 10% fetal calf serum (FBS) while H1299 cells were inoculated in RPMI-1640 medium supplemented with 10% FBS, and cultured at 37°C with 5% CO2, as described previously.35
Expression and Purification of Different Domains of P53 Protein
The cDNA encoding different domains of p53 was subcloned into the pET28a vector, which was then transformed into E. coli BL21 (DE3) and the recombinant proteins were purified using Ni-NTA affinity chromatography (GE Healthcare).
Co-Immunoprecipitation Assay
HEK293A cells were transfected with Flag-p55PIK plasmid, 48 hrs later the cells were washed twice with PBS and NP40 buffer, and lysed on ice for 30 min. The supernatant was centrifuged at 4°C to obtain the protein extract, which was incubated with Anti-Flag M2 Affinity Gel at 4°C overnight. The beads were washed 4 times with NP40 split buffer and subjected to Western blot analysis.
Pull-Down Experiments
Pull-down experiments were carried out with N24-agar beads to pulldown p53 in cancer cell lines and colon cancer tissues. The plasmids harboring p55PIK or GFP-p53 were transiently transfected into SW480 cells using Lipofectamine 2000 and pUC19 was used as a control, 48 hrs later cell lysates were collected for pulldown assay with N24-agar beads.
N24-agar beads were also used to test the interaction of N24 with different domains of p53 protein, ie, p531-93 (transactivation domain, TAD), p5394-293 (DNA binding domain, DBD) and p53293-393 (the oligomerization and ring domains, OD and RD, 294–360 amino acid residues).36
Western Blot Assay
Western blot assay was carried out according to the method described previously by Zhang et al.37 Total protein extracts were subjected to SDS-PAGE assay and transferred into PVDF membranes, which were blocked with a 5% skimmed milk powder dissolved in TBST buffer at room temperature for 1 hr, and then incubated with primary antibodies for 2 hrs at room temperature or overnight at 4°C. The membranes were washed 3 times with TBST at room temperature and then incubated with secondary antibody for 1 hr at room temperature. The membranes were then washed with TBST buffer three times, and protein signal was scanned using Odyssey dual-color infrared laser imager (LICOR).
Immunofluorescence Assay
Cells were evenly spread on glass slides and fixed with 4% paraformaldehyde (PFA) at 37°C for 20 min. The slides were washed three times at 37°C with PBS containing 0.5% Triton-100. The sample was further washed three times with 37°C PBS containing 3% BSA and 0.5% Triton-100 and blocked for 1 hr at room temperature. The slides were washed three times with PBS and incubated with primary antibody at 4°C overnight. Then, the cells on the slide were rinsed with the secondary antibody at room temperature for 1 hr in the dark. Nuclei were stained with DAPI at 37°C for 10 mins. The cells were observed under a Zeiss 710 inverted confocal microscope with a 40 ×1.3 NA oil lens. The excitation wavelength for DAPI was set at 405 nm. The excitation wavelength of red light was set at 543 nm. The excitation wavelength of green light was set at 488 nm.
siRNA Transfection
The siRNAs were designed with the following sequences: siRNA_P55PIK 5ʹ-GGACUUGCUUUAUGGGAAATT-3ʹ and 5ʹ- UUUCCCAUAAAGCAAGUCCTT-3ʹ; siRNA_p53 5ʹ- GAUGUUUUGCCAACUGGCCTT-3ʹ and 5ʹ-GGCCAGUUGGCAAAACAUCTT-3ʹ; siRNA Negative control 5ʹ-UUCUCCGAACGUGUCACGUTT-3ʹ and 5ʹ- ACGUGACACGUUCGGAGAATT-3ʹ. SW480 cells were cultured in serum-free OPTI-DMEM medium and transfected with siRNAs using lipofectamine-RNAiMAX. The cells were collected 72 hrs later for analysis.
Real-Time PCR
Total RNAs were extracted with a standard Trizol-chloroform protocol. The concentration of RNAs was measured with Nanodrop and reverse transcription was performed with Transtran® One-step gDNA Removal and cDNA Synthesis SuperMix kit (Quanshijin, Beijing). Real-time PCR was performed on a CFX96 qPCR instrument (BioRad, USA).
Flow Cytometric Assay
Cells were washed twice with cold PBS buffer and resuspended in 1x Annexin-binding buffer at 1x106 cells/mL. The cell samples were incubated with Alexa Fluor488 Annexin V and PI work solution (Dead Cell Apoptosis Kit) at room temperature for 15 min. The stained cells were immediately detected on a BD Accuri Flow Cytometer.
Statistical Analysis
The data were expressed as mean ± standard error (SEM), and statistical analysis was performed using GraphPad Prism 5 Software. Differences between two independent samples were analyzed using Student’s t-test. A p-value of less than 0.05 was considered significant.
Results
N24 Domain of p55PIK Interacts with DNA-Binding Domain of P53 in Cancer Cells
To investigate whether p55PIK interacts with p53, we first explored their localization in cancer cells using HeLa cell as a model. As shown in Figure 1A, p55PIK was highly expressed and mainly localized in the nucleus, while p53 was expressed at very low level in Hela cells under normal conditions. Upon UV-irradiation for 3 min, both the expression of p55PIK and p53 increased in the nucleus (Figure 1B). Furthermore, Co-IP assay showed that Flag-p55PIK could bind endogenous p53 protein in HEK293A cells (Figure 1C).
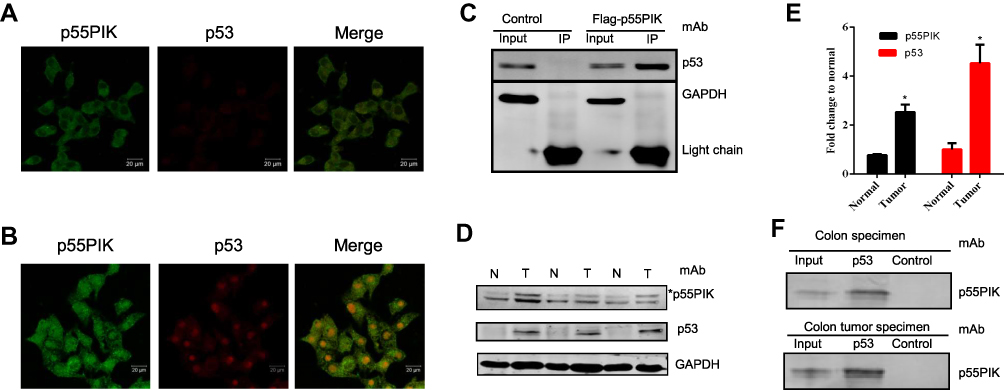 |
Figure 1 Interaction of p55PIK with p53 in cancer cells. (A and B) p55PIK protein and p53 protein are co-localized in the nucleus in HeLa cells without or with UV treatment for 3 min, respectively. (C) Co-immunoprecipitation of transiently expressed flag-p55PIK and p53 protein in HEK293A cells. Control: the cells were transfected with default plasmid; Flag-p55PIK: the cells were transfected with a plasmid harboring Flag-p55PIK. (D) Detection of p55PIK and p53 protein expression in normal tissue samples and tumor samples of colon cancer patients Notes: *The band indicated p55PIK protein. (E) Comparison of p55PIK and p53 expression in normal colon tissue samples and colon tumor samples. Data are mean ± SEM. N = 3 pairs of samples; *p < 0.05 for normal vs tumor samples. (F) p53 protein interacted with p55PIK protein in tumor tissue samples and normal tissue samples from colon cancer patients.Abbreviations: N, normal cells; T, tumor cells. |
Next, we detected p55PIK and p53 proteins in colon tumor cells and normal colon tissue cells. As shown in Figure 1D and E, both p55PIK and p53 proteins were significantly overexpressed in colon tumor cells, compared with normal colon tissue cells. Furthermore, pulldown assay with Protein G beads revealed that p53 protein bound to endogenous p55PIK protein in colon tumors and normal colon tissues (Figure 1F).
Since N24 domain is important for nuclear localization of p55PIK, we used N24-Agar beads to perform pulldown assay for extracts from SW480, HCT116, P6C, HeLa and HEK293A cells. To ensure whether the cell lines used in this study contain p55PIK and p53 or not, we evaluated individual cell line for the protein expression levels of p55PIK and p53 using Western blotting(Figure 2A).When N24-agar beads were applied to the cell extracts of these cell lines, the beads could pulldown endogenous p53 proteins from these cells, as shown inFigure 2B, indicating that N24 domain was able to interact with p53. Furthermore, N24-Agar beads could pulldown endogenous p53 proteins from the extracts of both colon tissues and colon tumor tissues(Figure 2C).
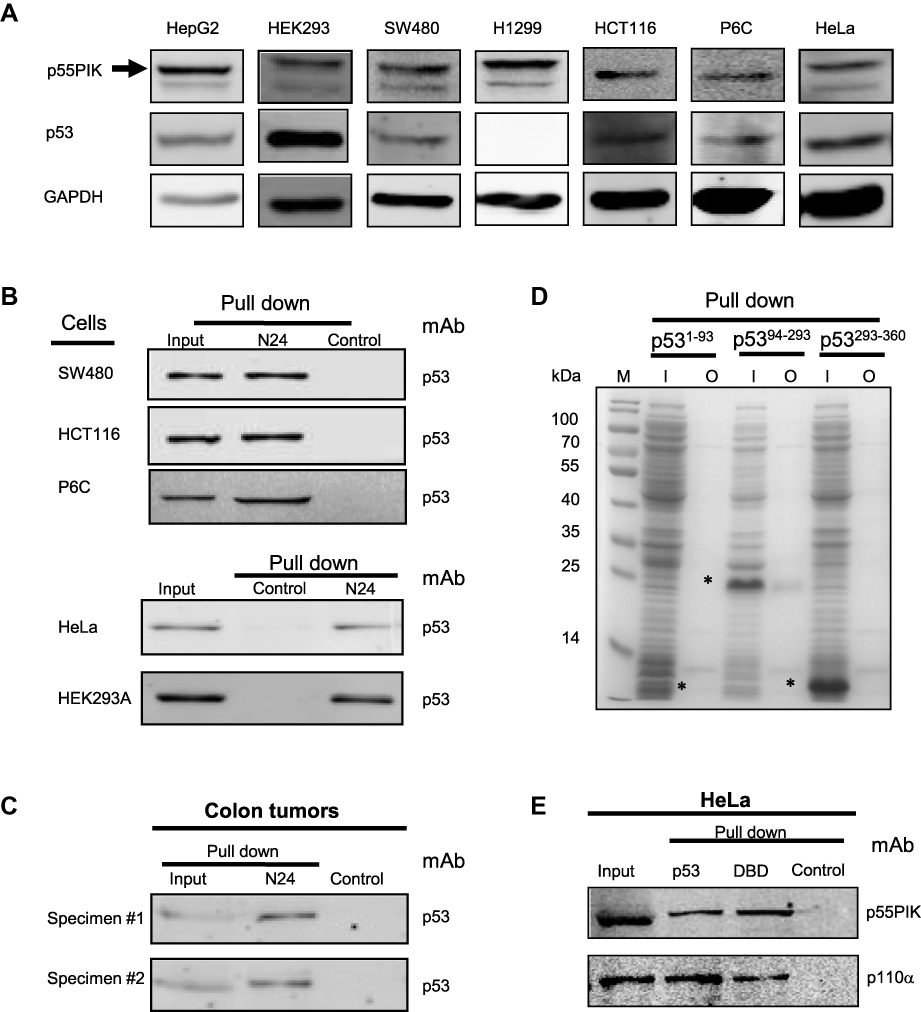 |
Figure 2 Interaction of N24 domain with DBD domain of p53 in cancer cells. (A) Evaluation of the levels of p53 and p55PIK in all cell lines used in this study. (B) N24-agar beads pull down endogenous p53 protein in SW480, HCT116, P6C, HeLa and 293A cells. Control: agar beads. (C) N24-agar beads pull down endogenous p53 protein in colon specimen (#1) and colon tumor specimen (#2). Control: agar beads. (D) N24-agar beads pull down the DBD domain of p53recombinantly expressed in E. coli cells. M: molecular weight marker; I: inputs of the extracts from the cells expressing p53 domains, ie, p531−93,p5394−293and p53293−360. O: output of p53 domains with N24-agar beads. Asterisks indicate p53 domains. (E) The pulldown of endogenous p55PIK and p110α proteins simultaneously from HeLa cells using agar-beads coupled with full-length p53 and the DBD domain. Control: agar beads. |
To determine which domain of p53 protein is responsible for the interaction with N24, we performed pulldown assay with N24-Agar beads for three domains of p53 expressed in E. coli BL21 cells. The results showed that only DBD domain of p53 (p5394−293) was pulled down by N24-Agar beads (Figure 2D), indicating that N24 could bind to the DBD domain of p53.
To confirm whether the DBD domain has the same affinity as the full-length p53 protein to interact with p55PIK, we prepared p53-agar beads and DBD-agar beads, and mixed them with HeLa cell extracts. The results indicated that both full-length p53 protein and p53-DBD domain exhibited the same binding affinity to endogenous p55PIK protein (Figure 2E). Importantly, the pulldown complexes contained p110α (Figure 2E), indicating that catalytic domain of PI3K was recruited.
Knockdown of p55PIK Upregulates the Expression of Endogenous P53
To explore whether p55PIK and p53 proteins mutually regulate each other in cancer cells, the plasmids harboring p55PIK and p53 (in form of GFP fusion protein) were transfected into SW480 cells. As shown in Figure 3A, the overexpression of p55PIK protein had no significant effect on the expression of endogenous p53 protein, or vice versa. Notably, knockdown of endogenous p55PIK led to increased expression level of endogenous p53, although knockdown of endogenous p53 did not significantly affect expression level of endogenous p55PIK (Figure 3B). Quantitative analysis showed that when the expression level of p55PIK protein was decreased by 30%, the expression level of endogenous p53 protein increased 2.2-folds (Figure 3C). When p53 protein was overexpressed by 2-folds, no significant change in the expression level of p55PIK protein was observed (Figure 3D). When p53 protein was decreased by 60%, the expression level of p55PIK protein only slightly reduced by around 20% (Figure 3D).
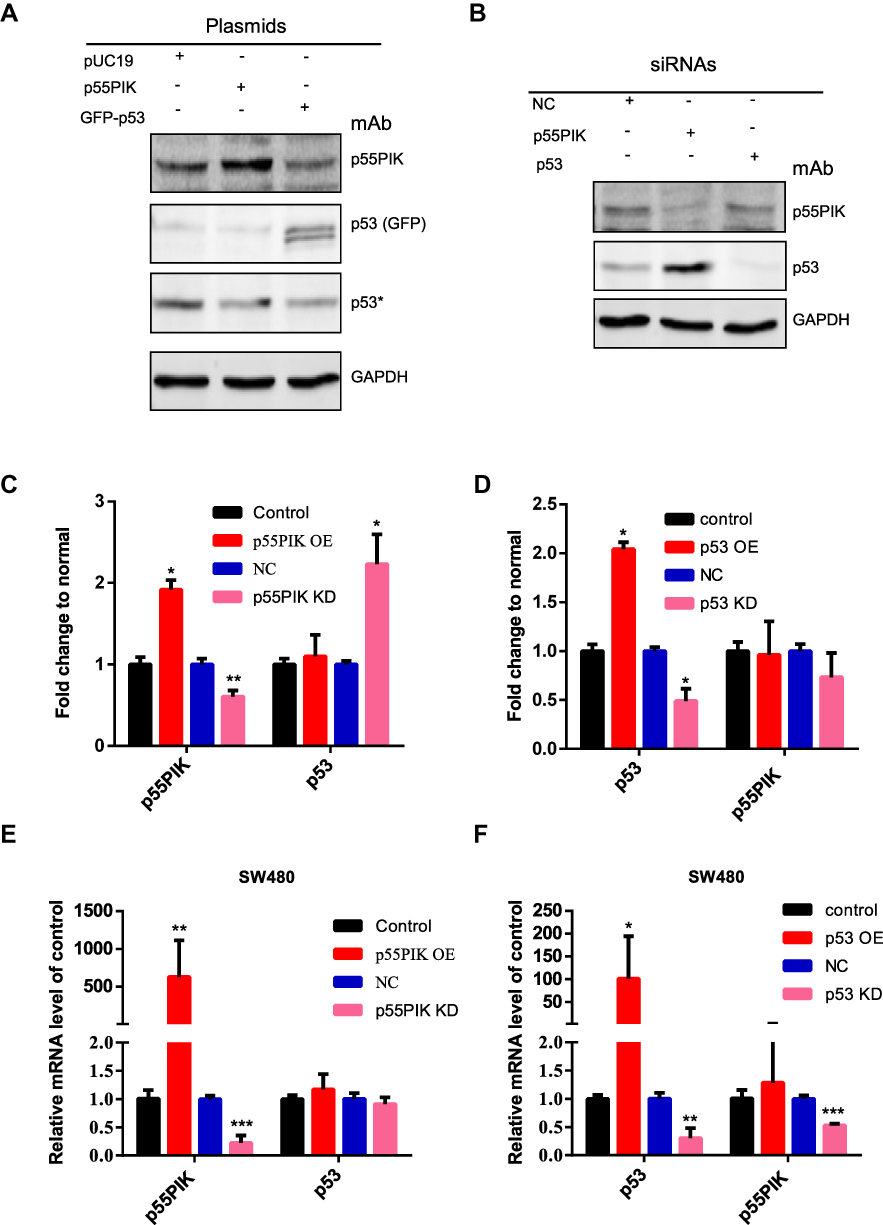 |
Figure 3 Correlation of gene expression between p55PIK and p53. (A) Western blotting of either p55PIK or p53 in SW480 cells. The plasmids harboring p55PIK or p53 were used to upregulate the expression of p55PIK and GFP-p53, respectively. Asterisk indicated endogenous p53 protein. (B) Specific siRNAs targeting p55PIK mRNA or p53 mRNA were used to knockdown p55PIK and p53. NC: negative control. (C) The effects of p55PIK up-regulation and down-regulation on p53 protein expression. Data are mean ± SEM. N = 3 samples per group; *p < 0.05; **p < 0.01, compared with the control group. (D) The effects of p53 up-regulation and down-regulation on p55PIK protein expression. Data are mean ± SEM. N = 3 samples per group; *p < 0.05, compared with the control group. (E) The effect of p55PIK up-regulation and down-regulation on p53 mRNA level in SW480 cells. Data are mean ± SEM. N = 4 independent experiments; **p < 0.01; ***p < 0.005, compared with the control group. (F) The effect of p53 up-regulation and down-regulation on p55PIK mRNA level in SW480 cells. Data are mean ± SEM; N = 4 independent experiments; *p < 0.05; **p < 0.01; ***p < 0.005, compared with the control group. |
To explore whether p55PIK and p53 mutually regulate each other at the transcriptional level, we performed real-time PCR. As shown in Figure 3E, transcription level of p55PIK in SW480 cells had no significant effect on transcription level of p53. On the other hand, the downregulation of p53 transcription significantly reduced transcription level of p55PIK, although high transcription level of p53 had no significant effect on transcription level of p55PIK (Figure 3F).
Interaction of p55PIK with P53 Regulates the Expression of Genes Related to P53-Dependent Apoptosis
To understand the functional significance of p55PIK and p53 interaction, we focused on genes related to p53-dependent apoptosis, including GADD45α, S100A9, Bax, AIP1 and MDM2.36,38 As shown in Figure 4A, when p55PIK was upregulated in SW480 cells, the mRNA levels of GADD45α, S100A9, AIP1 and MDM2 were significantly upregulated, while the mRNA level of Bax was unaffected. When p55PIK was downregulated, the mRNA levels of GADD45α, S100A9, Bax, AIP1 and MDM2 were significantly downregulated.
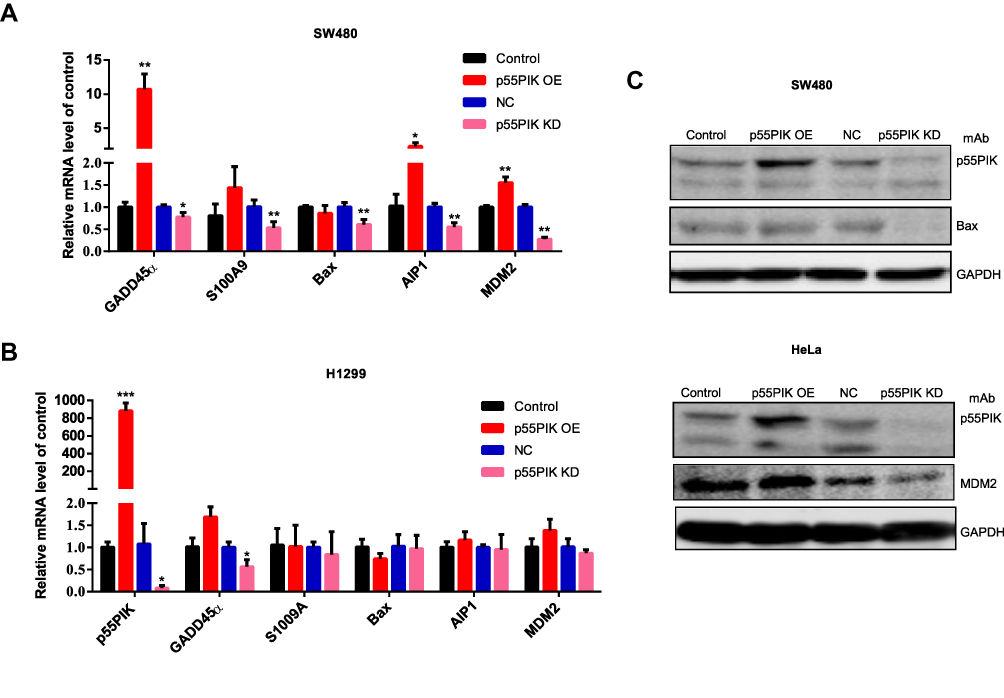 |
Figure 4 The interaction of p55PIK with p53 regulates the downstream genes of p53. (A) Real-time PCR assay of mRNA levels of GADD45α, S100A9, Bax, AIP1 and MDM2 in SW480 cells upon the up-regulation and down-regulation of p55PIK. Data are mean ± SEM. N = 4 independent experiments; *p < 0.05; **p < 0.01, compared with the control group. (B) The mRNA levels of GADD45α, S100A9, Bax, AIP1, and MDM2 in p53-null H1299 cells upon the up-regulation and down-regulation of p55PIK. Data are mean ± SEM. N = 4 independent experiments; *p < 0.05; ***p < 0.005 compared with the control group. (C) Western blotting assay on the regulation of protein expression of Bax and MDM2 by p55PIK. |
Next, we used H1299 cells as control experiments because H1299 cells are in lack of p53 genes. We quantitated the mRNA levels of aforementioned p53-regulated downstream genes in p53-null H1299 cells upon increasing and decreasing p55PIK gene expression. As indicated in Figure 4B, whether p55PIK expression was increased or decreased, the mRNA levels of S100A9, Bax, AIP1 and MDM2 changed insignificantly except that of GADD45α (Figure 4B). These results suggest that p55PIK regulates transcription levels of p53-dependent apoptosis genes via interacting with p53, specifically with DNA-specific binding domain.
Furthermore, to provide a proof concept for the protein levels of down-stream products that contributed to apoptosis enhancement, we examined two typical p53-related signaling proteins, ie, Bax and MMD2, in SW480 and HeLa cell lines. As shown in Figure 4C, when the p55PIK was downregulated, the protein level of Bax significantly decreased in comparison with controls. The similar down-regulation of MDM2 protein level was also observed in HeLa cells when p55PIK was downregulated. These results support that p55PIK regulates transcription levels of p53-dependent apoptosis genes. The data also suggested that the GADD45α gene expression can be also regulated by p55PIK via a p53-dependent pathway.
Synthetic N24 Peptide Efficiently Inhibits Cancer Cell Proliferation
Since N24 domain of p55PIK can interact with DBD domain of p53, it is possible for N24 to interrupt the interaction between p55PIK and p53 to restore the activity of p53. N24 was fused with a well-known protein transduction domain of TAT,31 and the fusion peptide, namely, TAT-N24 should function as an inhibitor of p55PIK-p53 interaction. As shown in Figure 5A, TAT-N24 could be efficiently translocated to the nucleus of HeLa cells. Translocated TAT-N24 exhibited significant inhibitory effect on cell growth (Figure 5B). Quantitative analysis showed that TAT-N24 inhibited the proliferation of Hela cells and HepG2 cells in a time- and dose-dependent manner (Figure 5C and D).
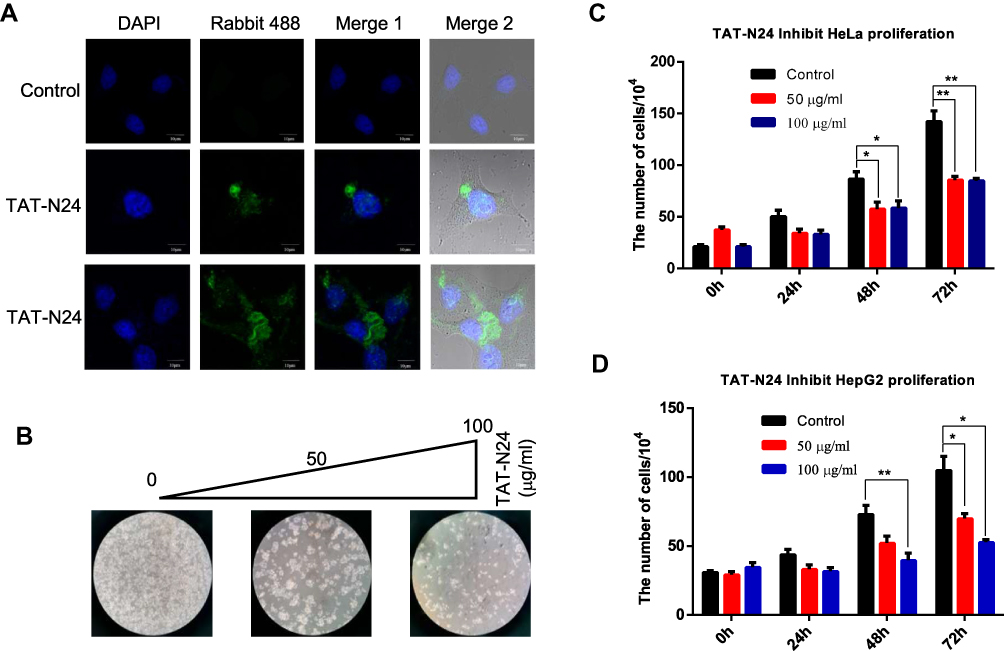 |
Figure 5 TAT-N24 inhibits cancer cell proliferation. (A) Fluorescence microscopic imaging confirmed that N24 was efficiently localized into the nucleus. (B) HeLa cell colonies were monitored by light microscopy after the cells were treated with 0, 50 and 100 mg/mL of TAT-N24 peptide, respectively, for 72 hrs. (C) The number of cells after treatment with different concentration of TAT-N24 peptide for 3 days. Data are mean ± SEM. N = 4 independent experiments; *p < 0.05; **p < 0.01, compared with the control group. (D) The number of cells after treatment with different concentrations of TAT-N24 peptide for 3 days. Data are mean ± SEM. N = 4 independent experiments; *p < 0.05; **p < 0.01, compared with the control group. |
Synthetic N24 Peptide Enhanced MMS-Mediated P53-Dependent Apoptosis
To determine whether the inhibitory effect of N24 on cell proliferation was related to p53-dependent apoptosis, we treated SW480 cells with 100 µg/mL TAT-N24 but found no significant apoptosis by flow cytometry (Figure 6A and B). Nevertheless, we observed synergetic effect on apoptosis by combining MMS with TAT-N24 on SW480 cells. While the dosage of 200 µM MMS only induced 15% apoptosis (Figure 6C), the addition of 10 μg/mL TAT-N24 led to 2-fold increase of apoptosis (Figure 6D). Such synergetic effect was significant at lower concentration of MMS and started to attenuate when the concentration of MMS was large than 200 µM (Figure 6E). When the concentration of TAT-N24 was increased to more than 50 µg/mL, the enhancement on MMS-induced apoptosis reached the maximum (Figure 6F). Moreover, the enhancement of MMS-inducing apoptosis by TAT-N24 was time dependent (Figure 6G). In addition, the enhancement of MMS-inducing apoptosis by TAT-N24 was observed in Hela cells and HepG2 cells (Figure S1). To further confirm whether TAT-N24 enhances MMS-induced apoptosis in p53-dependent manner, we examined the effect of TAT-N24 on MMS-inducing apoptosis in p53-null H1299 cells. H1299 cells treated with 100 µg/mL TAT-N24 showed no significant apoptosis assayed by flow cytometry (Figure 7A and B and E). Because H1299 cells are in lack of p53 genes, TAT-N24 exhibited no enhancement on apoptosis of these cells, as expected (Figure 7C–E).
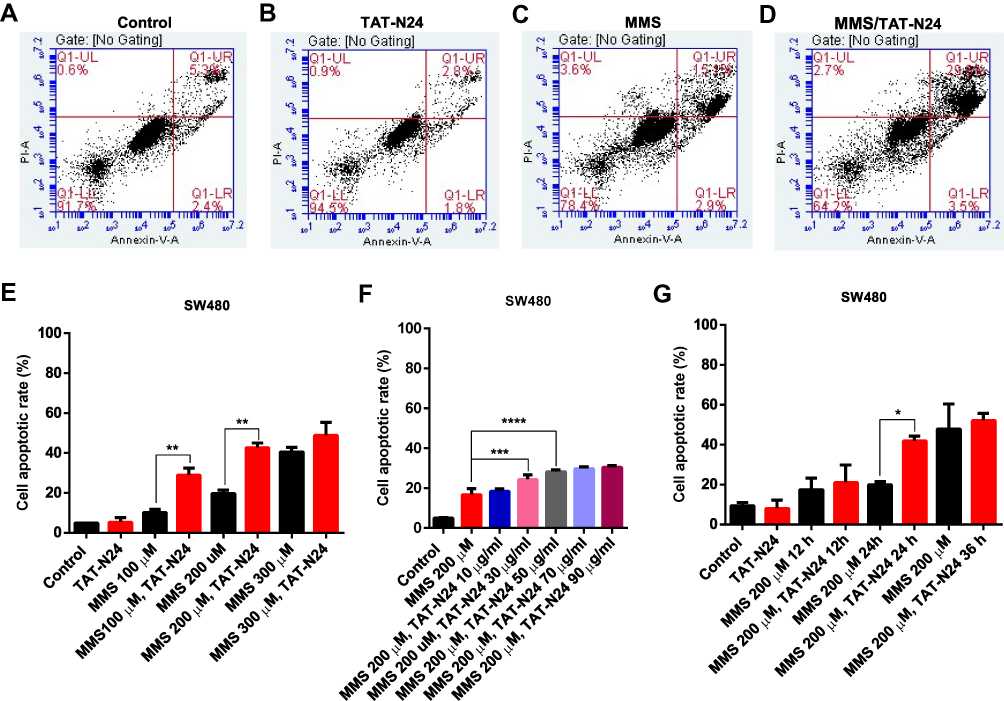 |
Figure 6 The enhancement of MMS-induced apoptosis by TAT-N24 peptide in SW480 cells. (A–D) Flow cytometric assays of MMS-treated SW480 cells in the presence of different concentrations of TAT-N24 peptide. Control: TAT. (E) Effects of N24 on MMS-induced apoptosis in the presence of different MMS concentrations. Cells were cultured for 36 hrs. Data are mean ± SEM. N = 3–5 independent experiments; **p < 0.01, compared to the control group. (F) Effects of TAT-N24 on MMS-induced apoptosis in the presence of 200 µM MMS. Data are mean ± SEM. N = 3–5 independent experiments; ***p < 0.005; ****p < 0.001, compared to the control. (G) Effects of TAT-N24 on MMS-induced apoptosis in the presence of 200 µM MMS and 50 µg/mL TAT-N24. Data are mean ± SEM. N = 3–5 independent experiments; *p < 0.05, compared with the control group. |
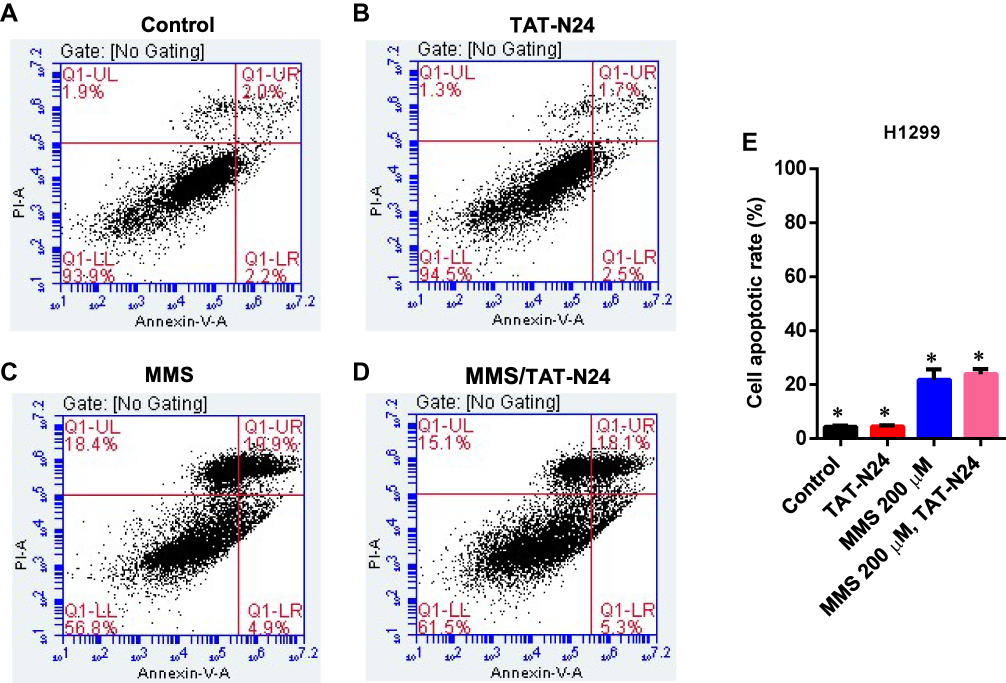 |
Figure 7 The enhancement of MMS-induced apoptosis by TAT-N24 peptide in H1299 cells. (A–D) Flow cytometric assays of MMS-treated H1299 cells in the presence of different concentrations of TAT-N24 peptide. Control: TAT. (E) Significant difference analysis on effects of TAT-N24 on MMS-induced apoptosis in the presence of 200 µM MMS and 50 µg/mL TAT-N24 in the cells cultured for 36 hrs. Data are mean ± SEM. N = 3 independent experiments; *p < 0.05 compared with the control group. |
Furthermore, we investigated gene expression of p53-dependent apoptosis. As shown in Figure 8A, when SW480 cells was treated with TAT-N24, transcription level of p55PIK was unaffected, while transcription level of p53 and genes related to p53-dependent apoptosis was decreased. When SW480 cells were treated with MMS, transcription level of p55PIK was slightly upregulated while transcription level of p53 was slightly downregulated. Transcription level of GADD45α was significantly downregulated and transcription level of AIP1 was significantly upregulated, while transcription levels of S100A9, MDM2 and Bax were unaffected, compared with the control. On the other hand, when SW480 cells were treated with TAT-N24 in addition to MMS, transcription levels of both p55PIK and p53 were significantly upregulated, and transcription levels of p53-dependent apoptosis-related genes were significantly upregulated except GADD45α.
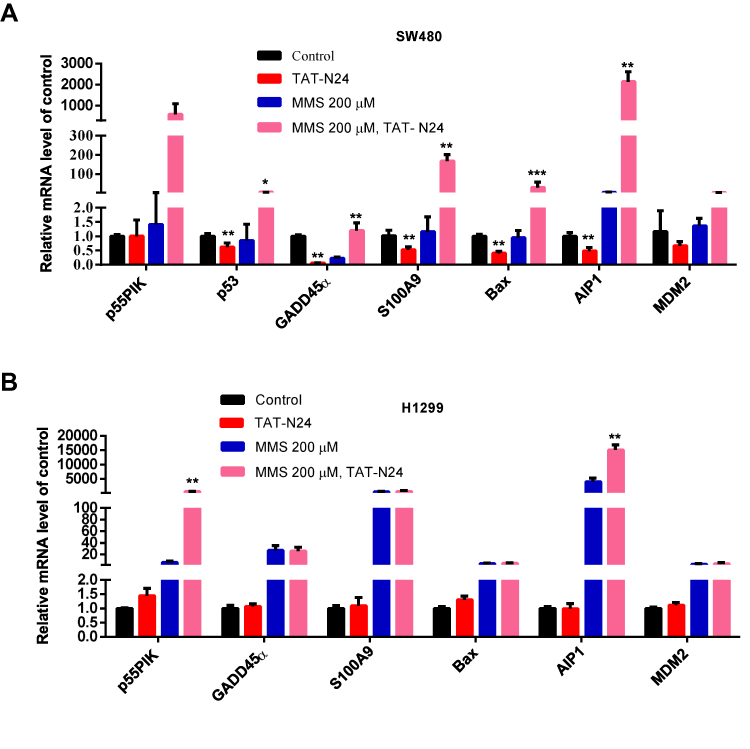 |
Figure 8 The interaction of N24 with p53 regulates downstream genes of p53 involved in apoptosis. (A) Real-time PCR assay of mRNA levels of GADD45α, S100A9, Bax, AIP1 and MDM2 in MMS-treated SW480 cells upon the addition of N24 peptide. Data are mean ± SEM. N = 3–4 independent experiments; *p < 0.05; **p < 0.01, ***p < 0.005, compared with the control group. (B) Real-time PCR assay of mRNA levels of GADD45α, S100A9, Bax, AIP1 and MDM2 in MMS-treated H1299 cells upon the addition of TAT-N24 peptide. Data are mean ± SEM. N = 3–4 independent experiments; **p < 0.01, compared with the control group. |
In contrast, transcription levels of all tested genes were not significantly changed in H1299 cells treated with TAT-N24 (Figure 8B). When H1299 cells were treated with MMS, transcriptional levels of all tested genes were significantly increased. When H1299 cells were treated with TAT-N24 in addition to MMS, transcription levels of all tested genes exhibited insignificant difference from these treated with MMS alone, except p55PIK. Taken together, these data suggest that the enhancement of p53-dependent apoptosis by TAT-N24 is dependent on the interaction of N24 with p53.
Discussion and Conclusion
As a regulatory subunit of PI3K, p55PIK is similar to p85 but has a different structure. Therefore, the function of p55PIK may not completely depend on PI3K/AKT pathway. In this study, we provide the first evidence that p55PIK regulates transcription levels of p53-dependent apoptosis genes via interacting with p53, specifically with DNA-specific binding domain. N24 domain of p55PIK can bind to DBD domain of p53 to inhibit the expression of endogenous p53. The downregulation of p55PIK increased the expression of p53. Conversely, the downregulation of p53 did not affect the expression of p55PIK. These observations suggest that p55PIK inhibits endogenous p53 in a unidirectional mode. Previously, Wang et al found that p53 binds to the promoter of miR-148b and transcriptionally upregulates miR-148b, causing the downregulation of p55PIK, tested in a variety of cell lines.24 Considering that both p55PIK and p53 are important signaling proteins in the regulation of cell proliferation and cell cycle, previous discovery and our work provided two different p53-involved signaling pathways, the p55PIK/miR-148b/p53 axis and p55PIK/p53 axis. The first axis is probably responsible for the regulation of p55PIK gene expression, whereas the second axis likely to regulate p53 functions. Therefore, ian integrated signaling map was generated by combining the data from this work with previously published data,24,39,40 indicating that the interaction of p55PIK with p53 functions as a new mode for the regulation of p53-dependent apoptosis, independent of p53/miR-148b/p55PIK axis, as depicted in Figure 9. On the other hand, the upregulation of p55PIK increased the transcription of p53-dependent apoptosis-related genes, such as GADD45α, S100A9, MDM2 and AIP1. Interestingly, the gene expression of GADD45α in p53-null cells can be regulated by p55PIK (Figure 4B), suggesting that there exists a p53-dependent pathway. Therefore, further investigation is required to understand the molecular mechanism of p55PIK-p53 interaction and beyond. Taken together, our data suggest that the regulation of p53 by p55PIK occurs at translation level, while the regulation of p55PIK by p53 occurs at transcription level.
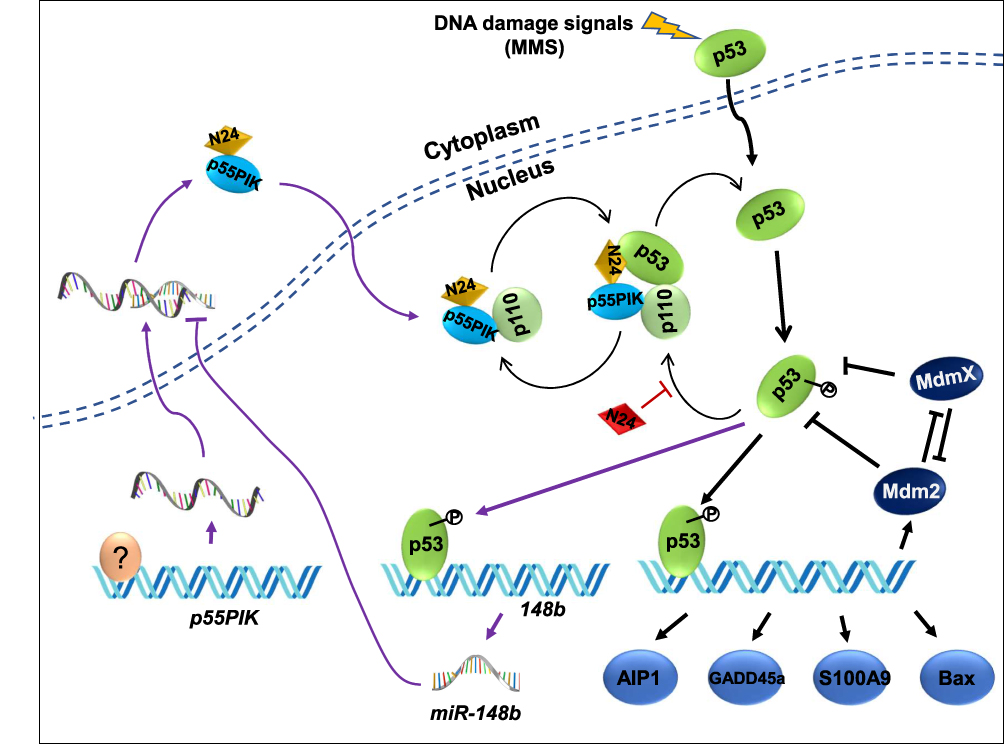 |
Figure 9 P55IPIK-p53 interaction plays an important role in cell apoptosis and N24 functions as a synergetic enhancer by inhibiting the interaction. Particularly the p53/miR-148b/p55PIK axis is integrated with p55PIK-p53 interaction forming an interactive loop. A question marker indicates transcription factors regulating p55PIK gene expression. |
Both p55PIK and p53 are co-localized in the nucleus and they are overexpressed in colon tumor cells. The role of p55PIK-p53 interaction in p53-dependent apoptosis can be illustrated using a membrane-permeable N24 peptide (TAT-N24). This synthetic N24 analogue (TAT-N24) functions as a p55PIK-specific inhibitor that can downregulate the transcription of p53-dependent apoptotic genes. Importantly, we found that TAT-N24 synergized with MMS, a DNA alkylating agent-inducing apoptosis in cancer cells, to promote cancer cell apoptosis. Up DNA damage in cells, p55PIK intrinsically regulates p53 apoptosis pathway for DNA damage or repair and N24 peptide enables to interrupt the p55PIK-p53 interaction. It is still unclear how the mutual regulation between p55PIK and p53 can affect the decision for the cells to undergo DNA damage, repair and apoptosis. On the other hand, our current study suggested that N24 peptide may also regulate the stability of p53 (Figure 9). Considering the complexities of p53 and p55PIK signaling pathways, the investigation on its anti-tumor effect will be a continuous work. Thus, an X-ray protein crystal structural investigation on p53-DBD/N24 complexes and cell biological study on the p53 signal transduction pathways are being carried out in our groups. On the other hand, since p55PIK also interacts with Rb, it is also worthy to investigate the crosstalk among PI3K/AKT, p53 and Rb pathways in regulating cell proliferation and apoptosis. Given important roles of PI3K and p53 signaling in the regulation of cell cycle, proliferation and apoptosis, p55PIK should be a promising target for cancer treatment, and synthetic N24 peptide as p55PIK-specific inhibitor may be a potential antitumor therapeutic agent.
Acknowledgments
We thank Professor Heping Cheng of the Institute of Molecular Medicine of Peking University for his permission to conduct experiments in his laboratory. We thank Professor Quan Chen from the Institute of Zoology, Chinese Academy of Sciencs for cell lines and Professor Xiaotao Duan in Academy of Military Medical Sciences for his valuable disscusion in protein profiling. This work was partially supported by the National Natural Science Foundation of China (YQH, 21603121). Dr. Xianmin Xia is currently working in Wuhan Yicheng Biotechnology Inc.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170(4):605–635. doi:10.1016/j.cell.2017.07.029
2. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15(1):7–24. doi:10.1038/nrc3860
3. Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield M. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17(1):615–675. doi:10.1146/annurev.cellbio.17.1.615
4. Pons S, Asano T, Glasheen E, et al. The structure and function of p55PIK reveal a new regulatory subunit for phosphatidylinositol 3-kinase. Mol Cell Biol. 1995;15(8):4453–4465. doi:10.1128/MCB.15.8.4453
5. Inukai K, Anai M, Van Breda E, et al. A novel 55-kDa regulatory subunit for phosphatidylinositol 3-kinase structurally similar to p55PIK Is generated by alternative splicing of the p85alpha gene. J Biol Chem. 1996;271(10):5317–5320. doi:10.1074/jbc.271.10.5317
6. Xia X, Serrero G. Multiple forms of p55PIK, a regulatory subunit of phosphoinositide 3-kinase, are generated by alternative initiation of translation. Biochem J. 1999;341(3):831–837. doi:10.1042/bj3410831
7. Hu J, Xia X, Cheng A, et al. A peptide inhibitor derived from p55PIK phosphatidylinositol 3-kinase regulatory subunit: a novel cancer therapy. Mol Cancer Ther. 2008;7(12):3719–3728. doi:10.1158/1535-7163.MCT-08-0499
8. Zhang L, Huang J, Yang N, et al. Integrative genomic analysis of phosphatidylinositol 3′-kinase family identifies PIK3R3 as a potential therapeutic target in epithelial ovarian cancer. Clin Cancer Res. 2007;13(18):5314–5321. doi:10.1158/1078-0432.CCR-06-2660
9. Wang G, Chen C, Yang R, et al. p55PIK-PI3K stimulates angiogenesis in colorectal cancer cell by activating NF-κB pathway. Angiogenesis. 2013;16(3):561–573. doi:10.1007/s10456-013-9336-y
10. Wang G, Deng Y, Cao X, et al. Blocking p55PIK signaling inhibits proliferation and induces differentiation of leukemia cells. Cell Death Differ. 2012;19(11):1870–1879. doi:10.1038/cdd.2012.70
11. Lai S, Wang G, Cao X, et al. KIT over-expression by p55PIK-PI3K leads to Imatinib-resistance in patients with gastrointestinal stromal tumors. Oncotarget. 2016;7(2):1367–1379. doi:10.18632/oncotarget.6011
12. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329–341. doi:10.1038/nrm2882
13. Deng Y, Wang J, Wang G, et al. p55PIK transcriptionally activated by MZF1 promotes colorectal cancer cell proliferation. Biomed Res Int. 2013;2013:868131. doi:10.1155/2013/868131
14. Yeates LC, Gallegos A, Kozikowski AP, Powis G. Down regulation of the expression of the p110, p85 and p55 subunits of phosphatidylinositol 3-kinase during colon cancer cell anchorage-independent growth. Anticancer Res. 1999;19(5B):4171–4176.
15. Mallory JC, Crudden G, Oliva A, Saunders C, Stromberg A, Craven RJ. A novel group of genes regulates susceptibility to antineoplastic drugs in highly tumorigenic breast cancer cells. Mol Pharmacol. 2005;68(6):1747–1756. doi:10.1124/mol.105.016519
16. Zhou Y, Li F, Tian X, Wang B, Ding M, Pang H. Changes in phosphatidylinositol 3-kinase 55 kDa gamma expression and subcellular localization may be caspase 6 dependent in paraquat-induced SH-SY5Y apoptosis. Hum Exp Toxicol. 2014;33(7):761–771. doi:10.1177/0960327113499044
17. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6(3):184–192. doi:10.1038/nrc1819
18. Sun Y-S, Thakur K, Hu F, Zhang J-G, Wei Z-J. Icariside II inhibits tumorigenesis via inhibiting AKT/Cyclin E/CDK 2 pathway and activating mitochondria-dependent pathway. Pharmacol Res. 2020;152:104616. doi:10.1016/j.phrs.2019.104616
19. Molinari F, Frattini M. Functions and regulation of the PTEN gene in colorectal cancer. Front Oncol. 2013;3:326.
20. Papa A, Pandolfi PP. The PTEN–PI3K axis in cancer. Biomolecules. 2019;9(4):153. doi:10.3390/biom9040153
21. Endersby R, Baker SJ. PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene. 2008;27(41):5416–5430. doi:10.1038/onc.2008.239
22. Venkatesan S, Lamfers ML, Dirven CM, Leenstra S. Genetic biomarkers of drug response for small-molecule therapeutics targeting the RTK/Ras/PI3K, p53 or Rb pathway in glioblastoma. CNS Oncol. 2016;5(2):77–90. doi:10.2217/cns-2015-0005
23. Abraham AG, O’Neill E. PI3K/Akt-mediated regulation of p53 in cancer. Biochem Soc Trans. 2014;42(4):798–803. doi:10.1042/BST20140070
24. Wang G, Cao X, Lai S, et al. Altered p53 regulation of miR-148b and p55PIK contributes to tumor progression in colorectal cancer. Oncogene. 2015;34(7):912–921. doi:10.1038/onc.2014.30
25. Ma M, Wang X, Chen X, et al. MicroRNA-432 targeting E2F3 and P55PIK inhibits myogenesis through PI3K/AKT/mTOR signaling pathway. RNA Biol. 2017;14(3):347–360. doi:10.1080/15476286.2017.1279786
26. Latonen L, Kurki S, Pitkänen K, Laiho M. p53 and MDM2 are regulated by PI-3-kinases on multiple levels under stress induced by UV radiation and proteasome dysfunction. Cell Signal. 2003;15(1):95–102. doi:10.1016/S0898-6568(02)00044-X
27. Lv F, Yu Y, Wang G, et al. Mechanisms by which the N-terminal 24 amino acids of the p55 regulatory subunit of phosphatidylinositol 3-kinase affect endotoxin-induced cytokine release in human keratinocytes. Mol Med Rep. 2015;11(5):3753–3759. doi:10.3892/mmr.2015.3184
28. Ye G, Sun G, Cheng Z, et al. p55PIK regulates alpha-fetoprotein expression through the NF-κB signaling pathway. Life Sci. 2017;191:104–110. doi:10.1016/j.lfs.2017.09.043
29. Xia X, Cheng A, Akinmade D, Hamburger AW. The N-terminal 24 amino acids of the p55 gamma regulatory subunit of phosphoinositide 3-kinase binds Rb and induces cell cycle arrest. Mol Cell Biol. 2003;23(5):1717–1725. doi:10.1128/MCB.23.5.1717-1725.2003
30. Wang G, Cao X, Lai S, et al. PI3K stimulates DNA synthesis and cell-cycle progression via its p55PIK regulatory subunit interaction with PCNA. Mol Cancer Ther. 2013;12(10):2100–2109. doi:10.1158/1535-7163.MCT-12-0920
31. Bechara C, Sagan S. Cell-penetrating peptides: 20years later, where do we stand? FEBS Lett. 2013;587(12):1693–1702. doi:10.1016/j.febslet.2013.04.031
32. Sun XJ, Zhao M, Yuan XH, et al. Inhibitory effect of N-terminal of p55PIK–regulatory subunit of phosphoinositide-3 kinase–on proliferation of gastric cancer cell line MGC803 and its mechanism. Ai Zheng. 2006;25(3):264–268.
33. Lv F, You W, Yu Y, Hu JB, Zhang B, Wang J. Effects of the 24 N-terminal amino acids of p55PIK on endotoxinstimulated release of inflammatory cytokines by HaCaT cells. J Huazhong Univ Sci Technolog Med Sci. 2013;33(4):587–593. doi:10.1007/s11596-013-1163-2
34. Du L, Rao G, Wang H, et al. CD44-positive cancer stem cells expressing cellular prion protein contribute to metastatic capacity in colorectal cancer. Cancer Res. 2013;73(8):2682–2694. doi:10.1158/0008-5472.CAN-12-3759
35. Wang J, Liao A-M, Thakur K, Zhang J-G, Huang J-H, Wei Z-J. Licochalcone B extracted from glycyrrhiza uralensis fisch induces apoptotic effects in human hepatoma cell HepG2. J Agric Food Chem. 2019;67(12):3341–3353. doi:10.1021/acs.jafc.9b00324
36. Kubbutat MHG, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387(6630):299–303. doi:10.1038/387299a0
37. Zhang -Y-Y, Zhang F, Zhang Y-S, et al. Mechanism of juglone-induced cell cycle arrest and apoptosis in ishikawa human endometrial cancer cells. J Agric Food Chem. 2019;67(26):7378–7389. doi:10.1021/acs.jafc.9b02759
38. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88(3):323–331. doi:10.1016/S0092-8674(00)81871-1
39. Brugarolas J, Moberg K, Boyd SD, Taya Y, Jacks T, Lees JA. Inhibition of cyclin-dependent kinase 2 by p21 is necessary for retinoblastoma protein-mediated G1 arrest after -irradiation. Proc Natl Acad Sci. 1999;96(3):1002–1007. doi:10.1073/pnas.96.3.1002
40. Mayo LD, Donner DB. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci. 2002;27(9):462–467. doi:10.1016/S0968-0004(02)02166-7
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.